

Abstract
The innate immune system senses danger signals via evolutionary conserved receptors. The nucleotide-binding domain leucine-rich repeat containing receptors (NLR) family is a group of intracellular receptors that drive a wide variety of inflammatory responses. A number of the NLR family members can form inflammasomes, which are multiprotein complexes that can activate caspase-1 and ultimately lead to the processing and secretion of interleukin (IL)-1β, IL-18 and IL-33. One of the best-studied members of the NLR family is NLRP3 for which a number of divergent activators have recently been described. These and other studies examining the NLRP3 inflammasome will be discussed in this review.
Keywords: Inflammasome, NLRP3, Caspase-1, Interleukin-1β
Introduction
The success of the mammalian immune system lies in the complementation of its two arms, the innate and adaptive systems. While the adaptive immune system has the ability to refine its response through exposure to challenges, these modifications and expansions are relatively slow to develop. Thus, were the adaptive immune system to exist independently, an invasive organism could compromise the host before the creation of a controlling immune response. The innate immune system fills this void with a more immediate response that is genetically programmed, or “hardwired”, and thus does not change in response to repeated activation. The advantage to the innate immune system is its ability to respond rapidly and consistently to adverse conditions. In addition, activation of the innate immune system acts as a direct trigger for the adaptive immune system, and is thereby critical, not only in providing early management for infections, but also for driving the subsequent acquired immune response.
Recognition by the innate immune system occurs via families of germline encoded receptors [1]. These receptors include Toll-like receptors (TLRs), nucleotide-binding domain leucine-rich repeat containing receptors (NLRs), RIG-I-like RNA helicases (RLHs) and C-type lectin receptors (CLRs). These receptors, known collectively as pattern recognition receptors (PRRs) are specific for conserved moieties associated with cellular damage (DAMPs; Danger Associated Molecular Patterns) or invading organisms (PAMPs; Pathogen Associated Molecular Patterns), which are composed of a wide and divergent group of molecules including proteins, lipids, carbohydrates and nucleic acids. Activation of these receptors ultimately leads to the production of molecules that drive the inflammatory response.
While each receptor has a unique ligand, the accessibility of these ligands to their receptors shares an invariant association with either an infecting microbe in the case of PAMPs or cellular damage for DAMPs. PAMPS are generally molecules critical for the function of the microbe; in the case of cellular damage, DAMPS are modified host molecules or endogenous molecules normally found in a sequestered location. The presence of a DAMP in an atypical location is a reliable alarm signal for cellular dysfunction. As intracellular receptors, the NLR family is in a prime location to detect danger signals and therefore play a critical role in recognizing a wide variety of molecules associated with host stress. The NLR family is found within the cytoplasm of the cell and contains 22 members including 14 NLRP members, 5 members of the NLRC subfamily, NAIP, NLRX, and CIITA [2–4]. Recent work has elucidated the conditions under which some of these family members are activated, which will be discussed below.
The NLR family
The NLR family members have similar structures with an effector domain at the amino terminus consisting of either a pyrin domain (PYD), a caspase activation and recruitment domain (CARD) or a baculovirus inhibitor of apoptosis protein repeat (BIR). NLR family members also contain a ligand binding leucine rich repeat domain (LRR) at the carboxy terminus and a central nucleotide binding and oligomerization domain (NACHT). The NLR domain structure is reminiscent of plant R proteins, which are known to be involved in pathogen recognition [5, 6]. Similarly, the NLR molecules have been shown to have a crucial role in alerting the mammalian immune system to the presence of pathogens and danger conditions.
A number of NLR molecules have been demonstrated to activate caspase-1 within multiprotein complexes termed inflammasomes. The NLRP1 (also known as NALP1 or DEFCAP), NLRP3 (also known as Cryopyrin, CIAS1 and NALP3) and NLRC4 (also known as CARD12 or IPAF) inflammasomes are amongst the best characterized and have all been shown to have clear physiological roles in vivo. However NLRP2, NLRP6, NLRP7, NLRP10 and NLRP12 have also been demonstrated to modulate caspase-1 activity in vitro although the significance of these interactions remains to be elucidated [7].
The NLRP1 inflammasome (a complex comprising of NLRP1, caspase-1, caspase-5 and the adaptor protein ASC) [8] has been revealed as the key mediator of cell death due to anthrax lethal toxin. Lethal toxin activates the inflammasome, which in turn activates caspase-1 leading to IL-1β processing and secretion and rapid cell death [9, 10]. Additionally, autoimmune and autoinflammatory diseases associated with vitiligo have been mapped to the NLRP1 genetic locus [11]. The NLRC4 inflammasome can be activated by numerous Gram-negative bacteria that possess either a type III or type IV secretion systems [12–14]. NLRC4 activation leads to the production of pro-inflammatory cytokines and similarly to NLRP1, ultimately results in cell death. The mechanism by which the NLRC4 inflammasome is activated remains controversial. NLRC4 has been shown to be activated in response to cytosolic delivery of bacterial flagellin [15, 16]. The NLRC4 inflammasome can also be activated independently of flagellin. The non-flagellated bacterium S. flexneri [17] and the P. aeruginosa mutant PAKΔfliC [14], which is deficient in flagellin, are still capable of activating caspase-1 in an NLRC4-dependent manner. Furthermore, NLRC4 may possibly interact with another cytosolic NLR, Naip5 that may play a role in NLRC4 inflammasome activation [13]. A recent study by Lightfield and colleagues suggests that activation of the NLRC4 inflammasome by flagellin requires the carboxy terminus of flagellin and is also dependent on Naip5 [18].
The NLRP3 inflammasome
One of the best characterized NLR family members is NLRP3 which is linked to the human diseases Muckle-Wells syndrome, familial cold autoinflammatory syndrome and NOMID [19–22]. These disorders share similar clinical findings of skin rashes and a spectrum of other symptoms associated with generalized inflammation. These multi-system syndromes involve the musculoskeletal system with the development of arthralgias ranging from mild to debilitating joint damage, the central nervous system leading to headaches, elevated spinal fluid pressure, cognitive deficits and sensorineural deafness, and renal amyloidosis. These disorders are associated with mutations within the NLRP3 gene leading to a constitutively active form of the molecule. This in turn results in uncontrolled IL-1β production, which is the mediator of the clinical syndrome.
NLRP3 has been shown to act in a multiprotein complex termed the NLRP3 inflammasome, composed of NLRP3, the adaptor molecule ASC as well as the cysteine protease caspase-1 [23]. Cardinal is part of the NLRP3 inflammasome complex in humans [23] although no mouse homologue of Cardinal has been identified to date and its specific function is unknown. The recruitment of the molecules to form the inflammasome is regulated by their effector domains. It is believed that activation of NLRP3 leads to the association of the PYD of NLRP3 with the PYD of ASC. This causes the CARD of ASC to associate with the CARD of caspase-1, which completes the assembly of the NLRP3 inflammasome (Figure 1). This association and resultant activation of the inflammasome leads to the activation of caspase-1, heralded by its self-cleavage from the inactive precursor pro-caspase-1 to the active form. Active caspase-1 cleaves the pro-forms of the cytokines IL-1β, IL-18 and IL-33 to their active and secreted forms (Figure 1). Recent studies have suggested caspase-1 may also have additional functions including regulation of glycolysis pathways [24] and unconventional protein secretion [25].
Figure 1.
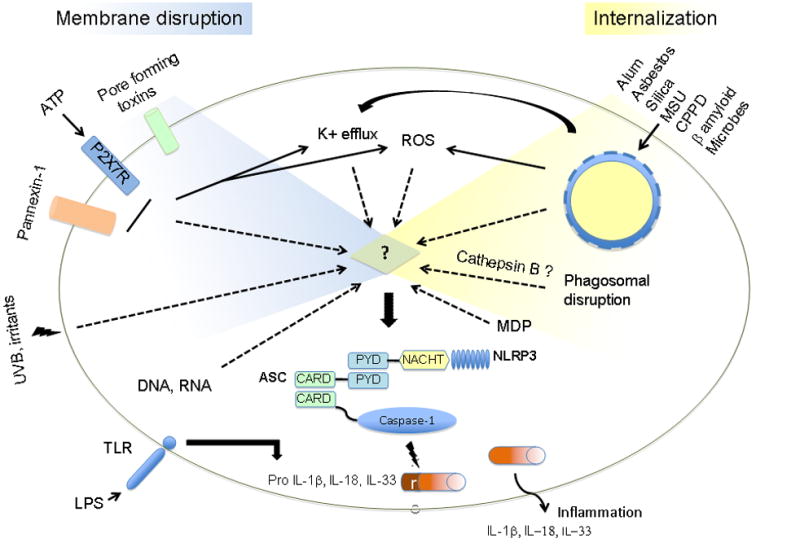
Model of NLRP3 inflammasome activation. Endogenous and exogenous danger signals engage various cytosolic events towards the assembly of a multiprotein complex composed of the cytosolic nucleotide-binding domain leucine-rich repeat containing receptor NLRP3, the adaptor molecule ASC and the cysteine protease caspase-1. Once activated, the NLRP3 inflammasome results in caspase-1 activation leading to the processing and secretion of the proinflammatory cytokines IL-1β, IL-18 and IL-33. The commonality between signals involved in pathways as distinct as cell surface mediated events and lysosomal disruption is yet to be discovered, however, a unique molecule at the crossroads of these pathways is likely involved.
The NLRP3 inflammasome can be activated in response to a number of diverse stimuli including those of microbial, endogenous and exogenous origins. However, the common pathway at which these stimuli converge to activate NLRP3 remains unclear. Microbes that can activate the NLRP3 pathway include Sendai virus [26], influenza virus [26, 27], adenovirus [28], Staphylococcus aureus [29], Listeria monocytogenes [29, 30] and Shigella flexneri [31]. Bacterial pore forming toxins can also activate this pathway; nigericin (Streptomyces hygroscopicus), maitotoxin (Gambierdiscus toxicus), aerolysin (Aeromonas hydrophila) and listeriolysin O (L. monocytogenes) have all been shown to activate caspase-1 in a NLRP3 dependent manner [29, 32]. The pannexin hemichannel has also been shown to play an important role in activation of the NLRP3 inflammasome particularly in response to ATP, through the P2X7 receptor, nigericin and maitotoxin [33, 34]. However, the role for pannexin in NLRP3 activation in response to other stimuli remains unclear. DNA, bacterial RNA and the antiviral imidazoquinoline compounds R837 and R848 can also activate the NLRP3 inflammasome independently of TLRs and RIG-I [26, 28]. Interestingly, various immune cells differentially regulate IL-1β production. Netea et al. have shown that while macrophages required a two signal process (TLR ligand and NLRP3 stimuli), monocytes only require stimulation with TLR2 or TLR4 ligands since they possess a constitutively activated form of caspase-1 and release endogenous ATP [35]. Due to their obvious role in innate responses to invading microorganism, most studies have focused on the role of NLRP3 in immune cells, especially monocytes and macrophages. However, recent studies have examined the role of the NLRP3 inflammasome in other cell types and consequently other tissues and diseases. Indeed, skin keratinocytes have been shown to mediate a NLRP3-dependent response to various exogenous sources of skin irritants such as UVB and chemicals inducing contact hypersensitivity [36–38]. The number of reports describing molecules that activate NLRP3 to generate inflammation is drastically increasing. Recently, exogenous as well as endogenous molecules have been implicated in activation of this pathway, which we will discuss in further detail below.
Particulate activators of the NLRP3 inflammasome
Uric acid
A number of recent studies have associated the activation of the NLRP3 inflammasome with exposure to crystalline particles. Uric acid was the first of these crystalline molecules demonstrated to activate the NLRP3 inflammasome [39]. Uric acid is not only the mediator of the inflammatory condition gout, but has also been postulated to be an endogenous danger signal released from dying cells [40]. Monosodium urate (MSU, uric acid) as well as calcium pyrophosphate dihydrate (CPPD), the causative agents of gout and pseudogout respectively, were both shown to activate the NLRP3 inflammasome and elicit IL-1β release from macrophages [39]. Based on these findings a pilot study to examine the efficacy of blockade of this pathway in acute gout was carried out. Ten patients with acute gout who could not tolerate or had failed standard anti-inflammatory therapies all responded to the IL-1R antagonist Anakinra [41]. These encouraging preliminary studies suggest that treatment modalities aimed at IL-1β and the NLRP3 inflammasome may be an avenue to provide effective new therapies for numerous inflammatory diseases, especially autoimmune diseases.
Acetaminophen-induced hepatotoxicity results in substantial necrosis of the liver. Inhibition of the NLRP3 inflammasome pathway either by neutralizing antibodies against IL-1β or utilizing mice deficient in components of the NLRP3 inflammasome resulted in decreased hepatic injury and improved survival in response to acetaminophen challenge [42]. Overall this study underlines the importance of the NLRP3 pathway in mediating a response to sterile inflammatory insults. However in this particular context, the role of endogenous molecules such as uric acid or ATP in the activation of the NLRP3 inflammasome have yet to be demonstrated. In a recent study by Gasse and colleagues it was shown that bleomycin-induced pulmonary injury and fibrosis were dependent on the NLRP3 inflammasome [43]. Uric acid released from injured cells played a central role in mediating pulmonary injury and inhibitors of uric acid synthesis, such as allopurinol or uricase, decreased bleomycin-induced pulmonary damage in vivo [43].
Silica and asbestos
It is known that IL-1β is a critical cytokine in the development of disease in response to inhaled silica or asbestos particles. Three recent studies [44–46] expanded this understanding and showed that asbestos and silica crystals activate the NLRP3 inflammasome to produce IL-1β. Using in vivo models of asbestosis and silicosis respectively, these groups were also able to show an amelioration of the inflammatory diseases in NLRP3 deficient mice [44–45]. This was shown for asbestosis via a decrease in the immediate inflammatory cellular influx into the lungs following inhalation of asbestos and for silicosis in impairment in the chronic inflammation and fibrosis associated with inhaled silica crystals. In addition to the study demonstrating the role of the NLRP3 inflammasome in bleomycin-induced lung injury [43], these studies show that NLRP3 plays a central role in mediating a number of pulmonary diseases culminating in pulmonary fibrosis. It will be interesting to expand these studies and determine if other fibrotic diseases are also dependent upon the activity of the NLRP3 inflammasome.
Vaccine adjuvants
Alum, an aluminum vaccine adjuvant, has been an important agent for physicians and immunologists alike for many years. It is a key ingredient for the development of an inflammatory response to an adsorbed protein, but its mechanism of action has remained unknown. Recent studies have revealed that alum acts via activation of the NLRP3 inflammasome [46–49]. Eisenbarth et al. demonstrated that the adjuvant activity of alum requires the NLRP3 inflammasome using a mouse model of allergic airway disease in which alum is administered intraperitoneally along with the protein ovalbumin [47]. Deficiency in components of the NLRP3 inflammasome resulted in abrogation of the cellular influx into the airway as well as loss of specific IgG1 antibody production to ovalbumin [47]. Independently, Li et al. [49] as well as Kool et al. [48] also demonstrated a role for NLRP3 in the generation of specific antibody responses to antigen challenge with the adjuvant alum. Interestingly Li and colleagues found that the adjuvants QuilA and chitosan also activated the NLRP3 inflammasome in vitro suggesting that particulate adjuvants may share a common mechanism of action [49]. Recently, Sharp et al. demonstrated further that enhancement of IL-1β secretion through NLRP3 in dendritic cells was likely a general property shared by particulate adjuvants [50]. In addition they demonstrated that contrary to results obtained using alum, the generation of a potent antigen-specific antibody response with PLG (poly (lactide-co-glycolide)) microparticles was not dependent on NLRP3. However, NLRP3-deficient mice presented defective antigen specific cell mediated immunity when PLG was used as an adjuvant [50].
Amyloid-β
Amyloid-β is an endogenous peptide that constitutes the main component of amyloid plaques in Alzheimer’s disease. The insoluble fibrillar form, as opposed to non-fibrillar, has also been implicated in the activation of NLRP3 [51]. The authors showed that phagocytosis of amyloid-β by mouse microglia resulted in IL-1β secretion via the NLRP3 inflammasome. Moreover, this study suggested that the presence of cytoplasmic cathepsin B, as a result of destabilization and dysfunction of amyloid-β containing lysosomes, was responsible for NLRP3 inflammasome activation. They further showed that secretion of neurotoxic mediators, such as TNF-α and nitric oxide, is associated with IL-1β production elicited via NLRP3 activation. Injection of amyloid-β resulted in recruitment of microglia in wild type mice, but the migration was impaired in mice lacking either components of the NLRP3 inflammasome or the IL-1 receptor [51]. Amyloid and structurally similar fibrillar molecules can be found in deposits at various sites of inflammation. It is intriguing to consider that these amyloid deposits may not only be a consequence of chronic inflammation but also act as endogenous danger signal themselves.
Mechanism of NLRP3 inflammasome activation
NLRP3 has been implicated in activation of innate immune responses to a wide range of seemingly unrelated danger signals. Given the structural dissimilarity of these stimuli, it is unlikely that NLRP3 is directly interacting with all of them. It is however plausible that a single ligand interacts with NLRP3 and the identified stimuli in fact serve to facilitate the release, modification or recognition of that ligand. Determining the direct ligand of NLRP3 requires understanding the common signaling pathways utilized by these divergent molecules (Figure 1).
Multiple studies reveal that phagocytosis of particulate activators of the NLRP3 inflammasome is necessary for activation [39, 44–47, 51]. However, while this activity is clearly required for particulate activators, such as MSU, silica, asbestos, and alum, it is not required for ATP, which binds the P2X7 receptor and triggers the formation of a pannexin pore. Similarly, bacterial pore forming toxins that activate the NLRP3 inflammasome also do not require internalization by the macrophage for their ability to activate caspase-1. One event that is required for NLRP3 inflammasome activation by all known activators is a shift in potassium down its concentration gradient and out of the cell [12, 52]. This movement of potassium can be prevented by equilibrating intra- and extra-cellular potassium in the cell culture media. Under these conditions, the potassium shift is abrogated as is the activation of the inflammasome in response to silica, asbestos, alum and ATP [44, 45, 47]. The exact role of the potassium efflux has yet to be explained; however the assembly of the NLRP3 inflammasome may require a low potassium environment.
Another common cellular event shared by a number of NLRP3 inflammasome activators is the generation of reactive oxygen species (ROS). It has been shown that the activation of caspase-1 in response to ATP is dependent upon the intervening step of ROS generation [53]. The critical role for ROS in stimulating caspase-1 has been expanded to other activators of the NLRP3 inflammasome via the use of inhibitors [44, 45, 52]. The source of the ROS remains unclear; studies utilizing macrophages derived from mice deficient in components of the NADPH oxidase system retained the ability to activate caspase-1 in response to NLRP3 inflammasome activators [46, 54]. These data suggest that the source of ROS required to activate the NLRP3 inflammasome may be of mitochondrial origin. A recent study by Meissner and colleagues examined the role for superoxide dismutase 1 (SOD1) in the regulation of caspase-1 activation [54]. Higher superoxide production in SOD1-deficient macrophages resulted in inhibition of caspase-1 activation by reversible oxidation and glutathionylation of the redox-sensitive cysteine residues in caspase-1 [54]. Although this study does not directly explain how ROS are required for NLRP3 inflammasome activation it does demonstrate that the redox potential of caspase-1 and possibly the NLRP3 inflammasome plays an important role in its activation.
The uptake of particulates, such as amyloid-β, silica or alum, by phagocytosis results in lysosomal disruption [46, 51]. Release of the lysosomal protease cathepsin B into the cytosol has been shown to be necessary for IL-1β release in response to a number of the phagocytosis dependent mediators [46, 51]. This intriguing role of cathepsin B, though, is not required for all the activators of NLRP3, as ATP dependent release of IL-1β is not impacted by inhibition of cathepsin B.
Conclusion
Despite extensive progress in our understanding of the assembly and activation of the NLRP3 inflammasome, a central molecule at the crossroads of each pathway implicated in NLRP3 inflammasome activation has yet to be identified. Common pathways uniting all activators remain elusive, which is not unexpected given the marked diversity of the individual activators. It is attractive to focus on the role of the phagosome and the theory of phagosome disruption as a common thread. The difficulty therein, though, is the inherent exclusion of activation by ATP, bacterial toxins, UVB or chemical irritants that occur independently from phagocytosis. The key seems to be held in one or both of the two aspects of activation which remain conserved to numerous classes of NLRP3 inflammasome activators, that is the potassium efflux and the generation of ROS. Studies on the molecular and signal transduction pathways leading to NLR activation and resulting downstream events are likely to emerge as a next step for the inflammasome research. The unfolding of this conundrum undoubtedly will be a focus of this field for a time to come.
Acknowledgments
This work was supported by National Institutes of Health grants K08 AI065517 (F.S.S.) and K08 AI067736 (S.L.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 2.Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr Opin Immunol. 2008;20:3–9. doi: 10.1016/j.coi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Dostert C, Meylan E, Tschopp J. Intracellular pattern-recognition receptors. Adv Drug Deliv Rev. 2008;60:830–40. doi: 10.1016/j.addr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He P, Shan L, Sheen J. Elicitation and suppression of microbe-associated molecular pattern-triggered immunity in plant-microbe interactions. Cell Microbiol. 2007;9:1385–96. doi: 10.1111/j.1462-5822.2007.00944.x. [DOI] [PubMed] [Google Scholar]
- 6.Altenbach D, Robatzek S. Pattern recognition receptors: from the cell surface to intracellular dynamics. Mol Plant Microbe Interact. 2007;20:1031–9. doi: 10.1094/MPMI-20-9-1031. [DOI] [PubMed] [Google Scholar]
- 7.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 8.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-1β. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 9.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–44. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 10.Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1β secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A. 2008;105:7803–8. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med. 2007;356:1216–25. doi: 10.1056/NEJMoa061592. [DOI] [PubMed] [Google Scholar]
- 12.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 13.Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol. 2006;7:318–25. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 14.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–45. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat Immunol. 2006;7:576–82. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 16.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol. 2006;7:569–75. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol. 2008;9:1171–8. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002;70:1498–506. doi: 10.1086/340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006;6:183–95. doi: 10.1038/nri1788. [DOI] [PubMed] [Google Scholar]
- 23.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 24.Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem. 2007;282:36321–29. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- 25.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 26.Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281:36560–8. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- 27.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–7. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 29.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 30.Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol. 2008;180:7558–64. doi: 10.4049/jimmunol.180.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willingham SB, Bergstralh DT, O’Connor W, Morrison AC, Taxman DJ, Duncan JA, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–59. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126:1135–45. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 33.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. Embo J. 2006;25:5071–82. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelegrin P, Surprenant A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1β release through a dye uptake-independent pathway. J Biol Chem. 2007;282:2386–94. doi: 10.1074/jbc.M610351200. [DOI] [PubMed] [Google Scholar]
- 35.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood. 2008 doi: 10.1182/blood-2008-03-146720. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, et al. Activation of the IL-1β-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 2007;127:1956–63. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- 37.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr Biol. 2007;17:1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 38.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 40.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 41.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–14. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, et al. Uric Acid is a Danger Signal Activating NALP3 Inflammasome in Lung Injury Inflammation and Fibrosis. Am J Respir Crit Care Med. 2009 doi: 10.1164/rccm.200808-1274OC. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 44.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–40. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–6. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, et al. Cutting Edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–9. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–5. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 53.Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–9. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol. 2008;9:866–72. doi: 10.1038/ni.1633. [DOI] [PubMed] [Google Scholar]