

Summary
Mdmx is a critical negative regulator of the p53 pathway that is stoichiometrically limiting in some tissues. Post-translational modification and degradation of Mdmx after DNA damage have been proposed to be essential for p53 activation. We tested this model in vivo, where critical stoichiometric relationships are preserved. We generated an Mdmx mutant mouse in which three conserved serines (S341, S367, S402) targeted by DNA damage-activated kinases were replaced by alanines to investigate whether modifications of these residues are important for Mdmx degradation and p53 activation. The mutant mice were remarkably resistant to radiation, and very susceptible to Myc-induced lymphomagenesis. These data demonstrate that Mdmx down-regulation is crucial for effective p53-mediated radiation responses and tumor suppression in vivo.
Significance
P53 function is sensitive to the levels of its negative regulators, Mdm2 and Mdmx. Cell culture studies have suggested the importance of post-translational modifications in Mdm2 and Mdmx for p53 activation, but this has not been rigorously tested in vivo. This work shows that DNA damage and activated c-Myc both require phosphorylation of Mdmx in residues targeted by the damage kinases ATM and Chk2 for robust p53 activation. Preventing Mdmx post-translational modification stabilizes this negative regulator, which mitigates p53 activation, and presumably enables c-Myc to drive tumor cells with defective genomes into cycle in vivo. The data also stress the relevance of Mdmx as a potential therapeutic target.
Introduction
The p53 tumor suppressor is a transcription factor that is activated by diverse conditions to induce or repress hundreds of protein and miRNA encoding genes (Toledo and Wahl, 2006). Its activation elicits responses ranging from reversible cell cycle arrest, to cellular senescence, cell death, blastocyst implantation, or cytokine induction to recruit the immune system (Riley et al., 2008). Its critical role in tumor suppression is evident from the tumor predisposition of mice and humans harboring germ line p53 mutations (reviewed by Varley, 2003), and its mutation and loss of heterozygosity in 50% of human cancers (Vousden and Lu, 2002).
p53 function is also compromised in tumors encoding wild type p53. This typically occurs by overexpression of either one of its two negative regulators, the oncoproteins Mdm2 and Mdmx (Marine and Jochemsen, 2005), or by loss of the tumor suppressor Arf (Eischen et al., 1999), which antagonizes Mdm2 (see below). Mice lacking either Mdm2 or Mdmx die during embryogenesis, and the embryonic lethality is rescued by p53 deletion (Jones et al., 1995; Montes de Oca Luna et al., 1995; Parant et al., 2001; Migliorini et al., 2002). This demonstrates that Mdm2 and Mdmx serve non-redundant functions in p53 regulation. Both proteins bind to the same region in the p53 transcription activation domain, and inhibit p53-dependent transcription (reviewed by Marine et al., 2007). In addition, Mdm2 functions as a RING domain E3 ubiquitin ligase to mediate p53 degradation (Fang et al., 2000; Michael and Oren, 2003). While Mdmx is also a RING domain protein that is highly homologous to Mdm2, it does not exhibit detectable E3 ligase activity (Stad et al., 2001). However, several studies have indicated that Mdmx can heterodimerize with Mdm2 and enhance or even rescue Mdm2 ligase activity (Uldrijan et al., 2007; Poyurovsky et al., 2007; Singh et al., 2007; Kawai et al., 2007). As with other RING domain E3 ligases such as BRCA1 and BARD1 (Christensen et al., 2007), it is likely that an Mdmx/Mdm2 heterodimer forms a more optimal interface to recruit one or more E2 ubiquitin transferases required to ubiquitylate p53 (Linke et al., 2008).
It has long been known that DNA double-strand breaks (DSBs) activate the ATM and Chk2 protein kinases to induce multiple post-translational modifications on p53 and Mdm2 (Shieh et al., 1997; Prives, 1998; Maya et al., 2001). In vitro data suggest that the N-terminal modifications of p53 near the Mdm2/Mdmx binding site may serve to reduce p53 interaction with these negative regulators, as well as enhancing association with the histone acetyl transferase co-activators that bind to the same region (Grossman et al., 1998). In mice, p53 post-translational modifications serve the important function of fine-tuning p53 activity in different tissues (reviewed by Toledo and Wahl, 2006).
Another important contribution to p53 activation is the DNA damage-induced, accelerated degradation of Mdm2 and Mdmx, which limits their ability to antagonize p53. We previously showed that phosphorylation of Mdm2 by DNA damage-induced kinases accelerates Mdm2 auto-degradation (Stommel and Wahl, 2004). Similarly, double-strand DNA breaks result in phosphorylation of human Mdmx at Ser342 and Ser367 by Chk2 (Okamoto et al., 2005; Chen et al., 2005; LeBron et al., 2006; Pereg et al., 2006) and Ser403 by ATM (Pereg et al., 2005), all of which are needed for efficient Mdmx degradation. These phosphorylations reduce binding of HAUSP, a ubiquitin specific protease, (Meulmeester et al., 2005), which appears to render Mdmx vulnerable to Mdm2-mediated ubiquitylation and proteasomal degradation (Kawai et al., 2003; de Graaf et al., 2003; Pan et al., 2003). Phosphorylations at Ser342 and Ser367 of Mdmx are also necessary for interaction with 14-3-3 proteins, which affect Mdmx nuclear accumulation and degradation (Okamoto et al., 2005; LeBron et al., 2006; Pereg et al., 2006; Jin et al., 2006), perhaps by serving as a steric barrier to limit HAUSP binding.
The molecular intermediates that transduce signals from activated oncogenes are beginning to be understood. As one example, c-Myc is a transcription factor that activates numerous genes including those involved in cell cycle entry and progression, DNA replication, and metabolism (reviewed by Eilers and Eisenman, 2008). While it is clear that c-Myc overexpression robustly activates p53, whether DNA damage is involved remains controversial (e.g., see Vafa et al., 2002; Maclean et al., 2003; Wade and Wahl, 2006). c-Myc overexpression results in induction of the INK4a/Arf tumor suppressor (Zindy et al., 1998). Arf binds to and inhibits Mdm2, which, as a consequence, leads to p53 activation and induction of p53-mediated downstream responses (Kamijo et al., 1998). The importance of the Arf-Mdm2-P53 pathway in Myc-induced tumorigenesis is evident in the Eμ-Myc lymphoma model where multiple copies of the c-Myc gene are overexpressed in the B cell lineage (Adams et al., 1985). Recently, an alternative mouse model (iMycEμ) was generated by inserting a single c-Myc gene into the immunoglobulin heavy-chain enhancer (Eμ) (Park et al., 2005). This mimics the human t(8;14)(q24;q32) translocation that results in activation of c-Myc in human endemic Burkitt’s lymphomas (Alitalo et al., 1987), and makes iMycEμ a better model for this disease. In both models, lymphomagenesis progresses when p53 function is abrogated by loss of Arf, overexpression of Mdm2, or mutation of p53 (Eischen et al., 1999; Park et al., 2005). Consistent with these observations, deleting either the p53 or Arf gene in Eμ-Myc mice accelerates the onset of lymphomas to the same extent (Eischen et al., 1999; Schmitt et al., 1999; Martin et al., 2006). In contrast, losing one copy of Mdm2 increases lymphoma latency (Eischen et al., 2004; Terzian et al., 2007), while overexpressing Mdm2 in Eμ-Myc mice decreases tumor latency (Wang et al., 2008). Recently, Terzian et al showed that Eμ-Myc;Mdmx+/− mice also exhibited delayed lymphomagenesis, suggesting a role for Mdmx in Myc-induced tumorigenesis (Terzian et al. 2007). However, the mechanism by which Mdmx contributes to Myc-induced tumorigenesis remains unclear.
Understanding the factors that control p53 activation requires that they be analyzed in physiologically relevant settings given the exquisite sensitivity of p53 to the levels of its negative regulators. We report the analysis of an Mdmx mutant mouse (hereafter Mdmx3SA) designed to test the importance of three highly conserved serine residues targeted by the DNA damage-activated kinases ATM (Ser402) and Chk2 (Ser341, Ser367) with regards to irradiation and c-Myc driven tumorigenicity.
Results
Generation of Mdmx3SA mice
We generated a mouse model in which the two highly conserved Chk2 sites at serines 341, 367 and the ATM site at Ser402 of Mdmx were mutated to alanines to begin to evaluate the importance of Mdmx stability regulation in different tissues in vivo. The targeting construct (Figure 1A) was electroporated into the Mdmx locus of PrmCre ES cells, allowing for direct transfer of the edited allele upon mating of transmitting males (O’Gorman et al., 1997). Allele-specific PCR (Figure S1) and Southern blotting (Figure 1B), demonstrated that 5–10% of all G418-resistant clones had undergone homologous recombination. Four independent homologous recombinant ES clones were injected into C57BL/6 blastocysts to produce Mdmx3SA chimeras, and 80% (4/5) of male chimeras transmitted the mutant allele through the germ line. Mdmx3SA homozygotes were obtained by intercrossing heterozygous mutant mice and were generated at Mendelian ratios typical for the transmission of an autosomal allele lacking adverse developmental effects (Figure 1C). Mdmx transcripts expressed in thymus isolated from Mdmx3SA mice were sequenced, and found to be wild type for all codons except the three encoding the targeted mutations (Figure 1D and 1E). The genetic background for all studies is 50% 129/Sv and 50% C57BL/6. We have followed Mdmx3SA homozygotes for more than 18 months, and have observed no obvious tumor predisposition, or differences in vitality compared to WT mice (data not shown).
Figure 1. Generation of Mdmx3SA allele and genotype analysis of Mdmx3SA.
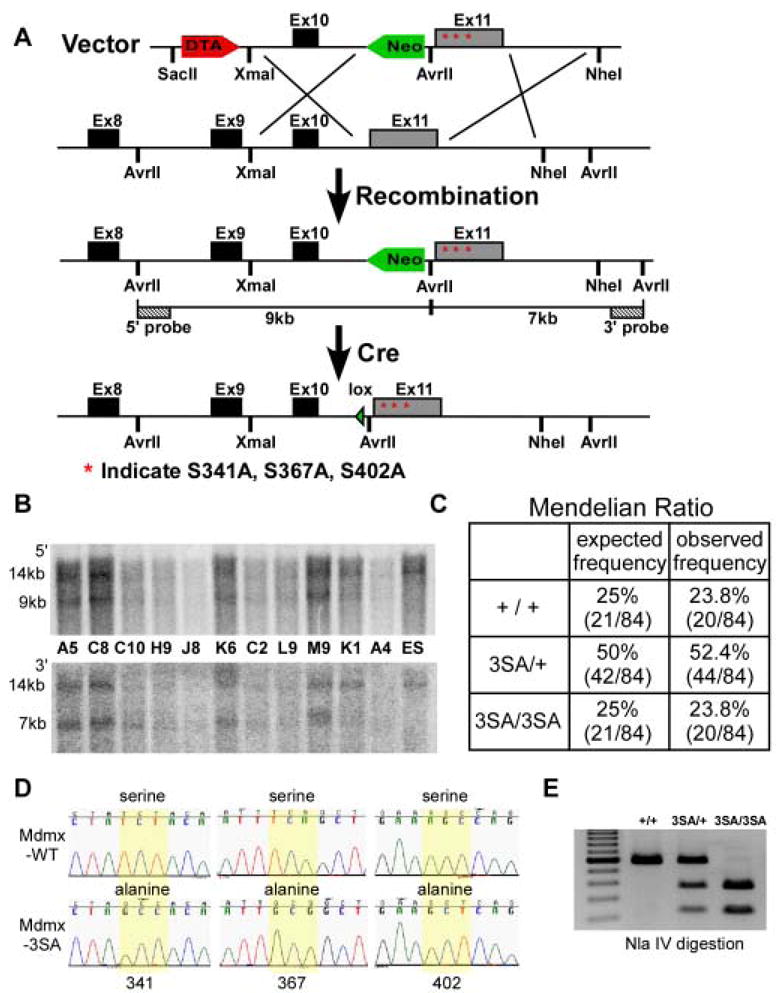
(A) Schematic representation of the 3′ end of the mouse Mdmx locus, the targeting vector and homologous recombined Mdmx3SA allele, followed by excision of loxP-flanked Neo cassette. The targeting construct contains a DTA negative selection marker, 5′ homology region with exon10, a loxP flanked Neo positive selection cassette, followed by the exon11 with Mdmx mutations indicated as stars and 5′ homology region. The insertion of Neo introduces an additional Avr II site that was used for Southern analysis with the indicated external 5′ and 3′ probes. Cre expression in the male germline subsequently allows excision of Neo. (B) Southern blot analysis of targeted ES clones. The 14kb band represents the Avr II fragments from the wild type allele using both 5′ and 3′ probes. The recombined allele generates a 9kb fragment detected by the 5′ probe and a 7kb fragment detected by the 3′ probe. (C) The expected and observed Mendelian ratios from Mdmx3SA heterozygote breeding. (D) Sequences of Mdmx transcript expressed from MdmxWT and Mdmx3SA thymus. Yellow highlight shows that the codons that encode S341, S367 and S402 of the wild type Mdmx are mutated in the Mdmx3SA allele. (E) PCR Genotype of the Mdmx3SA mutant. PCR amplifies the region surrounding the mutations to produce a 579bp fragment. The introduction of mutations generates an Nla IV site that results in 366bp and 213bp fragments when PCR product is digested with Nla IV.
MEFs were isolated from embryonic day 13.5 (E13.5) to determine the biochemical consequences of DNA damage on cells expressing MdmxWT or Mdmx3SA. Using an antibody specifically recognizing phospho-S367 of Mdmx, we detected robust accumulation of a doublet band in MdmxWT MEFs when cells were treated with the radiomimetic agent neocarzinostatin (NCS) plus proteasome inhibitors (PI) (Figure 2A). Note, a weak signal of phospho-S367 was detected in MdmxWT cells in the absence of exogenous damage (Figure 2A, lane 1), indicating that an intrinsic stress occurring during cell culture activates kinases that target this phosphorylation site in Mdmx. The slower migrating band likely derives from the phosphorylation at Ser402 (Pereg et al., 2005) since an anti-phospho-S402 antibody only recognizes the slower migrating band in NCS plus PI treated MdmxWT MEFs (Figure 2B). Importantly, the single Mdmx band observed in Mdmx3SA MEFs did not react with any of phospho-specific antibodies (Figure 2A, 2B), which confirms the sequencing results (Figure 1D), and further shows that one or more of the three mutated serines engender the damage-induced mobility shift of the endogenous gene product.
Figure 2. Mdmx3SA mutations impair DNA damage-induced Mdmx degradation.
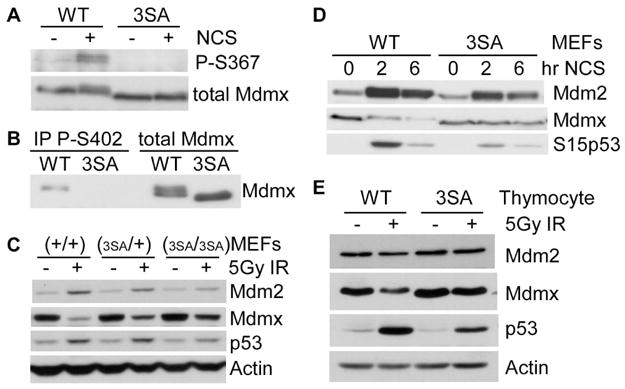
(A) The MdmxWT and Mdmx3SA MEFs were pre-incubated with MG132 (10 μM) for 30 minutes, followed by 2 hrs of NCS (200 ng/ml) incubation. Lysates were immunoprecipitated with anti-Mdmx antibody, and blotted with anti-phospho-S367 antibody. (B) MEFs were treated as described in (A). Lysates were immunoprecipitated with anti-phospho-S402 Mdmx antibody, and blotted with anti-Mdmx antibody. (C) MEFs were irradiated with 5Gy of γ-irradiation. Four hours later, cells were lysed for western analysis. (D) MEFs were treated with 200ng/ml of NCS for 0, 2 and 6 hrs. Lysates were analyzed by western blotting using antibodies against Mdm2, Mdmx and phospho-S15 of p53. (E) Thymocytes were irradiated with 5Gy of γ-irradiation. Four hours later, cells were lysed for western analysis.
The Mdmx3SA mutations impair DNA damage-induced Mdmx degradation and p53 stabilization
Previous studies in human cell lines suggest that Mdmx is a stable protein in cells not exposed to exogenous challenges. Upon DNA damage, Mdmx is modified by kinases and degraded at the proteasome (Okamoto et al., 2005; Pereg et al., 2005; Chen et al., 2005; LeBron et al., 2006; Pereg et al., 2006). Consistent with previous studies, induction of DNA damage reduced the abundance of MdmxWT in MEFs (Figure 2C, 2D), which can be prevented by incubating cells with proteasome inhibitors (Figure S2). This indicates accelerated MdmxWT degradation following DNA damage. By contrast, the 3SA mutations significantly reduced DNA damage-induced Mdmx degradation (Figure 2C, D). No further accumulation of Mdmx3SA in the presence of proteasome inhibitors was observed (Figure S2), indicating that Mdmx3SA was quite resistant to DNA damage–induced degradation. The mutations stabilize Mdmx in other cell types as well, since Mdmx3SA in thymocytes was significantly more resistant to damage-induced degradation than MdmxWT after irradiation (Figure 2E). We also noticed that Mdmx3SA is slightly more abundant than MdmxWT in the cells that were not exposed to exogenous DNA damage. Concomitant with a more stable Mdmx3SA after DNA damage, we observed lower accumulation of p53 in Mdmx3SA cells, consistent with a model in which Mdmx phosphorylation at these residues decreases its stability while increasing p53 stability (Pereg et al., 2005 and Figure S2).
The effect of Mdmx phosphorylations on protein interactions
We next determined whether mutation of endogenous S341, 367 and 402 would affect 14-3-3 binding after DNA damage, as this has been suggested to contribute to Mdmx stability control (Okamoto et al., 2005; Pereg et al., 2006; LeBron et al., 2006) Cell lysates from MdmxWT, Mdmx3SA and p53−/−;Mdmx−/− (2KO) MEFs were incubated with GST-14-3-3 recombinant proteins; glutathione beads were then used to isolate proteins that bound to GST-14-3-3. Figure 3A shows that MdmxWT bound to 14-3-3 when cells were treated with NCS. In the absence of NCS, a small portion of MdmxWT was also pulled down by GST-14-3-3 (Figure 3A, lane 3, compare Mdmx to Total Mdmx), which is consistent with the observation in Figure 2A that Mdmx is phosphorylated at low levels under our cell culture conditions. On the other hand, no interaction was detected with Mdmx3SA. Subcellular fractionation analyses indicated that Mdmx was predominantly cytoplasmic in untreated MdmxWT and Mdmx3SA MEFs (Figure S3). Notably, the abundance of nuclear Mdmx3SA was not affected by DNA damage (Figure S3).
Figure 3. Analyses of protein interactions with Mdmx3SA in response to DNA damage.

(A) Cell lysates from MdmxWT, Mdmx3SA, 2KO (p53−/−;Mdmx−/−) MEFs either untreated or treated with NCS (200 ng/ml) in the presence of MG132 (10 μM) for 2 hrs were incubated with glutathione beads pre-bound with GST-14-3-3ε proteins. Bead-bound proteins were eluted and analyzed by western blotting. (B) MEFs were either untreated or treated with MG132 (10 μM) or treated with MG132 plus NCS (200 ng/ml) and lysed 2hrs after. Cell lysates were immunoprecipitated with anti-Mdmx antibody, blotted with anti-Mdm2, anti-Mdmx and anti-p53 antibodies. Anti-phospho-S15 of p53 antibody was used as a control for detecting the DNA damage signal induced by NCS.
We next investigated whether S341, S367, and S402 phosphorylations affect binding to Mdm2, as Mdmx degradation upon DNA damage is mediated by Mdm2 (Kawai et al., 2003). MdmxWT and Mdmx3SA MEFs were left untreated, or exposed to NCS in the presence of proteasome inhibitors to maintain sufficient Mdmx to enable detection, and then immunoprecipitated with Mdmx-specific antibodies (Figure 3B) The data demonstrate that similar amounts of Mdm2 were co-immunoprecipitated with MdmxWT and Mdmx3SA following DNA damage. Mdm2 was also co-immunoprecipitated with Mdmx3SA following treatment with the topoisomerase I inhibitor, etoposide (Figure S4). These data lead us to conclude that DNA damage-induced modifications on Mdmx have little effect on its interaction with Mdm2 when both proteins are expressed at endogenous levels.
p53 activity is reduced in Mdmx3SA MEFs and thymocytes
Mdmx antagonizes p53 –induced transcription by binding the p53 transactivation domain (reviewed by Marine and Jochemsen, 2005; Toledo and Wahl, 2006). Since Mdmx degradation tracks with p53 activation (Wang et al., 2007), and Mdmx3SA is more stable than MdmxWT after DNA damage, we predicted that Mdmx3SA would compromise p53 activation. We therefore compared the expression of the p53 target genes p21 and puma in MEFs and thymocytes. Under normal cell culture conditions, p53 abundance in MdmxWT and Mdmx3SA cells was not significantly different (Figure S9). However, we did observe a lower basal level of p21 transcription in the Mdmx3SA expressing cells (Figure 4A). Upon DNA damage, the fold induction of p21 was slightly lower than that in MdmxWT cells (Figure S5), but the absolute level of induced p21 transcript was 50% of the wild type level (Figure 4A). The reduced transcription of p21 was manifested functionally by a reduced efficiency of DNA damage-induced arrest in MEFs (Figure 4B, 4C).
Figure 4. p53 activity is reduced in Mdmx3SA mutant.

(A) MEFs were treated with 500ng/ml of NCS or 5 week-old mice were exposed to 5Gy of γ-irradiation. Six hours after NCS incubation or 4 hrs after irradiation, cells or thymus removed from MdmxWT (WT) and Mdmx3SA (3SA) mice were lysed for RNA isolation. p21 and puma gene expression was analyzed by RT-qPCR. Results were normalized to hprt mRNA. (B) Cell cycle arrest analysis. MEFs were either untreated or exposed to 5Gy or 10Gy of γ-irradiation. Twenty-three hours after irradiation, 10μM of BrdU was added in the cell culture for one hour before FACS analysis. BrdU-positive cells that represent cells in S-phase during BrdU incorporation were quantified in (C). Error bars represent +/−SD from three independent experiments. (D) Analysis of apoptosis in vivo. Mice were untreated or exposed to 5Gy of γ-irradiation. Four hours later thymocytes were isolated from mice and stained with Annexin V-FITC for FACS analysis. Annexin V positive cells that represent apoptotic cells were quantified in (E). Error bars represent +/−SD from three independent experiments. (F) Apoptosis in splenocytes and thymocytes. Mice were treated as described in (D). Splenocytes and thymocytes were isolated and analyzed for the expression of active form of Caspase-3 by western blotting with high exposure (hi) or low exposure (lo).
Induction of apoptosis is a second critical function of p53, and induction of the pro-apoptotic gene puma is required for damage-induced thymic apoptosis (Erlacher et al., 2005). Consistently, we observed a lower basal level of puma transcription in Mdmx3SA thymocytes and a slightly lower fold-induction of puma after exposing mice to 5Gy of whole-body irradiation (Figure 4A and S5). Importantly, we observed a significantly reduced abundance of puma transcripts in Mdmx3SA thymocytes, and this correlated with a significant decrease in the number of Mdmx3SA apoptotic thymocytes after ionizing radiation (Figure 4D–F). To summarize, although the fold activation of p53-dependent transactivation is similar in MdmxWT and Mdmx3SA tissues (Figure S5), the absolute mRNA level following stress is much lower in Mdmx3SA expressing cells. We suggest that this absolute level (rather than the fold change compared to basal expression) is what ultimately dictates the biological response.
Mdmx3SA mice show increased resistance to death induced by ionizing radiation
Irradiation of mice at 10Gy induces a p53-dependent hematopoietic syndrome characterized by severe bone marrow depletion, which results in death within 2 weeks of exposure (Komarova et al., 2004). We therefore determined whether changes in Mdmx stability affect the p53 pathway and its impact on radio-sensitivity in the hematopoietic compartment by comparing the effect of 10Gy irradiation in MdmxWT and Mdmx3SA mice.
As expected, a single dose of 10Gy whole body ionizing irradiation in MdmxWT 129/sv/C57BL6 mice resulted in death of all animals within 18 days (Figure 5A). Surprisingly, 60% of the irradiated Mdmx3SA homozygous mice lived at least 100 days post-irradiation (Figure 5A). We performed complete pathological analysis on a subset of MdmxWT and Mdmx3SA mice 13 days post-irradiation at the time when the first death was observed in MdmxWT mice. Complete blood count (CBC) revealed severe leucopenia, anemia and thrombocytopenia in MdmxWT mice 13 days post-irradiation, while Mdmx3SA mice exhibited only moderate leucopenia and thrombocytopenia and mild anemia (Figure 5C). Histopathological analysis did not show lesions in any other tissues, including the gastrointestinal tract in either MdmxWT or Mdmx3SA mice (Figure 5B). This suggests that Mdmx3SA mitigates p53 activity and confers radiation resistance predominantly by affecting the hematopoietic compartment in vivo. Consistent with these data, the majority of Mdmx3SA mice show long-term survival following irradiation at a dose that is lethal to WT animals. A subset of these mice exhibited moderate anemia, leucopenia and thrombocytopenia, which allowed them to survive the acute mortality phase, but died of anemia 20 to 60 days post irradiation (data not shown).
Figure 5. Mdmx3SA mice are resistant to a lethal dose of ionizing irradiation.
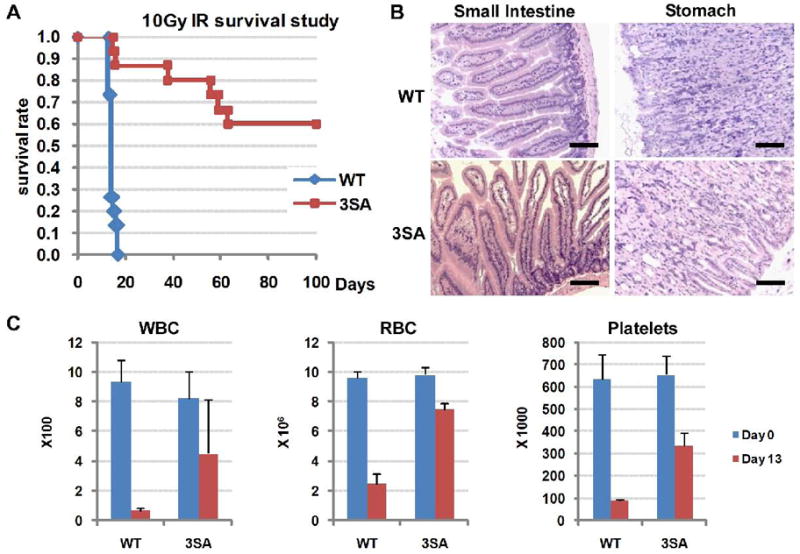
(A) Kaplan-Meier survival curves of fifteen age-matched MdmxWT (WT, n=15) and Mdmx3SA homozygote (3SA, n=15) mice following exposure to 10Gy of whole-body γ-irradiation. (B) Histology of the gastrointestinal tract from MdmxWT and Mdmx3SA mice showing no lesions associated with irradiation (H&E, Bar = 100 μm). (C) Complete blood count analysis of white blood cells (WBC), red blood cells (RBC) and platelets from MdmxWT and Mdmx3SA mice, 13 days post 10Gy of whole-body γ-irradiation. Error bars are +/−SD from 3 animals.
μ
Acceleration of c-Myc-induced lymphomagenesis in Mdmx3SA mice
Loss of one mdmx allele enhances p53-dependent apoptosis and significantly delays the onset of c-Myc-induced lymphoma (Terzian et al., 2007). However, whether modification of Mdmx affects Myc-induced tumorigenesis is unclear. We have begun to evaluate whether Mdmx phosphorylation modulates oncogene-induced tumorigenicity using the iMycEμ lymphoma model. These mice develop late onset B-cell lymphomas between 6 and 21 months of age, which often exhibit changes in the Arf-Mdm2-p53 tumor suppression pathway (Park et al., 2005). Mdmx3SA mice were bred with heterozygous iMycEμ mice to generate iMycEμ;+/+ and iMycEμ;3SA/3SA mice. Figure 6A shows that Mdmx3SA significantly accelerated lymphomagenesis with 50% survival at 3 months of age. Tumors arising from iMycEμ;3SA/3SA mice presented with severe generalized lymphadenopathy and hepatosplenomegaly (Figure 6B). Histologically, most lymphomas were diffuse, B220+IgM+IgD+ (data not shown), high grade B-cell lymphoma with intermediate sized cells, moderate to marked anisocytosis/anisokaryosis, high mitotic index (8–16/High Power Field) and abundant apoptotic cells and large macrophages containing tingible bodies producing a typical “starry-sky” effect (Figure 6C). Tumors were highly infiltrative in several tissues including liver, kidneys, lungs, meninges, eyes and, occasionally, the gastrointestinal tract (Figure S6). Consistent with previous studies, iMycEμ;+/+ mice started to die by about 6 months of age with 50% survival about a year of age (Figure 6A; Park et al., 2005). On gross examination, iMycEμ;+/+ mice presented with marked hepatosplenomegaly and variable lymphadenopathy. Histologically, lymphomas had similar features as those observed in the iMycEμ;3SA/3SA mice (data not shown).
Figure 6. Myc-induced tumorigenesis is accelerated in Mdmx3SA mice.

(A) Kaplan-Meier survival curves of iMycEμ;+/+ (n=14) and iMycEμ;3SA/3SA (n=18) mice. (B) Early onset lymphoma in a 113 day-old iMycEμ;3SA/3SA mouse: dissection reveals severe enlargement of the spleen, liver and most lymph nodes (notably, the mesenteric, cervical, brachial, axillary, mediastinal, pancreatic, renal, inguinal and lumbar nodes). (C) High grade B-cell lymphoma in iMycEμ;3SA/3SA mice. Upper panel: the lymph node architecture is replaced with solid sheets of intermediate size lymphocytes admixed with apoptotic tumor cells and large macrophages containing tingible bodies producing a typical “starry-sky” effect (H&E, Bar = 50 μm). Lower panel: Intermediate size lymphocytes with moderate to marked anisocytosis/anisokaryosis, high mitotic index, abnormal mitosis and apoptotic tumor cells (H&E, Bar = 20 μm). (D) Apoptosis analysis. Splenocytes were isolated from 8 week-old littermate mice and stained with Annexin V-FITC for FACS analysis. Annexin V positive cells were quantified. Error bars represent +/−SD from analysis. Eight week-old littermate mice that contain were i.p. injected with 100mg/kg of BrdU. Five hours later, and stained with anti-BrdU antibody for FACS analysis represent cells in S-phase were quantified. Error bars animals. (F) Levels of Mdm2, Mdmx, p53, Arf assessed extracts from iMycEμ;+/+, iMycEμ;3SA/+ and iMycEμ;3SA/3SA mice. p53−/− Mdm2−/− MEFs(2KO) and thymus irradiated in vivo (IR-thymus) were panel shows the p53 status in those tumors (w indicates mutated p53). Tumor 1 from iMycEμ;+/+ and Tumor 2 from red contain p53 mutations at R172H (CGC → CAC) and K129T (AAG → ACG) respectively (see Figure S8 for details).
We further investigated whether changes in Myc-induced apoptosis or cell proliferation accompanied the early onset of lymphomas in iMycEμ;3SA/3SA mice. Using age-matched pre-tumor littermates, we observed similar increases in apoptosis in iMycEμ;+/+ and iMycEμ;3SA/3SA splenocytes when compared to their littermates that did not contain iMycEμ (Figure 6D). However, we did detect significantly more iMycEμ;3SA/3SA cells in the S-phase (Figure 6E). Additionally, we detected phosphorylation of histone H2AX in the iMycEμ;3SA/3SA splenocytes and phosphorylation of Ser15 of p53 in iMycEμ induced tumors, consistent with an ongoing DNA damage response and activated ATM kinase pathway in these cells (Figure S7).
Tumors arising from overexpression of c-Myc require inactivation of p53 pathway by either p53 mutation, loss of Arf, or overexpression of Mdm2 (Eischen et al., 1999). We therefore determined whether Mdmx3SA was sufficient to blunt the p53 response to c-Myc without additional disruption of Arf-Mdm2-p53 pathway. We observed increased levels of p53 and Arf in tumors arising from iMycEμ;+/+ and in one iMycEμ;3SA/+ mouse (Figure 6F). This is consistent with the presence of one or more mutant p53 alleles, since p53 negatively regulates Arf (Stott et al., 1998). Indeed, one out of three iMycEμ;+/+ tumors and one out of four iMycEμ;3SA/+ tumors that were analyzed contained p53 mutations at R172H and K129T, respectively (Figure S8). By contrast, no p53 mutations were detected in 10 iMycEμ;3SA/3SA tumors that were analyzed (data not shown).
Discussion
Mdmx turnover is a critical component for p53 activation in vivo
The Mdmx3SA mouse model presented here establishes post-translational modification of Mdmx as an important component of the damage- and oncogene-responsive mechanisms that activate p53 (Figure 7). Reducing Mdmx degradation compromised p53 transactivation of its target genes, and induction of cell cycle arrest and apoptosis. The Mdmx3SA mice exhibit significantly reduced sensitivity of hematopoietic cells to radiation, which renders Mdmx3SA mice extraordinarily resistant to 10Gy of ionizing radiation (a myeloablative, lethal dose for wild type mice). Conversely, Mdmx3SA mice expressing the iMycEμ oncogene die rapidly of widely disseminated lymphoma (see below). Thus, Mdmx regulation is involved in cellular responses to both DNA damage and some oncogenic stimuli.
Figure 7. Mechanism for the role of Mdmx modification in DNA damage and oncogenic signaling pathway.
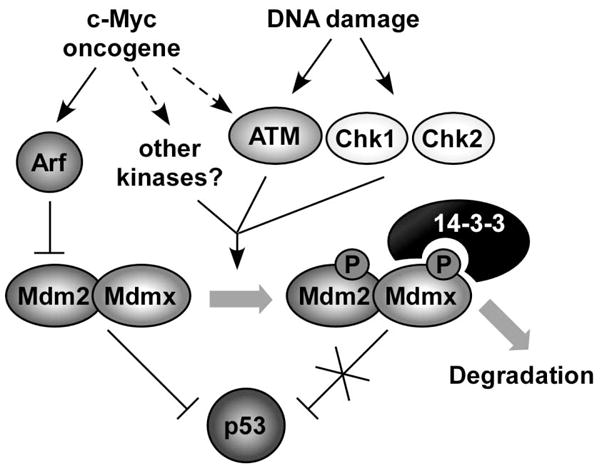
Phosphorylations of Mdmx can be initiated by either DNA damage signaling or by activated oncogenes. Phosphorylations of Mdmx modulate its interaction with other proteins, such as 14-3-3, the protein analyzed in this study. Interaction of Mdmx and 14-3-3 is required for accelerated Mdmx and Mdm2 degradation after DNA damage, probably by limiting Mdm2/Mdmx interaction with the deubiquitylase HAUSP (see text). Preferential degradation of Mdm2 and Mdmx reduces their ability to interact with and antagonize p53 function, leading to p53 activation. The 3SA mutations introduced in this study prevent 14-3-3 interaction after DNA damage, leading to Mdmx-Mdm2 stabilization, and a corresponding increased ability to degrade p53.
Our data show that Mdmx3SA is slightly more abundant than MdmxWT before DNA damage, suggesting the mutations render Mdmx more stable during homeostatic growth. We infer that a low level of stress during cell culture conditions is responsible for this effect. This is supported by the detection of phospho-S367 in the absence of exogenous DNA damage (Figure 2A, lane 1). This ‘constitutive’ Mdmx phosphorylation is likely responsible for degradation of MdmxWT, and thus contributes to the increase in basal levels of Mdmx3SA. This may lead to the lower basal p53 activity in Mdmx3SA, since Mdmx3SA is now refractory to intrinsic damage induced modification/degradation and can limit p53-dependent transactivation. Nevertheless, the functional consequence of this difference in p53 basal activity appears subtle for the following reasons: (1) Mdmx3SA exhibits cell cycle (Figure 4C) and apoptosis (Figure 4E) profiles identical to MdmxWT in the absence of exogenous DNA damage and (2) there is no increased spontaneous tumor formation in Mdmx3SA mice.
Interestingly, the fold induction of p53 target genes in response to DNA damage was similar between MdmxWT and Mdmx3SA cells, although the absolute level of p21 and puma transcripts was lower in Mdmx3SA. The basal levels of p53 are similar in Mdmx3SA and MdmxWT, but damage-induced p53 levels are consistently lower in Mdmx3SA (Figure S9). It is important to note that the net activity of p53 results from a combination of its abundance, the interaction with and stability of its negative regulators, and the effects of stress on other factors involved in p53 activation such as chromatin modifiers, etc. Note that after stress, Mdm2 level is typically lower in the mutant due to reduced gene transcription. Thus, in the Mdmx3SA cells after DNA damage, there is generally less Mdm2 and more Mdmx than in MdmxWT cells. This results in somewhat lower p53 accumulation. The NET of all these effects is the observed 2-3-fold decrease in activity which is apparently sufficient to prevent the biological responses required for extensive hematopoietic system apoptosis or tumor suppression. These data emphasize the importance of Mdmx regulation for setting basal and stress-induced p53 activity, and the associated biological responses.
We previously showed that p53-Mdm2 or p53-Mdmx interactions subsequent to DNA damage are readily detectable as long as proteasome inhibitors are present (Stommel and Wahl, 2004; Wang et al., 2007), implying that p53 post-translational modifications alone are insufficient to prevent interactions with Mdm2/Mdmx. We suggest that, in addition to p53 phosphorylation, damage-induced post-translational Mdm2/Mdmx modifications are required to enable their accelerated degradation, and maximal activation of p53. Previous in vitro data showed that when Mdmx is phosphorylated, HAUSP’s affinity for Mdmx is reduced, and 14-3-3′s affinity is increased (Meulmeester et al., 2005; Okamoto et al., 2005; Pereg et al., 2006; LeBron et al., 2006). Consistent with this, data herein indicate that 14-3-3 binds only to phosphorylated Mdmx. Both basal and damage-induced associations of Mdmx with 14-3-3 (Figure 3) were abolished by the 3SA mutation. This indicates that cultured cells experience sufficient stress to induce a damage response. Additionally, it may provide an explanation for the slightly elevated basal levels of Mdmx3SA compared to MdmxWT cells, since Mdmx3SA is refractory to the endogenous damage signaling and can no longer bind 14-4-3. Loss of interaction between Mdmx3SA and 14-3-3 leads us to speculate that competition between 14-3-3 and HAUSP for Mdmx binding after DNA damage contributes to Mdmx stability control. Furthermore, we infer that 14-3-3 binding is not required for Mdmx nuclear localization per se, but that it can regulate the abundance of nuclear Mdmx following DNA damage as indicated by previous studies (LeBron et al., 2006; Pereg et al., 2006). Whether this is due to perturbed nuclear degradation of Mdmx, or is a consequence of altered nucleo-cytoplasmic shuttling remains to be determined.
Mdmx regulation contributes to oncogene-mediated activation of p53
Oncogene overexpression can activate p53 by several mechanisms (reviewed by Halazonetis et al., 2008). As one example, expressing c-Myc under the control of the Eμ immunoglobulin enhancer induces Arf expression, which antagonizes Mdm2-mediated p53 ubiquitylation, resulting in p53 activation (Zindy et al., 1998). It has been suggested that Arf does not interact with Mdmx directly (Wang et al., 2001; Li et al., 2002; Laurie et al., 2006). However, Mdmx and Mdm2 form a heterodimer and Mdmx haploinsufficiency clearly delays the onset of Myc-induced lymphomas (Terzian et al., 2007), implicating the involvement of Mdmx in the Arf-Mdm2-P53 tumor suppressor pathway. Note that most of the tumors arising from both Eμ-Myc mice and iMycEμ mice exhibit loss of Arf, p53 mutation or Mdm2 overexpression (Eischen et al., 1999; Park et al., 2005). By contrast, we found no evidence of these changes in tumors arising from iMycEμ;3SA/3SA mice. Although Mdm2 levels appear slightly elevated in some tumors arising from iMycEμ;3SA/3SA mice compared to iMycEμ;+/+ mice, this probably reflects the lower expression of Mdm2 in WT tumors that have suffered p53 mutations. Together these data indicate that alterations in Mdmx (either modification or stability) are sufficient to mitigate p53 function in Myc-induced lymphomagenesis.
Tumors isolated from patients often exhibit markers of double stranded DNA damage, including phospho-ATM, phospho-Chk2 and phospho-histone H2AX (Bartkova et al., 2005), suggesting that ongoing DNA damage is a hallmark of many tumor types. Our attempts to study Mdmx phosphorylation in Myc-induced tumors by immunofluorescence were unsuccessful due to the difficulty of preserving the unstable phosphorylated form of Mdmx in vivo and current technical limitations of the available antibodies (data not shown). However, we did detect phospho-H2AX in iMycEμ;3SA/3SA splenocytes, and phosphorylation of S15p53 in iMycEμ induced tumors, suggesting the presence of double strand breaks and an activated ATM kinase pathway. Consistent with this inference, Myc-induced lymphomagenesis is accelerated in ATM deficient mice (Maclean et al., 2007). This provides a strong link between the DNA damage response and Myc-induced lymphomas. Whether the damage is triggered through Myc induced replication stress or occurs as a consequence of lymphomagenesis itself remains unclear. It has also been suggested that Ser367 of Mdmx is a target of the Akt kinase (Lopez-Pajares, et al., 2008), and therefore we cannot exclude the possibility that members of other kinase families may also mediate the phosphorylations of Mdmx in response to Myc activation. Interestingly, we observed similar levels of Myc-induced apoptosis in iMycEμ;+/+ and iMycEμ;3SA/3SA mice, while increased proliferation of lymphocytes was detected in iMycEμ;3SA/3SA mice. Thus Mdmx3SA may blunt the p53 response to activated Myc, allowing more cells to enter S-phase. This could explain why lymphomas develop with a shorter latency, and appear more aggressive in the Mdmx3SA background. Indeed, in the absence of iMycEμ, no increased spontaneous tumorigenesis was observed in Mdmx3SA mice. This indicates that accelerated lymphomagenesis in iMycEμ;3SA/3SA mice is via mitigation of an oncogene-induced anti-proliferative response, rather than simple attenuation of basal p53 activity. In summary, our data clearly implicate Mdmx phosphorylation(s) as a determinant of oncogene-induced tumorigenesis.
Mdmx modification in tumorigenesis and as a therapeutic target
Our findings indicate Mdmx modification is another example of the much-wielded “double-edged sword”. On the one hand, phosphorylation of Mdmx appears to be critical for Mdmx degradation and tumor suppression. Thus, the function of factors controlling this process may be attenuated in cancer, and their (re)activation may be beneficial for cancer therapy. Conversely, damage-induced modification of Mdmx enhances p53-dependent radio-sensitivity. In this case, inhibition of Mdmx phosphorylation may be a strategy to prevent cytotoxicity in normal tissues exposed to environmental or chemotherapeutic genotoxins. A precedent for this approach is the use of the p53 inhibitor, pifithrin-α, as a radio-protective agent in vivo (Burdelya et al., 2006). Although challenging, finding ways to promote selective Mdmx degradation in tumor versus normal cells may increase the efficacy of current p53-targeted therapies.
Experimental procedures
Targeting construct
Fragments of mouse genomic Mdmx gene, extending from intron 9 through the 3′ UTR, were cloned into pACYC177 (NEB) using ET recombination (Genebridge). Mutations that substitute serines 341, 367 and 402 with alanines were introduced by site-directed mutagenesis using the QuikChange kit (Stratagene). A positive selection marker, the neomycin (Neo) resistance gene, driven by the PGK promoter and flanked by loxP sites, was introduced into an Avr II site upstream of exon 11. A negative selection marker, Diptheria Toxin A (DTA), driven by the PGK promoter was inserted into the targeting vector upstream of exon 10. The exons, the intron-exon boundary, Neo and DTA were sequenced to ensure no unexpected mutations were introduced during the cloning.
Generating and genotyping Mdmx3SA mice
The targeting vector was linearized with Not I and electroporated into PrmCre 129/Sv ES cells before being selected for neomycin resistance. Homologous recombination was confirmed by PCR screening and Southern blotting with Avr II digested genomic DNA. Southern probes were generated from PCR amplification in the regions external to the targeting vector. Four independent ES clones containing the targeted Mdmx3SA allele were injected into C57BL/6 blastocysts, which were then implanted into pseudo-pregnant females. Germ-line transmission was confirmed by breeding chimeras with C57BL/6 mice. The offspring were PCR genotyped using a primer set (3SA-fw: AAT TTG TTC AGG TCT CAG GTT GG and 3SA-rv: CAT AAG CTA CAC GGC TTC AAG AC), followed by Nla IV digestion. Heterozygous mutant mice were interbred to produce homozygous mutant mice. All animals described are on a mixed 129/Sv X C57BL/6 background.
Protein analysis
Cells or tissues were lysed in RIPA buffer (50mM Tris-HCl, 150 mM NaCl, 0.1 % SDS, 0.5% Na. Deoxycholate, 1% NP40) or in Giordano buffer (50 mM Tris- HCl, pH 7.4, 250 mM NaCl, 0.1% Triton X-100, 5 mM EDTA). Protein extracts were either analyzed by direct western blotting, or by IP/western. Mdmx IPs were performed with mixture of rabbit polyclonal antibodies, p55 and p56 (Ramos et al., 2001). Blots were probed with antibodies specific for Mdm2 (4B2 and 2A10), Mdmx (MX-82, Sigma), P-S367Mdmx and P-S402Mdmx (both gifts from Yosef Shiloh), p53 (1C12, Cell Signaling Technologies), P-S15p53 (Cell Signaling Technologies), Arf (Ab80, Abcam), active Caspase3 (cleaved-Caspase3 Asp175, Cell Signaling Technologies), PARP (Santa Cruz), Tubulin (Sigma) and Actin (Sigma).
Mdmx/14-3-3 interaction
Protein extracts made in Giordano buffer were incubated for 2 hours with gluthatione beads to which bacterially produced GST-14-3-3ε was coupled. Subsequently, beads were washed four times with Giordano buffer, bound proteins eluted with sample buffer and analyzed by western blotting.
QPCR
RNA was isolated and subjected to real time quantitative PCR as described previously (Krummel et al., 2005).
Flow cytometry
For cell cycle analysis, MEFs were irradiated with 5 or 10Gy γ-IR and incubated for 23 hours, followed by 1hr incubation with 10 μM of BrdU (Sigma). For cell proliferation analysis in the iMycEμ study, mice were i.p. injected with 100mg/ml of BrdU (Sigma). Five hours later, splenocytes were isolated. Cells were then fixed in 70% ethanol, and stained with FITC anti-BrdU and propidium iodide. For apoptosis assay, mice were exposed to 5Gy of whole-body irradiation. Four hours later, mice were sacrificed and thymocytes or splenocytes were isolated and stained with Annexin-V-FITC (BD Biosciences) for analysis. For both cell cycle and apoptosis, the cells were sorted using a Becton-Dickinson FACScan machine and data were analyzed using CellQuest Pro.
Animal studies
All procedures were approved by the Salk IACUC. For the irradiation study, age and gender-matched mice were exposed to a whole body irradiation with a 60Co γ-irradiator. For the tumorigenicity study, Mdmx3SA/+ mice were bred with iMycEμ mice to generate iMycEμ;+/+, iMycEμ;3SA/+ and iMycEμ;3SA/3SA mice. Animals were observed daily for signs of morbidity and tumor development. Tumor development was monitored by palpation of the abdomen and cervical, axillary and inguinal regions. Mice were euthanized humanely when moribund or when reaching tumor specific endpoints. Portions of all tumors and major organs were fixed in formalin for histopathology and snap frozen for protein and RNA extraction.
Supplementary Material
Acknowledgments
We thank Tomiko Velasquez and Yelena Dayn for their help with generating Mdmx3SA mice, Dr. Inder Verma’s lab for providing iMycEμ mice, Beejal Ruparelia and Daphne Chen for mouse colony assistance and genotyping and Rose Rodewald for technical assistance. We also wish to thank Dr. Yosef Shiloh for the phospho-Mdmx specific antibodies and Ari Elson for GST-14-3-3 expression vectors. This work was supported by grants from NCI (grants CA100845, CA61449 to G. M. W.) and the Cancer Center Core Grant for Core Facility support (grant 5 P30 CA014195).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams J, Harris A, Pinkert C, Corcoran L, Alexander W, Cory S, Palmiter R, Brinster R. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318(6046):533–8. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Alitalo K, Koskinen P, Mäkelä TP, Saksela K, Sistonen L, Winqvist R. myc oncogenes: activation and amplification. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1987;907(1):1. doi: 10.1016/0304-419x(87)90016-3. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Burdelya LG, Komarova EA, Hill JE, Browder T, Tararova ND, Mavrakis L, DiCorleto PE, Folkman J, Gudkov AV. Inhibition of p53 Response in Tumor Stroma Improves Efficacy of Anticancer Treatment by Increasing Antiangiogenic Effects of Chemotherapy and Radiotherapy in Mice. Cancer Res. 2006;66(19):9356–9361. doi: 10.1158/0008-5472.CAN-06-1223. [DOI] [PubMed] [Google Scholar]
- Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. Embo J. 2005;24(19):3411–22. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol. 2007;14(10):941. doi: 10.1038/nsmb1295. [DOI] [PubMed] [Google Scholar]
- de Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG. Hdmx protein stability is regulated by the ubiquitin ligase activity of Mdm2. J Biol Chem. 2003;278(40):38315–24. doi: 10.1074/jbc.M213034200. [DOI] [PubMed] [Google Scholar]
- Eilers M, Eisenman RN. Myc’s broad reach. Genes & Development. 2008;22(20):2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13(20):2658–69. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Alt JR, Wang P. Loss of one allele of ARF rescues Mdm2 haploinsufficiency effects on apoptosis and lymphoma development. Oncogene. 2004;23(55):8931. doi: 10.1038/sj.onc.1208052. [DOI] [PubMed] [Google Scholar]
- Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, Adams JM, Strasser A, Villunger A. BH3-only proteins Puma and Bim are rate-limiting for {gamma}-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106(13):4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275(12):8945–51. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2(4):405–15. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science. 2008;319(5868):1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Jin YDM, Lu SZ, Xu Y, Luo Z, Zhao Y, Lu H. 14-3-3gamma binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. EMBO. 2006;25(6):1207–18. doi: 10.1038/sj.emboj.7601010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378(6553):206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(14):8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278(46):45946–53. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING Domain-Mediated Interaction Is a Requirement for MDM2’s E3 Ligase Activity. Cancer Res. 2007;67(13):6026–6030. doi: 10.1158/0008-5472.CAN-07-1313. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, Gudkov AV. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23(19):3265. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci U S A. 2005;102(29):10188–93. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444(7115):61. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- LeBron C, Chen L, Gilkes DM, Chen J. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. EMBO. 2006;25:1196–1206. doi: 10.1038/sj.emboj.7601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chen L, Chen J. DNA Damage Induces MDMX Nuclear Translocation by p53-Dependent and -Independent Mechanisms. Mol Cell Biol. 2002;22(21):7562–7571. doi: 10.1128/MCB.22.21.7562-7571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2//MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15(5):841. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- Lopez-Pajares V, Kim MM, Yuan Z-M. Phosphorylation of MDMX Mediated by Akt Leads to Stabilization and Induces 14-3-3 Binding. J Biol Chem. 2008;283(20):13707–13713. doi: 10.1074/jbc.M710030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc Augments Gamma Irradiation-Induced Apoptosis by Suppressing Bcl-XL. Mol Cell Biol. 2003;23(20):7256–7270. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean KH, Kastan MB, Cleveland JL. Atm Deficiency Affects Both Apoptosis and Proliferation to Augment Myc-Induced Lymphomagenesis. Mol Cancer Res. 2007;5(7):705–711. doi: 10.1158/1541-7786.MCR-07-0058. [DOI] [PubMed] [Google Scholar]
- Marine JC, Jochemsen AG. Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun. 2005;331(3):750–60. doi: 10.1016/j.bbrc.2005.03.151. [DOI] [PubMed] [Google Scholar]
- Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13(6):927. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- Marine JCW, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci. 2007;120(3):371–378. doi: 10.1242/jcs.03362. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the Therapeutic Efficacy of p53 Restoration in Tumors. 2006;127(7):1323. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15(9):1067–77. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulmeester EMM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Molecular Cell. 2005;18(5):565–76. doi: 10.1016/j.molcel.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13(1):49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22(15):5527–38. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378(6553):203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- O’Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci U S A. 1997;94(26):14602–7. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, Teunisse A, Migliorini D, Kitabayashi I, Marine J-C, et al. DNA Damage-Induced Phosphorylation of MdmX at Serine 367 Activates p53 by Targeting MdmX for Mdm2-Dependent Degradation. Mol Cell Biol. 2005;25(21):9608–9620. doi: 10.1128/MCB.25.21.9608-9620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Chen J. MDM2 Promotes Ubiquitination and Degradation of MDMX. Mol Cell Biol. 2003;23(15):5113–21. doi: 10.1128/MCB.23.15.5113-5121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29(1):92–5. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, Tae Chung S, Torrey TA, Cheung WC, Polakiewicz RD, et al. Insertion of c-Myc into Igh Induces B-Cell and Plasma-Cell Neoplasms in Mice. Cancer Res. 2005;65(4):1306–1315. doi: 10.1158/0008-5472.CAN-04-0268. [DOI] [PubMed] [Google Scholar]
- Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, Teunisse AF, Lehmann WD, Jochemsen AG, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci U S A. 2005;102(14):5056–61. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereg Y, Lam S, Teunisse A, Biton S, Meulmeester E, Mittelman L, Buscemi G, Okamoto K, Taya Y, Shiloh Y, et al. Differential Roles of ATM- and Chk2-Mediated Phosphorylations of Hdmx in Response to DNA Damage. Mol Cell Biol. 2006;26(18):6819–6831. doi: 10.1128/MCB.00562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, Pavletich N, Prives C. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO. 2007;26(1):90–101. doi: 10.1038/sj.emboj.7601465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prives C. Signaling to p53: breaking the MDM2-p53 circuit. Cell. 1998;95(1):5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- Ramos YFM, Stad R, Attema J, Peltenburg LTC, van der Eb AJ, Jochemsen AG. Aberrant Expression of HDMX Proteins in Tumor Cells Correlates with Wild-Type p53. Cancer Res. 2001;61(5):1839–1842. [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402.27. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW. INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes & Development. 1999;13(20):2670–2677. doi: 10.1101/gad.13.20.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Singh RK, Iyappan S, Scheffner M. Hetero-oligomerization with MdmX Rescues the Ubiquitin/Nedd8 Ligase Activity of RING Finger Mutants of Mdm2. J Biol Chem. 2007;282(15):10901–10907. doi: 10.1074/jbc.M610879200. [DOI] [PubMed] [Google Scholar]
- Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP, Saville MK, Jochemsen AG. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001;2(11):1029–34. doi: 10.1093/embo-reports/kve227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004;23(7):1547–56. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott F, Bates S, James M, McConnell B, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden K, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17(17):5001–14. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in Tumorigenesis and Development. Mol Cell Biol. 2007;27(15):5479–5485. doi: 10.1128/MCB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6(12):909. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. EMBO J. 2007;26(1):102–12. doi: 10.1038/sj.emboj.7601469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9(5):1031–44. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Human Mutation. 2003;21(3):313–320. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- Wade M, Wahl GM. c-Myc, genome instability, and tumorigenesis: the devil is in the details. Curr Top Microbiol Immunol. 2006;302:169–203. doi: 10.1007/3-540-32952-8_7. [DOI] [PubMed] [Google Scholar]
- Wang X, Arooz T, Siu WY, Chiu CH, Lau A, Yamashita K, Poon RY. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001;490(3):202–8. doi: 10.1016/s0014-5793(01)02124-x. [DOI] [PubMed] [Google Scholar]
- Wang YV, Wade M, Wong E, Li YC, Rodewald LW, Wahl GM. Quantitative analyses reveal the importance of regulated Hdmx degradation for P53 activation. Proceedings of the National Academy of Sciences. 2007;104(30):12365–12370. doi: 10.1073/pnas.0701497104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Lushnikova T, Odvody J, Greiner TC, Jones SN, Eischen CM. Elevated Mdm2 expression induces chromosomal instability and confers a survival and growth advantage to B cells. Oncogene. 2008;27(11):1590. doi: 10.1038/sj.onc.1210788. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Development. 1998;12(15):2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.