

Abstract
Indocyanine Green (ICG) is a fluorescent probe used in various optically-mediated diagnostic and therapeutic applications. However, utility of ICG remains limited by its unstable optical properties and non-specific localization. We have encapsulated ICG within electrostatically-assembled mesocapsules (MCs) to explore its potential for targeted optical diagnosis and therapy. In this study, we investigate how the surface coating and size of the MCs influences ICG's biodistribution in vivo. ICG was administered intravenously to Swiss Webster mice as a free solution or encapsulated within either 100 nm diameter MCs coated with dextran; 500 nm diameter MCs coated with dextran; or 100 nm diameter MCs coated with 10 nm ferromagnetic iron oxide nanoparticles, themselves coated with polyethylene glycol. ICG was extracted from harvested blood and organs at various times and its amount quantified with fluorescence measurements. MCs containing ICG accumulated in organs of the reticuloendothelial system, namely the liver and spleen, as well as the lungs. The circulation kinetics of ICG remained unaffected by encapsulation; however, the deposition within organs other than the liver suggests a different biodistribution mechanism. Results suggest that the capsules' coating influences their biodistribution to a greater extent than their size. The MC encapsulation system allows for delivery of ICG to organs other than the liver, enabling the potential development of new optical imaging and therapeutic strategies.
Keywords: drug delivery, fluorescence imaging, ICG, nanoparticles, optical therapy, tumor imaging, vascular imaging
Introduction
Indocyanine Green (ICG) is a tricarbocyanine dye with substantial absorption and fluorescence in the near infrared wavelength (NIR) region1, where water and other intrinsic biomolecules display negligible absorption or autofluorescence. It has minimal toxicity, and the FDA has approved its use for ocular imaging, measurements of cardiac output, and assessment of hepatic function 2-5. These characteristics have motivated investigations into ICG's utility as a sensitizer for photothermal therapy of tumors and cutaneous blood vessels 6-8, photodynamic therapy (PDT) 9-13, tissue welding 14, estimation of burn depth 15, as well as imaging vascularization and bloodflow within breast tumors, skin, and the brain 16-19.
After a bolus injection of ICG solution, its total concentration within the bloodstream decreases at a reportedly bi-exponential rate with a half-life on the order of τ1/2 = 2-4 minutes 20. Previous studies indicate that ICG binds readily to albumin and high-density lipoproteins (HDLs) in blood plasma such as alpha-1 lipoprotein 21, 22. While the mechanism of ICG removal from blood is not clearly understood, it is thought that ICG is eliminated from the general circulation by the liver and excreted in unmetabolized form into the bile 20, 22. ICG's optical properties are sensitive to a variety of factors, including solvent, concentration, and temperature 23-26. It also undergoes a variety of physicochemical degradations in aqueous media over time, which reduces its absorption and fluorescence 26.
The utility of ICG for laser-mediated diagnostic and therapeutic applications could be vastly improved if ICG could preferentially accumulate in significant quantities at a targeted site. Procedures such as PDT and photothermal therapy generally require exposure times that can last several minutes. Typically for these applications, it is necessary to wait several hours or even days after a systemic injection to accumulate significant concentrations of the photosensitive drug at the target site.
A number of technologies have been developed and characterized for drug delivery purposes over the past three decades, such as polymeric nanoparticles (NPs), hollow liposomes, and vesicles 27 28. These materials allow for efficient delivery of therapeutic agents to targeted sites while protecting them from degradation and clearance. Investigators have reported on the use of NP systems to control the distribution and kinetics of therapeutic and diagnostic reagents 29. Often times, for NP-mediated tumor diagnosis and therapy, the accumulation of reagent at the tumor site is dependent on the enhanced permeability and retention (EPR) effect, in which the particles collect within tumor tissue after escaping from the bloodstream through leaky blood vessels 30. To enhance targeting of cancerous cells, NP surfaces are conjugated with antibodies or ligands that recognize receptors expressed in large amounts along the membranes of the targeted cells 29, 31.
ICG's limiting factors, described above, have motivated us and other investigators to develop encapsulation systems to stabilize its optical properties and control its circulation kinetics. A number of nanometer-sized encapsulation systems have been developed to protect ICG from degradation and potentially modulate its in vivo distribution and circulation kinetics, including poly (D,L-lactic-co-glycolic acid) (PLGA) nanoparticles, sol-gel matrices (PEBBLEs), phospholipid emulsions, and di-block copolymer micelles 32-37. We have previously reported on the synthesis and characterization of a sub-micron diameter capsule system, assembled by electrostatic interaction, to encapsulate ICG 38, 39. These capsules consist of a spherical, positively charged polymer-salt aggregate core, and can be coated with a variety of species, including various negatively charged NPs and biocompatible polymers such as dextran and polyethylene glycol (PEG). We refer to the capsules as mesocapsule constructs (MCs) to reflect the fact that the capsule diameters generally lie in an intermediate range, slightly larger than particles that are conventionally referred to as nanoparticles (< 100 nm) and smaller than microparticles (> 1 μm) 40.
Compared with other established drug delivery vehicle techonologies, our MCs differ both structurally and in the preparation process. Unlike polymeric nanoparticles, which are generally comprised of a solid colloidal polymer matrix in which a therapeutic drug is embedded throughout the particle 41, the ICG within our MCs remains confined primarily to the polymer-salt aggregate core by electrostatic attraction. Vesicular technologies, such as nanocapsules, micelles, and liposomes, also confine the active drug within their aqueous or oily cores. However, the surfaces of vesicles are generally a continuous polymeric or lipid membrane28, 29. Our MCs, by comparison, can be coated with either a porous shell of nanoparticles or a continuous film of polymer. A relatively new class of multilayered capsules also exists that can be synthesized using layer-by-layer assembly42. These capsules are created by repeatedly assembling NPs onto a colloidal polymer core and subsequently removing the core by calcination or exposure to solvents. By comparison, the MCs' outer shell is attached rapidly during a single step, and no calcination is required.
Among the primary advantages of our capsule system is its simple, fast, and inexpensive synthesis. Many other encapsulating technologies require considerable time, exposure to harsh environmental conditions that can damage the cargo, and involve several intermediate steps such as emulsion, spray drying, nozzle generation, and freeze-drying41. By comparison, synthesis of our MCs can be performed within minutes in aqueous solution near neutral pH and room temperature, and using inexpensive precursor materials38. Other advantages of the MC system include controllability of size (with mean diameters ranging from 100 nm to 5 μm), high loading efficiency (up to 85%), their ability to protect ICG from thermal and photo-damage, the ability to coat them with different materials, and functionalize the capsule surface with antibodies or other localizing agents 39, 43. Additionally, as liposomes and vesicles are known to dissolve in the presence of surface-active agents or collapse when dried 28, the MCs offer greater physical stability over these technologies, maintaining their integrity when dried or suspended in certain other solvents. With easily adjustable parameters such as size, coating material, and loading content, these MCs offer tremendous versatility, allowing for their use in a variety of optically mediated procedures and multimodal imaging methods. We have demonstrated previously that our MCs have potential for both photothermal therapy as well as in vivo fluorescence imaging44, 45.
In this study, we evaluate the biodistribution kinetics of various formulations of MCs containing ICG in an in vivo mouse animal model. We assess the influence of the coating material and capsule size on the tissue distribution in healthy Swiss Webster mice. Specifically, the biodistribution kinetics of 100 nm MCs coated either with dextran or magnetic NPs coated with PEG 46 are investigated. These coating materials were selected for their biocompatibility, ability to be functionalized with targeting ligands, consistency with our previous studies (in the case of the dextran-coated MCs) 43, 44 and, in the case of the NP-coated MCs, in efforts toward multimodal imaging strategies (as these MCs are both optically and magnetically active)47. To assess the influence of capsule size on MC distribution, we also determine the distribution of dextran-coated MCs with a diameter of 500 nm. The circulation of these MC systems within the bloodstream and its uptake by various organs are compared with that of freely dissolved ICG in mice. This work signifies the first evaluation of biodistribution properties of our MC systems in an animal model.
Experimental Section
MC preparation and characterization
Indocyanine green was purchased in powder form under the product name Cardiogreen from Sigma. Stock solutions of ICG (1 mg/ml) were prepared by dissolving the powder form into sterile, triple-distilled water, covered with foil, and refrigerated at 4° C to minimize degradation.
Disodium phosphate (Na2HPO47H2O), poly(allylamine hydrochloride) (PAH, 70kDa), and dextran (40 kDa) were purchased from Sigma-Aldrich (USA) in powder form and dissolved in deionized water (Diamond Nanopure water system) to the desired concentration.
Figure 1 illustrates the synthesis process for ICG-containing MCs. The process requires three simple steps similar to those described in previous reports 38, 39, 43-45. Briefly, we first prepared spherical, charged salt-bridged polymer aggregates by lightly mixing pre-cooled PAH in de-ionized water with Na2HPO4 (pH = 7.4) at room temperature for 15 seconds in a 15 ml centrifuge tube. The diameters of MCs were varied by adjusting the aggregate aging time as well as the R ratio, defined as the ratio of the total negative charge from salt anions to the total positive charge. The amount of charge was determined by the product of valence charge and total mass of each specie 38. In order to create 100 nm MCs, the concentrations of PAH and Na2HPO4 salt were 1 mg/ml (0.5 ml) and 0.025 M (3 ml), respectively, and the volumes were controlled such that the R ratio was maintained at 3. Alternatively, we created 500 nm MCs by using concentrations of 2 mg/ml for PAH and 0.01 M for Na2HPO4 to maintain the R ratio at 6.
Figure 1.
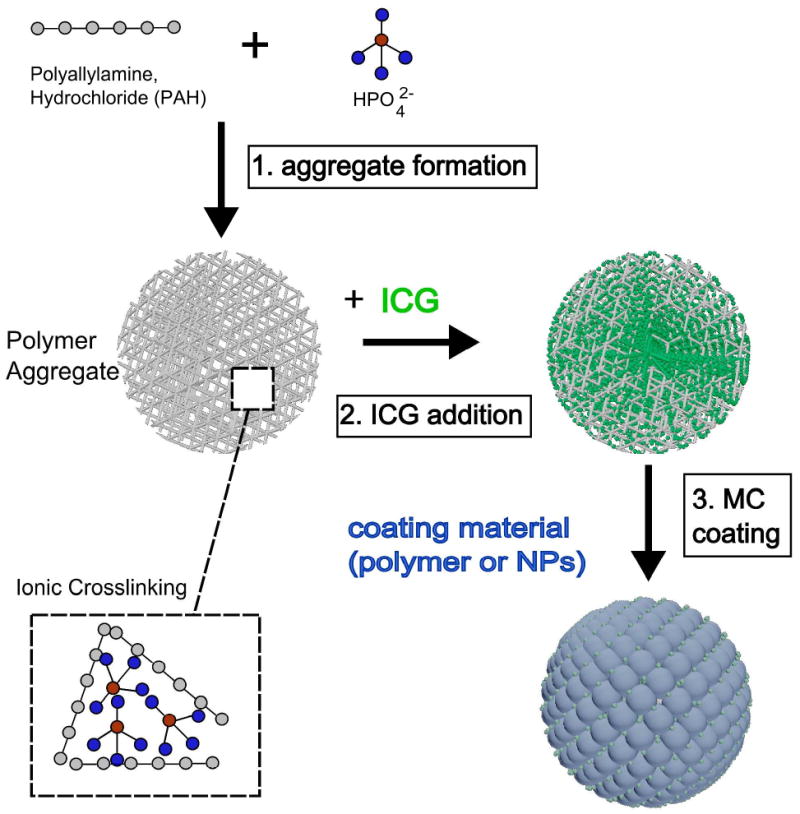
3-step synthesis process for self assembled MCs containing ICG. All steps were performed within minutes at 4 °C. In the final step, the coating material is a negatively charged specie, including polymers such as functionalized dextran, PEG, or NPs with negative surface charge.
Next, we immediately mixed the aggregate suspension with aqueous ICG solution (either 1 mg/ml or 0.4 mg/ml, 3 ml). The negatively charged ICG molecules penetrated into the MC aggregates composed of the polymer-salt matrix and remained there by electrostatic and hydrophobic interactions.
In the final step, the MC aggregates containing ICG were coated with either NPs or polymer. Specifically, 10 minutes after the ICG was added to the MC aggregates, either an additional 1.5 ml of dextran (2 mg/ml in H2O), or PEG-coated magnetite (Fe3O4) NPs (10 nm diameter, generously donated), was added to the mixture and mixed for 30 seconds. The negatively charged polymer molecules of the PEG-NPs formed a coating around the surface of the aggregate through electrostatic interactions. Dextran polymer does not contain charged groups. The mechanism by which it coats the aggregate core is currently under investigation; however, it is likely attributable to numerous hydrogen bonding interactions between the dextran polymer and the aggregate core molecules 48, 49. The MCs were aged for 2 hours at 4 °C, at which point they were centrifuged at 7,000 rpm for 90 min and re-suspended in phosphate-buffered saline (PBS) by sonication. The process was repeated twice to remove excess precursors. The resultant MCs remained suspended in PBS and stored in the dark at 4° C until performing the in vivo experiments.
In our experiments, we utilized three MC types: (1) 100 nm MCs containing ICG prepared at aqueous ICG concentration of 500 μg/ml, and coated with dextran (referred to as the D100 system); (2) 500 nm MCs containing ICG prepared at the aqueous ICG concentration of 200 μg/ml, and coated with dextran (referred to as the D500 system); and (3) 100 nm MCs containing ICG prepared at the aqueous ICG concentration of 500 μg/ml, and coated with PEG-coated 10 nm magnetite NPs (referred to as the Fe100 MCs).
We opted to use a lower ICG concentration for the D500 MCs when we observed that these MCs aggregated at the aqueous ICG concentration of 500 μg/ml. We found that diluting the concentration to 200 μg/ml greatly reduced the likelihood of aggregation. While we did not perform a systematic study to investigate this effect, it is likely that the aggregation occurred before the addition of the dextran coating. This explanation is consistent with previous studies, which have demonstrated that aggregate cores tend to grow in size by coalescence with aging time, leading to larger, more polydisperse MCs 38, 50. Additionally, the reports show that aggregate growth rate increases with higher R ratio. Although it is not clear why a lower ICG concentration prevented aggregation of the MCs, we suspect that with fewer bound ICG molecules, the D500 cores had more free charged groups. The repulsive forces among the cores likely counteracted the coalescence process.
MC size analysis and morphology was determined using scanning electron microscopy (SEM), as well as brightfield microscopy. Figure 2 displays an SEM image of dry D100 MCs. Using QCapture software (Qimaging, Surrey,BC Canada), size analysis was performed by measuring the diameters of 200 MCs in an SEM image (measuring the length in pixels of lines drawn across each capsule). As described in our earlier work 43, encapsulation efficiencies were determined by using absorption measurements (Shimadzu UV-2401PC) to quantify the amount of ICG in capsules disassembled with NaOH. MC zeta potentials were measured using phase analysis light scattering (PALS) in a ZetaPALS Zeta Potential Analyzer (Brookhaven Instruments Corporation) as a method to characterize the capsule surface charge.
Figure 2.

SEM image of D100 MCs. MCs had spherical conformation and were found to be monodisperse in suspension.
Physical Stability Assessment
To evaluate the effects of plasma proteins, lipids, and other biomolecules upon the stability of the MCs, we incubated 500 nm dextran-coated MCs under simulated in vivo conditions51, 52. For these stability experiments, capsules were coated with 40 kDa dextran fluorescently labeled with fluorescein isothiocyanate (FITC), a widely-used fluorophore that is spectrally distinct from ICG (excitation peak: ∼488 nm, emission peak: 515 nm). The MCs were suspended in cell culture medium containing fetal bovine serum, and were incubated at physiological temperature with no light exposure. The media and serum contained essential nutrients for cells such as glucose and amino acids as well as various dissolved proteins, hormones, and growth factors found in blood plasma.
At various time points, the capsules were centrifuged, and the supernatant was analyzed for both ICG content and FITC-dextran released from the capsules using fluorescence measurements (described in the Supplementary Information).
Biodistribution Studies
Female Swiss Webster mice (Charles River Laboratories) were utilized in this study under a protocol approved by the Institutional Animal Care and Use Committee at Rice University. The Swiss Webster strain is a widely-utilized general purpose animal model that has been used in pharmaceutical research for several decades 53. The mice were housed in ventilated polypropylene cages, at an average temperature of 22 °C, with exposure to 12 hours of light and 12 hours of darkness each day. All mice received a standard laboratory diet of food and water ad libitum and weighed between 20-35 g.
Preparations of either freely dissolved ICG or 100 nm MCs containing ICG were administered to the mice by caudal vein injection at a concentration of 2 mg/kg. For comparison with the MCs, the stock ICG aqueous solution was diluted with PBS and administered at a concentration matching the ICG concentration contained in the 100 nm dextran-coated and PEG-NP-coated MC suspensions. Since D500 MCs were prone to aggregation at high concentrations, they were injected at a concentration of 0.8 mg/kg. The mice were euthanized by anesthesia with 5% Isoflurane followed by cervical dislocation at various times (t = 2.5, 5, 10, 20, 30, and 60 min), and the blood (100 μl), heart, lungs, kidneys, liver, and spleen were harvested from each mouse. Three mice were used for each time-point.
To extract ICG, the organs were homogenized in 5ml of Dimethyl sulfoxide (DMSO) 34, and centrifuged at 9000 rpm for 15 minutes. DMSO is a small (78.13 Da), dipolar molecule. It is miscible with water and easily penetrates cell membranes. DMSO is frequently used as a solvent for drug molecules, transdermal delivery agent, and cryoprotectant 54, 55.
To quantify the extracted ICG, we took advantage of ICG's strong fluorescence activity in the NIR range. As intrinsic autofluorescent tissue biomolecules such as NADH, porphyrins, and collagen, display minimal excitability in this spectral range 56, the fluorescence signal collected from the DMSO extracts arises exclusively from ICG. ICG content was assessed for harvested organs and plasma samples through fluorescence measurements using a Fluoromax 3 Fluorimeter (JY-Horiba). The fluorimeter is equipped with a 150 W xenon arc lamp coupled to a monochromator for excitation light, and an emission monochromator coupled to a photomultiplier tube with extended red sensitivity (R928P, Hamamatsu). The samples were excited at λ = 780 nm, and the fluorescence spectra were obtained from λ = 795 nm to 900 nm. ICG concentration of each sample was determined by comparison of the peak fluorescence value (λ = 820 nm) to a standard curve made with ICG dissolved in DMSO at various concentrations. The fluorescence quantum yield of ICG decreases at higher concentrations due to molecular oligomerization and fluorescence quenching. Consequently, liver samples, in which large amounts of ICG accumulated for all ICG formulations, were diluted in DMSO by a factor of 20 to yield ICG concentrations within the range at which fluorescence is linearly proportional to concentration (10 – 250 ng/ml). Blood plasma samples were also diluted by a factor of 50 in order to yield sufficient sample volume for fluorescence measurement. The total ICG mass within blood was calculated by assuming that the total blood volume comprises 7 % of the mouse's mass 57, and also that the injected preparation was uniformly distributed within the bloodstream within 20 seconds following the injection.
To verify that DMSO dissolves the MCs and liberates ICG into solution, small volumes of DMSO were pipetted into samples of MCs suspended in PBS. An immediate loss in turbidity was observed, and MCs could not be recovered as a pellet by centrifugation, thus confirming that the MCs dissolved in DMSO. Extraction efficiencies of DMSO were determined by injecting known quantities of ICG or ICG-MCs into harvested blood and organs of non-injected control mice. After 30 minutes, the ICG was collected by DMSO extraction as described above. The fluorescence profile of supernatants collected from these samples was compared to the profile of ICG in DMSO at the same concentration.
The stability of ICG's fluorescence profile in DMSO was also evaluated. Solutions of ICG in DMSO at various concentrations (10 ng/ml - 1μg/ml) were prepared and stored in the dark at room temperature. The fluorescence spectra of these samples were collected at several time-points up to 48 hours.
ICG recovered from plasma and organs at different time points is presented as mean value ± standard deviation of the percentage original ICG injection mass. The values were checked for statistical significance in two different ways. First, in order to determine whether ICG deposition from the MCs was significantly different from freely dissolved ICG, a student-t test was performed for each MC formulation against ICG solution at each time point for each organ. Second, in order to determine whether each formulation was significantly different from all others, a one-way ANOVA test was conducted for each organ at each time-point for all different formulations. Tukey-Kramer's honestly significantly different criterion was applied to the ANOVA results to determine which formulations were statistically different (α = 0.05) from all others.
Results
MC Characterization
MCs had approximately spherical morphology, as determined by SEM imaging, as seen in Figure 2. Table 1 lists the coating material, diameter, encapsulation efficiencies, and surface charge, in terms of zeta potential, for each MC system. Neither the capsules' coating material nor size appeared to have a substantial influence on the MC's ICG encapsulation efficiency. In the case of the D500 MCs, the large variability in size is similar to polydispersity observed in previous studies, where it was shown that MCs synthesized with a higher R ratio were more susceptible to aggregation by coalescence 38, 50.
Table 1.
Physical Characteristics of the MC systems
| MC System Type | Coating Material | Diameter (mean ± std. dev) | ICG Encapsulation Efficiency (%) | Zeta Potential (mV) |
|---|---|---|---|---|
| D100 | Dextran (40 kDa) |
90 ± 12 nm | 61 ± 3 | -22 ± 4 |
| D500 | Dextran (40 kDa) |
465 ± 156 nm | 56 ± 3 | -23 ± 2 |
| Fe100 | Fe3O4 NPs (10 nm, coated with PEG) |
97 ± 13 nm | 60 ± 2 | -10 ± 1 |
MC Stability in Plasma
MCs coated with fluorescently labeled dextran were incubated in cell culture media containing fetal bovine serum with no light exposure for 24 hours. At various time points, samples were imaged with SEM and centrifuged. Fluorescence measurements of supernatant revealed that after 24 hours, nearly 90% of the initially encapsulated ICG mass remained in the capsules, as illustrated in Figure S1. This observavtion, coupled with the fact that after 24 hours the capsules could still be centrifuged down to a pellet confirmed our hypothesis that the MCs maintain their integrity and ICG is not released in substantial amounts from the MCs when exposed to in vivo-like conditions.
We postulate that for dextran-coated MCs, uncharged dextran adheres to the capsule surface by hydrogen bonding interactions. We investigated whether dextran remains attached to the capsule surface by quantifying the amount of FITC-dextran in the supernatants of MCs incubated with serum. As seen in Figure S2, over the course of 24 hours, a maximum of 8 % of the FITC dextran mass incorporated on the capsule surfaces was found in the supernatants. Therefore, MCs did not degrade and liberate substantial amounts of ICG or dextran in the presence of plasma proteins.
Biodistribution
The efficiency of the DMSO extraction process was calculated to be 86.9 ± 4.5 %. The fluorescence of ICG was also found to be stable in DMSO. When stored in the dark and at room temperature, the fluorescence profile of ICG solution in DMSO did not diminish up to 48 hours after preparation, indicating that ICG did not degrade when dissolved in DMSO over the course of our experiments (data not shown). Additionally, the fluorescence profiles of the extracted ICG appeared empirically similar to the profile of ICG dissolved in DMSO. This validated our assumption that the fluorescence measurements of ICG extracted from tissue were not corrupted by tissue autofluorescence. Figures 3 a-f display the ICG content within the plasma and organs at various post-injection times for free ICG solution and the three MC systems. Asterisks indicate which formulations were significantly different from ICG solution, as determined by student-t test. Triangles indicate which formulations were significantly different from all others, as determined by one-way ANOVA followed by application of the Tukey-Kramer conjecture. As illustrated in Figure 3a, the residence time of ICG within plasma was similar for ICG solution and all types of MC systems containing ICG, with plasma concentrations of ICG dropping below 10% of the original injection mass within 2.5 minutes following a bolus injection.
Figure 3.
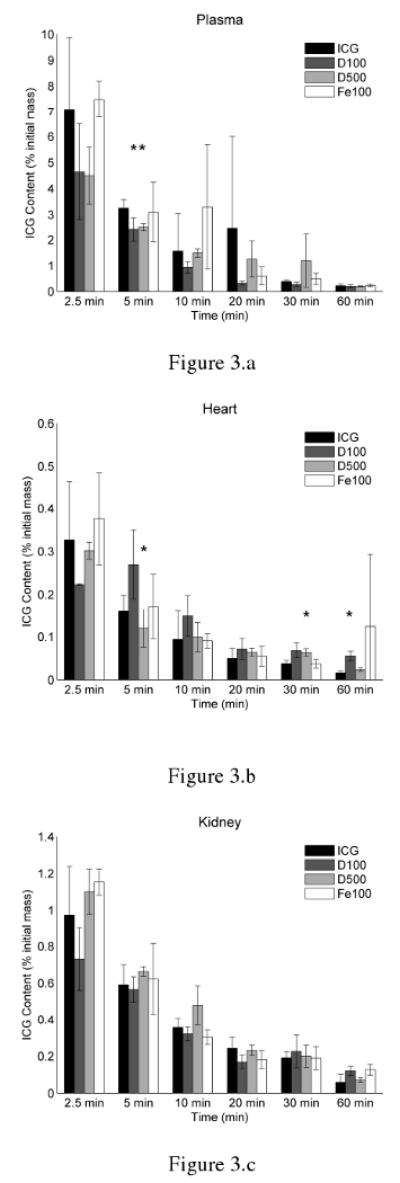
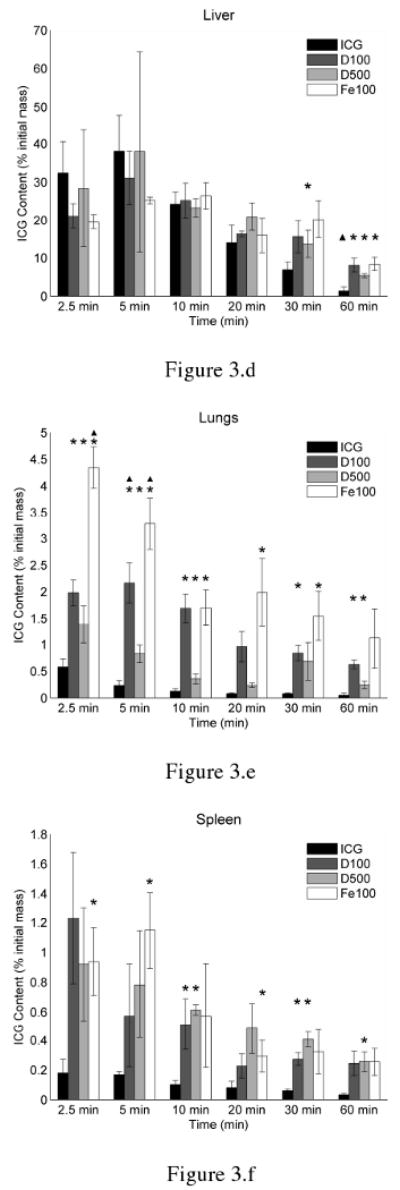
Biodistribution of ICG within (a) plasma, (b) heart, (c) kidneys, (d) liver, (e) lungs, and (f) spleen of healthy Swiss Webster mice at various post-injection times. ICG was delivered either in solution and non-encapsulated (ICG), or encapsulated within 100 nm MCs coated with dextran (D100), 500 nm MCs coated with dextran (D500), or 100 nm MCs coated with magnetite NPs (Fe100). The asterisks indicate a statistical difference (p < 0.05) from free ICG, as determined by student-t test analysis. Triangles indicate which formulations were significantly different from all others, as determined by one-way ANOVA (α = 0.05) followed by application of the Tukey-Kramer conjecture.
Figures 3b and 3c show that in all cases, negligible amounts of ICG were recovered from the heart or kidneys. For all formulations, less than 2% of the original ICG injection mass deposited within the heart and kidneys. Additionally, statistical analysis showed that, in general, the amount of ICG recovered from the plasma, heart, and kidneys was not significantly different for any of the MC systems at any time-point when compared to ICG solution.
As demonstrated in Figure 3d, the majority of ICG accumulated within the liver. Maximal peak values in the liver for various formulations ranged between 26 – 38 % of the original ICG injection mass. While at 10 minutes following injection, the total masses of ICG in liver are not statistically significant different from each other among the various formulations, it appears that freely dissolved ICG is transported to the bile ducts and mixes with the bile more readily than the MC formulations. This is especially evident at 60 minutes following injection, when ICG levels in the liver drop below 10 % of the original mass for all formulations. However, substantially more ICG is retained in liver at this time from the MC formulations when compared to free ICG.
Student t-test analysis showed significantly higher levels of ICG within the lungs and spleen when delivered in MC form as compared to ICG solution. In the lung, maximal ICG levels were nearly 2.5, 3.5, and 7.5 times higher than freely dissolved ICG when delivered through D500, D100, Fe100 MCs, respectively. There was a trend for both Fe100 and D100 MCs to deposit in the lungs in significantly greater amounts than ICG solution or D500 MCs, with Fe100 MCs remaining within the lung in significantly greater amounts until 30 minutes.
Interestingly, while in Figure 3e, it appears that the D100 MCs were taken up in greater amounts in the lungs than the D500 MCs, Figure 3f suggests that the same phenomenon was not observed in the spleen. While the amount of ICG recovered from the spleen was similar to those recovered from the kidneys, peak ICG levels in the spleen were 6.9, 5.1, and 5.2 times higher than ICG solution when delivered as D100, D500, and Fe100 MC systems. ICG levels in the liver, lungs, and spleen remained relatively higher for MC systems than ICG solution up to 60 minutes after injection.
The total amount of ICG recovered from each animal was considerably less than the initial administered dose. Further attempts to perform a more accurate mass balance were not performed in the present study. Nevertheless, this discrepancy was an issue of concern. Other investigators have accounted for this issue by harvesting samples of skin, muscle, and bone, and subsequently extrapolating levels in the entire animal carcass58. It is possible that a portion of our injected dose could reside within the muscle and bone, however, we feel it is more likely that the unaccounted ICG remained in the tail tissue. As the rapid infusion of a relatively large volume (up to 200μl) into the caudal vein could easily lead to ruptured blood vessels, extravasation of the ICG formulations could easily occur during the injections. Other investigators have recovered as much as 30% of their injected dose from harvested tails 59
In most cases, we believe that the variability and lack of statistical significance in our results can also be attributed to imperfect tail vein injections. Occasionally, the formulations were administered only after multiple injection attempts were made, causing us to slightly overestimate the total injected mass. In a few cases, we observed anomalous results, in which a specific tissue sample for a given subject contained much more ICG than the corresponding samples for other subjects in the same group. Examples include ICG solutions in plasma at t =20 min and Fe100 MCs in the heart at t = 60 minutes. These instances require further investigation.
Discussion
Before evaluating the biodistribution results, it is of critical importance to determine whether ICG remains encapsulated during the course of the in vivo experiments. Our supplementary results reveal that nearly 90% of ICG remains encapsulated, and over 90% of the dextran coating material remain on the MCs for up to 24 hours in the presence of plasma. Although we investigated only MCs with fluorescently labeled dextran coating, the results are nearly identical to results of similar experiments with dextran-coated capsules incubated in saline 43, a medium in which we have tested several capsule formulations and verified their stability 39, and they lead us to conclude that ICG remains encapsulated within our MCs over the course of our biodistribution experiments, which did not exceed 60 minutes for this study.
As Figures 3a and 3d demonstrate, after a bolus injection of ICG solution, ICG concentration in the plasma decreases rapidly with concurrent concentration rises within the liver. The data are in agreement with previous reports that state ICG is uptaken exclusively by the liver and excreted to bile 22. The results also validate ICG's utility in clinical practice for measuring hepatic function. Although curve-fitting data is not provided here because of the large variability in some samples, our results for freely dissolved ICG support pharmacokinetic reports of ICG, where the concentration of ICG within the bloodstream is modeled with a bi-exponential decay. The first exponential time constant represents the rapid uptake of ICG by the liver, while the second represents a slower more gradual uptake phase, also by the liver 20, 60.
Figure 3a suggests that the residence time of the MCs in the circulation is similar to that of ICG solution; however, the higher uptake of ICG by the lungs and spleen (as seen in Figures 3e and 3f) suggests a different biodistribution behavior for the MCs.
When delivered intravenously as a solution, ICG binds readily to albumin and high density lipoproteins (HDLs) in blood plasma such as alpha-1 lipoprotein, and moderately to low density lipoproteins (LDLs)21, 22. ICG solution is reportedly consumed exclusively by hepatic parenchymal cells lining the liver's sinusoids, and these hepatocytes eliminate ICG from the bloodstream unmetabolized through biliary excretion 20, 61, 62.
While the liver takes up the majority of MCs, the higher levels within the lungs and spleen suggest that one or more different mechanisms account for the removal of MCs from the bloodstream. When coupled with our previous findings45, these results suggest that, while liver hepatocytes remove some of the MCs from the blood and excrete them to bile, a fraction of the MCs are either engulfed by macrophages of the reticuloendothelial system (RES) or are taken up by the pulmonary endothelial cells.
The endothelial layers of organs comprising the RES, namely the liver, spleen, and bone marrow are equipped with mononuclear macrophages that filter the blood of non-native pathogens and particulate debris. Upon the introduction of foreign particles into the bloodstream, plasma proteins such as immunoglobulins, complement proteins, and soluble cell adhesion molecules, known as opsonins, adsorb to the foreign particle surface within seconds, a process known as opsonization. As receptors for various opsonin proteins are located on the surfaces of macrophages, opsonization facilitates recognition, phagocytosis, and subsequent digestion of foreign matter by macrophages 63-68.
We observed in our in vivo fluorescence imaging investigation that, following a bolus injection of either ICG solution or MCs containing ICG, the intestines emitted fluorescence signal that increased with time45. We attributed this increasing intestinal fluorescence signal to the secretion of bile containing ICG from the liver. This observation supports the idea that a portion of the MCs are removed from the blood by hepatic filtration and biliary excretion. The results of our current study, particularly the significantly higher uptake of MCs by the spleen than free ICG, and the seemingly longer retention time of MCs within the liver, suggest that some of the MCs are also removed from the bloodstream by the RES following opsonization. Both the deposition of MCs within the spleen and the longer retention within the liver is similar to reports of several polymer nanoparticulate systems, where injected nanoparticles containing encapsulated drug or imaging contrast agents reportedly undergo opsonization, phagocytosis by macrophages, and hepatic filtration 29, 64, 68.
Comparing the distributions of the three different MC systems within the spleen and lungs suggests that particle size has less influence on the biodistribution than the particle coating. The large variability in the data, indicated by the large standard deviations, prevent one from making definitive conclusions regarding these phenomena. However, the results suggest that in the case of the spleen, the capsules' size and coating appears to have minimal influence on its uptake, as they are all collected in similar amounts.
In the lungs, the smaller D100 MCs accumulated in greater amounts than the D500 capsules. This phenomenon appeared counterintuitive to our initial assumption that the MCs remain trapped by embolizing the pulmonary capillaries. If this were true, it seems that larger capsules would accumulate after becoming lodged within the pulmonary capillaries 69. As the pulmonary endothelium serves as a semipermeable membrane between the vasculature and the interstitium 70, the larger deposition of D100 capsules over D500 capsules is likely attributable to greater endocytic uptake of the smaller MCs. This observation is consistent with reports stating that smaller particles (100 nm) can more easily traverse the pulmonary endothelium than larger particles (500 nm) by either passing between endothelial cells or through endocytotic mechanisms 71. We suspect that endocytic uptake of MCs by the endothelial cells accounts for more of the removal than diffusion across the endothelium, as evidenced by the fact that less ICG was recovered from the lungs when administered as a solution. The Fe100 MCs clearly resulted in a greater accumulation of ICG in the lung than either dextran MC system. Statistical analysis of our results suggests that the uptake of particulate matter within the lungs is weakly dependent on size, but more dependent on coating. The increased uptake of the less negatively charged Fe100 MCs is consistent with reports that neutral and cationic particles are taken up by endothelial cells in the lung 71-73.
Other investigators have previously experimented with overcoming ICG's rapid circulation kinetics and optical instability by encapsulating it within PLGA NPs or di-block copolymer micelles, embedding ICG within a modified silicate matrix or a lipid suspension, or through noncovalent interactions of ICG with sodium polyaspartate 33-36, 74, 75. In all of these cases, ICG's optical properties appear to be stabilized. In most cases, these investigators also report that their encapsulation methods prolong the residence time of ICG within the bloodstream, and in the case of the studies by Saxena et al, a larger amount of ICG is also recovered from the heart, lungs, kidneys, liver, and spleen when administered in PLGA NP form. For each of these encapsulation formulations, a modest increase is observed for retention time of ICG within the circulation. In all cases, the majority of ICG when injected in encapsulated form is still removed from the bloodstream within the first 5 minutes, yet the amount of ICG that lingers remains slightly higher than when injected as free solution.
Like the other reported ICG encapsulated systems, our MC system also stabilizes ICG's optical properties 43. Additionally, the MCs have the added benefit of versatility in terms of size and coating material. Although the experiments described here use MCs coated with dextran polymer or PEG coated magnetic NPs, in principle, a variety of coating materials, including PEG, poly-lysine, and many other NPs can be utilized.
Many investigators rely on the enhanced permeability and retention (EPR) effect for treatment of malignant tumors, in which drug molecules accumulate at the tumor location by extravasation out of the tumors' poorly developed leaky blood vessels, to deliver drugs to tumor sites 30. Other strategies include functionalizing the particle surface with antibodies that would recognize receptors expressed in large amounts by the tumor, such as vascular endothelial growth factor receptor-1 or -2. For either of these strategies, it is necessary that the drug circulate within the bloodstream for several hours or days without being opsonized and subsequently consumed by the macrophages of RES. To increase circulation time of nanoparticles and facilitate delivery to tumors, it is suggested that hydrophilic polymers such as PEG and poloxamers be covalently attached to the particle surface to sterically hinder interactions between NPs and opsonin proteins 29, 63, 76-79. These flexible, hydrophilic, branched polymers form a cloud around the particle shielding it from opsonization.
The magnetite NPs coating our Fe100 MC system were coated with PEG prior to MC synthesis. These MCs are similar to another MC formulation coated with larger magnetite NPs utilized in our previous report45, in which we showed that the MCs have potential for fluorescence imaging of pulmonary pathologies. Interestingly, despite their PEG coating, the circulation time of the Fe100 capsules was not greater than other MC systems or of ICG solution. We observed similar results for an alternate MC system in which the 100 nm capsules were coated with PEG polymer (data not shown). Further investigation is required to explain why our PEG coatings did not more substantially influence the MCs biodistribution or prolong the circulation time of ICG within the bloodstream, but we suspect that covalently attaching branched PEG polymer instead of linear PEG may result in improved shielding effect of the aggregate core and enhanced protection against opsonization.
This work signifies our initial characterization of in vivo biodistribution behavior of our charge-assembled MC systems containing ICG. Motivated by these results, our future experiments will include attempts to covalently attach branched, functionalizable PEG and poloxamers to the MC surface to prolong the circulation time within the vasculature. Other experiments will also investigate the conjugation of antibodies to these polymers to actively target ligands on cells in specific tissues such as intercellular adhesion molecule on pulmonary endothelial cells80 and thereby increase tissue-targeting selectivity. As these current experiments did not explore toxicity concerns, experiments will also be conducted in the future to evaluate potential hepatotoxicity or pulmonary toxicity issues.
By enabling the delivery of ICG to organs other than the liver, namely the lungs and spleen, the MCs open up new possibilities for novel diagnostic and therapeutic techniques for treating tissue pathologies. In particular, the MCs coated with magnetic NPs are taken up in large quantities by the pulmonary endothelial cells, and they are both optically and magnetically active. These capsules therefore have potential use for multimodal imaging strategies for acute and chronic respiratory conditions. Lim et al have demonstrated the utility of their PLGA-matrix nanoparticles containing ICG and iron oxide NPs for tracking of dendritic cells in vivo using both NIR fluorescence imaging and magnetic resonance imaging (MRI) 32. We have demonstrated previously that laser irradiation of our MCs can induce noninvasive photothermal damage44, and additionally, MCs coated with iron oxide NPs deposit readily within the lungs and can be used for fluorescence imaging45. Future experiments will explore the use of magnetite coated MCs containing ICG for simultaneous multimodal imaging and photothermal therapy.
Conclusion
The MC system enhances the delivery of ICG to organs other than the liver, particularly the lung and spleen. These results suggest that MCs are removed from the bloodstream by hepatic filtration, phagocytosis by cells of the RES, and endocytosis by pulmonary endothelial cells. The polymer coating type and size of the MCs both influence the uptake by the lungs, whereas they have minimal influence on the uptake by the spleen. By manipulating these parameters, the uptake of MCs containing ICG, in particular by the lung may potentially be controlled. The collection of MCs containing ICG by the lungs and spleen offers potential for the development of optically mediated diagnostic and treatment applications for these organs.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health under grant No GMO 8362. It was also supported by the Student Research Award from the American Society for Laser Medicine and Surgery to M.A.Y. We wish to thank the Colvin Laboratory in the Chemistry Department at Rice University for the generous donation of PEG-coated magnetite nanoparticles. We also thank the Mikos laboratory in the Bioengineering Department for graciously permitting the use of the tissue homogenizer.
References
- 1.Landsman MLJ, Kwant G, Mook GA, Zijlstra WG. Light-absorbing properties, stability, and spectral stabilization of indocyanine green. J Appl Physiol. 1976;40(4):575–583. doi: 10.1152/jappl.1976.40.4.575. [DOI] [PubMed] [Google Scholar]
- 2.Benson RC, Kues HA. Fluorescence Properties of Indocyanine Green as Related to Angiography. Phys Med Biol. 1978;23(1):159–163. doi: 10.1088/0031-9155/23/1/017. [DOI] [PubMed] [Google Scholar]
- 3.El-Desoky A, Seifalian AM, Cope M, Delpy DT, Davidson BR. Experimental study of liver dysfunction evaluated by direct indocyanine green clearance using near infrared spectroscopy. Br J Surg. 1999;86:1005–1011. doi: 10.1046/j.1365-2168.1999.01186.x. [DOI] [PubMed] [Google Scholar]
- 4.Webster JG. Measurement of Flow and Volume of Blood. In: Webster G, editor. Medical Instrumentation: Application and Design. John Wiley & Sons, Inc; New York: 1998. [Google Scholar]
- 5.Shinohara H, Tankaka A, Kitai T, Yanabu N, Inomoto T, Satoh S, Hatano E, Yamaoka Y, Hirao K. Direct Measurement of Hepatic Indocyanine Green Clearance With Near-Infrared Spectroscopy: Separate Evaluation of Uptake and Removal. Hepatology. 1996;23:137–144. doi: 10.1053/jhep.1996.v23.pm0008550033. [DOI] [PubMed] [Google Scholar]
- 6.Chen WR, Adams RL, Heaton S, Dickey DT, Bartels KE, Nordquist RE. Chromophore-emhanced laser tumor tissue photothermal interaction using an 808-nm diode laser. Cancer Lett. 1995;88:15–19. doi: 10.1016/0304-3835(94)03609-m. [DOI] [PubMed] [Google Scholar]
- 7.Chen WR, Adams RL, Higgins AK, Bartels KE, Nordquist RE. Photothermal effects on murine mammary tumors using indocyanine green and 808-nm diode laser: an in-vivo efficacy study. Cancer Lett. 1995;98(2):169–173. [PubMed] [Google Scholar]
- 8.Babilas P, Shafirstein G, Baier J, Schacht V, Szeimies RM, Landthaler M, Bäumler W, Abels C. Photothermolysis of Blood Vessels Using Indocyanine Green and Pulsed Diode Laser Irradiation in the Dorsal Skinfold Model Chamber. Lasers Surg Med. 2007;39:341–352. doi: 10.1002/lsm.20483. [DOI] [PubMed] [Google Scholar]
- 9.Fickweiler S, Szeimies RM, Bäumler W, Steinbach P, Karrer S, Goetz AE, Abels C, Hofstädter F, Landthaler M. Indocyanine green: Intracellular uptake and phototherapeutic effects in vitro. J Photochem Photobiol B. 1997;38(23):178–183. doi: 10.1016/s1011-1344(96)07453-2. [DOI] [PubMed] [Google Scholar]
- 10.Abels C, Fickweiler S, Weiderer P, Bäumler W, Hofstädter F, Landthaler M, Szeimies RM. Indocyanine green (ICG) and laser irradiation induce photooxidation. Arch Dermatol Res. 2000;292(8):404–411. doi: 10.1007/s004030000147. [DOI] [PubMed] [Google Scholar]
- 11.Bäumler W, Abels C, Karrer S, Weiβ T, Messmann H, Landthaler M, Szeimies RM. Photooxidative killing of human colonic cancer cells using indocyanine green and infrared light. Br J Cancer. 1999;80(34):360–363. doi: 10.1038/sj.bjc.6690363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbanska K, Romnaowska-Dixon B, Matuszak Z, Oszajca J, Nowak-Sliwinska P, Stochel G. Indocyanine green as a prospective sensitizer for photodynamic therapy of melanomas. Acta Biochim Pol. 2002;49(2):387–391. [PubMed] [Google Scholar]
- 13.Tuchin VV, Genina EA, Bashkatov AN, Simonenko GV, Odoevskaya OD, Altshuler GB. A Pilot Study of ICG Laser Therapy of Acne Vulgaris: Photodynamic and Photothermolysis Treatment. Lasers Surg Med. 2003;33(5):296–310. doi: 10.1002/lsm.10211. [DOI] [PubMed] [Google Scholar]
- 14.Bass LS, Moazami N, Pocsidio J, Oz MC, LoGerfo P, Treat MR. Changes in Type I Collagen Following Laser Welding. Lasers Surg Med. 1992;12(5):500–505. doi: 10.1002/lsm.1900120508. [DOI] [PubMed] [Google Scholar]
- 15.Schomacker K, Torri A, Sandison DR, Sheridan RL, Nishioka NS. Biodistribution of Indocyanine Green in a Porcine Burn Model: Light and Fluorescence Mircoscopy. J Trauma Inj Infect Crit Care. 1997;43:813–819. doi: 10.1097/00005373-199711000-00013. [DOI] [PubMed] [Google Scholar]
- 16.Rübben A, Eren S, Krein R, Younoussi H, Böhler U, Wienert V. Infrared Videoangiofluorography of the Skin with Indocyanine Green- Rat Random Cutaneous Flap Model and Results in Man. Microvasc Res. 1994;47:240–251. doi: 10.1006/mvre.1994.1018. [DOI] [PubMed] [Google Scholar]
- 17.Ntziachristos V, Yodh AG, Schnall M, Chance B. Concurrent MRI and diffuse optical tomography of breast after indocyanine green enhancement. Proc Natl Acad Sci U S A. 2000;97:2767–2772. doi: 10.1073/pnas.040570597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung TS, Tachtsidis I, Tisdall M, Smith M, Delpy DT, Elwell CE. Theoretical investigation of measuring cerebral blood flow in the adult human head using bolus Indocyanine Green injection and near-infrared spectroscopy. Appl Opt. 2007;46:1604–1614. doi: 10.1364/ao.46.001604. [DOI] [PubMed] [Google Scholar]
- 19.Mudra R, Nadler A, Keller E, Niederer P. Analysis of near-infrared spectroscopy and indocyanine green dye dilution with Monte Carlo simulation of light propagation in the adult brain. J Biomed Opt. 2006;11:044009. doi: 10.1117/1.2341652. [DOI] [PubMed] [Google Scholar]
- 20.Desmettre T, Devoiselle JM, Mordon S. Fluorescence Properties and Metabolic Features of Indocyanine Green (ICG) as Related to Angiography. Surv Ophthalmol. 2000;45(1):15–27. doi: 10.1016/s0039-6257(00)00123-5. [DOI] [PubMed] [Google Scholar]
- 21.Yoneya S, Saito T, Komatsu Y, Koyama I, Takahashi K, Duvoll-Young J. Binding Properties of Indocyanine Green in Human Blood. Invest Ophthalmol Vis Sci. 1998;39(7):1286–1290. [PubMed] [Google Scholar]
- 22.Cherrick GR, Stein SW, Leevy CM, Davidson CS. Indocyanine Green: Observations on its Physical Properties, Plasma Decay, and Hepatic Extraction. J Clin Investig. 1960;39:592–600. doi: 10.1172/JCI104072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxena V, Sadoqi M, Shao J. Degradation Kinetics of Indocyanine Green in Aqueous Solution. J Pharm Sci. 2003;92(10):2090–2097. doi: 10.1002/jps.10470. [DOI] [PubMed] [Google Scholar]
- 24.Philip R, Penzkofer A, Bäumler W, Szeimies RM, Abels C. Absorption and fluorescence spectroscopic investigation of indocyanine green. J Photochem Photobiol A. 1996;96(13):137–148. [Google Scholar]
- 25.Bjornsson OG, Murphy R, Chadwick VS. Physicochemical Studies of Indocyanine Green(ICG): Absorbance/concentration relationship, pH tolerance and assay precision in various solvents. Experientia. 1982;38:1441–1442. doi: 10.1007/BF01955757. [DOI] [PubMed] [Google Scholar]
- 26.Holzer W, Mauerer M, Penzkofer A, Szeimies RM, Abels C, Landthaler M, Bäumler W. Photostability and thermal stability of indocyanine green. J Photochem Photobiol B. 1998;47(23):155–164. doi: 10.1016/s1011-1344(98)00216-4. [DOI] [PubMed] [Google Scholar]
- 27.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 28.Antonietti M, Förster S. Vesicles and Liposomes: A Self-Assembly Principle Beyond Lipids. Adv Mater. 2003;15:1323–1333. [Google Scholar]
- 29.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Delivery Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 30.Maeda H, Wu J, Sawa T, Matsamura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 31.Nie S, Xing Y, Kim GJ, Simons JW. Nanotechnology Applications in Cancer. Annu Rev Biomed Eng. 2007;9:257–288. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 32.Lim YT, Noh YW, Han JH, Cai QY, Yoon KH, Chung BH. Biocompatible Polymer-Nanoparticle-Based Bimodal Imaging Contrast Agents for the Labeling and Tracking of Dendritic Cells. Small. 2008;4:1640–1645. doi: 10.1002/smll.200800582. [DOI] [PubMed] [Google Scholar]
- 33.Saxena V, Sadoqi M, Shao J. Enhanced photo-stability, thermal-stability and aqueous-stability of indocyanine green in polymeric nanoparticulate systems. J Photochem Photobiol B. 2004;74(1):29–38. doi: 10.1016/j.jphotobiol.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Saxena V, Sadoqi M, Shao J. Polymeric nanoparticulate delivery system for Indocyanine green: Biodistribution in healthy mice. Int J Pharm (Kidlington) 2006;308:200–204. doi: 10.1016/j.ijpharm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Kim G, Huang SW, Day KC, O'Donnell M, Agayan RR, Day MA, Kopelman R, Ashkenazi S. Indocyanine-green-embedded PEBBLEs as a contrast agent for photoacoustic imaging. J Biomed Opt. 2007;12:044020. doi: 10.1117/1.2771530. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez VB, Henry SM, Hoffman AS, Stayton PS, Li X, Pun SH. Encapsulation and stabilization of indocyanine green within poly(styrene-alt-maleic anhydride) block-poly(styrene) micelles for near-infrared imaging. J Biomed Opt. 2008;13(1):014025. doi: 10.1117/1.2834296. [DOI] [PubMed] [Google Scholar]
- 37.Mordon S, Desmettre T, Devoiselle JM, Mitchell V. Selective Laser Photocoagulation of Blood Vessels in a Hamster Skin Flap Model Using a Specific ICG Formulation. Lasers Surg Med. 1997;21:365–373. doi: 10.1002/(sici)1096-9101(1997)21:4<365::aid-lsm8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 38.Rana RK, Murthy VS, Yu J, Wong MS. Nanoparticle Self-Assembly of Heirarchically Ordered Microcapsule Structures. Adv Mater. 2005;17(9):1145–1150. [Google Scholar]
- 39.Yu J, Yaseen MA, Anvari B, Wong MS. Synthesis of Near-Infrared-Absorbing Nanoparticle-Assembled Capsules. Chem Mater. 2007;19(6):1277–1284. [Google Scholar]
- 40.Kadali SB, Soultanidis N, Wong MS. Assembling Colloidal Silica into Porous Hollow Microspheres. Topics in Catalysis. 2008;49:251–258. [Google Scholar]
- 41.Couvreur P, Couarraze G, Devissaguet JP, Puisieux F. Nanoparticles: preparation and characterization. In: Benita S, editor. Microencapsulation: Methods and Industrial Application. Marcell Dekker, Inc.; New York: 1996. [Google Scholar]
- 42.Caruso F, Caruso RA, Mohwald H. Nanoengineering of Inorganic and Hybrid Hollow Spheres by Colloidal Templating. Science. 1998;282:1111–1114. doi: 10.1126/science.282.5391.1111. [DOI] [PubMed] [Google Scholar]
- 43.Yaseen MA, Yu J, Wong MS, Anvari B. Stability assessment of indocyanine green within dextran-coated mesocapsules by absorbance spectroscopy. J Biomed Opt. 2007;12(6):064031. doi: 10.1117/1.2821423. [DOI] [PubMed] [Google Scholar]
- 44.Yaseen MA, Yu J, Wong MS, Anvari B. Laser-Induced Heating of Dextran-Coated Mesocapsules Containing Indocyanine Green. Biotechnol Prog. 2007;23(6):1431–1440. doi: 10.1021/bp0701618. [DOI] [PubMed] [Google Scholar]
- 45.Yaseen MA, Yu J, Wong MS, Anvari B. In-vivo fluorescence imaging of mammalian organs using charge-assembled mesocapsule constructs containing indocyanine green. Optics Express. 2008;16:20577–20587. doi: 10.1364/oe.16.020577. [DOI] [PubMed] [Google Scholar]
- 46.Yu WW, Chang E, Sayes CM, Drezek R, Colvin VL. Aqueous dispersion of monodisperse magnetic iron oxide nanocrystale through phase transfer. Nanotechnology. 2006;17:4483–4487. [Google Scholar]
- 47.Levy L, Sahoo Y, Kim KS, Bergey EJ, Prasad PN. Nanochemistry: Synthesis and Characterization of Multifunctional Nanoclinics for Biological Applications. Chem Mater. 2002;14(9):3715–3721. [Google Scholar]
- 48.Hirata Y, Aoki M, Kobatake H, Yamamoto H. Insolubilization of water-soluble dextran. Biomaterials. 1999;20:303–307. doi: 10.1016/s0142-9612(97)00230-5. [DOI] [PubMed] [Google Scholar]
- 49.Stenekes RJH, Talsma H, Hennink WE. Formation of dextran hydrogels by crystallization. Biomaterials. 2001;22:1891–1898. doi: 10.1016/s0142-9612(00)00375-6. [DOI] [PubMed] [Google Scholar]
- 50.Murthy VS, Rana RK, Wong MS. Nanoparticle-Assembled Capsule Synthesis: Formation of Colloidal Polyamine-Salt Intermediates. J Phys Chem B. 2006;110:25619–25627. doi: 10.1021/jp061826b. [DOI] [PubMed] [Google Scholar]
- 51.Prencipe G, Tabakman SM, Welsher K, Liu Z, Goodwin AP, Zhang L, Henry J, Dai H. PEG Branched Polymer for Functionalization of Nanomaterials with Ultralong Blood Circulation. J Am Chem Soc. 2009;131:4783–4787. doi: 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang C, Bhattarai N, Sun C, Zhang M. Functionalized Nanoparticles with Long-Term Stability in Biological Media. Small. 2009;5(11) doi: 10.1002/smll.200801647. TBD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.NCI. APP - Cr:Sw (Swiss Webster) 2009 http://web.ncifcrf.gov/research/animal_production_program/strain_information/01S60.asp.
- 54.Yu ZW, Quinn PJ. Dimethyl Sulphoxide: A Review of Its Applications in Cell Biology. Biosci Rep. 1994;14:259–281. doi: 10.1007/BF01199051. [DOI] [PubMed] [Google Scholar]
- 55.Swanson BN. Medical use of dimethyl sulfoxide. Rev Clin Basic Pharm. 1985;5:1–33. [PubMed] [Google Scholar]
- 56.Bachmann L, Zezell DM, de Costa Ribeiro A, Gomes L, Ito AS. Fluorescence Spectroscopy of Biological Tissues- A Review. Applied Spectroscopy Reviews. 2006;41:575–590. [Google Scholar]
- 57.Hoff J. Methods of Blood Collection in the Mouse. Lab Animal. 2000;29:47–53. [Google Scholar]
- 58.Portet D, Denizot B, Rump E, Hindre F, Jeune JJL, Jallet P. Comparative Biodistribution of Thin-Coated Iron Oxide Nanoparticles TCION: Effect of Different Bisphosphonate Coatings. Drug Dev Res. 2001;54:173–181. [Google Scholar]
- 59.Gratton SEA, Pohlhaus PD, Lee J, Guo J, Cho MJ, DeSimone JM. Nanofabricated particles for engineered drug therapies: A preliminary biodistribution study of PRINT nanoparticles. J Control Release. 2007;121:10–18. doi: 10.1016/j.jconrel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sekimoto M, Fukui M, Fujita K. Plasma volume estimation using indocyanine green with biexponential regression analysis of the decay curves. Anaesthesia. 1997;52:1166–1172. doi: 10.1111/j.1365-2044.1997.249-az0389.x. [DOI] [PubMed] [Google Scholar]
- 61.Leevy CM, Bender J, Silverberg M, Naylor J. Physiology of dye extraction by the liver: comparative studies of sulfobromophtalein and indocyanine green. Ann N Y Acad Sci. 1963;111:161–175. doi: 10.1111/j.1749-6632.1963.tb36956.x. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka E, Choi HS, Humblet V, Ohnishi S, Laurence RG, Frangioni JV. Real-time intraoperative assessment of the extrahepatic bile ducts in rats and pigs using invisible near-infrared fluorescent light. Surgery. 2008;144:39–48. doi: 10.1016/j.surg.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moghimi SM. Mechanisms regulating body distribution of nanospheres conditioned with pluronic and tetronic co-polymers. Adv Drug Delivery Rev. 1995;16:183–193. [Google Scholar]
- 64.Owens DE, III, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm (Kidlington) 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 65.Guyton AC, Hall JE. Textbook of Medical Physiology. 10. W.B Saunders Co; Philadelphia: 2000. Resistance of the body to infection I; pp. 392–401. [Google Scholar]
- 66.Guyton AC, Hall JE. Textbook of Medical Physiology. 10. W.B Saunders Co; Philadelphia: 2000. Resistance of the body to infection II; pp. 402–412. [Google Scholar]
- 67.Li SD, Huang L. Pharmacokinetics and Biodistribution of Nanoparticles. Mol Pharm. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 68.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamm WJE, Bernard SL, Wagner WW, Glenny RW. Intravital microscopic observations of 15-μm microspheres lodging in the pulmonary microcirculation. J Appl Physiol. 2005;98:2242–2248. doi: 10.1152/japplphysiol.01199.2004. [DOI] [PubMed] [Google Scholar]
- 70.Dudek SM, Garcia JGN. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 71.Dziubla T, Muzykantov V. Nanocarriers for the Vascular Delivery of Drugs to the Lungs. In: Torchilin VP, editor. Nanoparticulates as Drug Carriers. Imperial College Press; London: 2006. pp. 499–526. [Google Scholar]
- 72.Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, Torchilin VP, Munn LL. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62(23):6831–6836. [PubMed] [Google Scholar]
- 73.le Masne de Chermont Q, Chanéac C, Seguin J, Pellé F, Maîtrejean S, Jolivet JP, Gourier D, Bessodes M, Scherman D. Nanoprobes with near-infrared persistent luminescence for in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:9266–9271. doi: 10.1073/pnas.0702427104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rajagopalan R, Uetrecht P, Bugaj JE, Achilefu SA, Dorshow RB. Stabilization of the Optical Tracer Agent Indocyanine Green Using Noncovalent Interactions. Photochem Photobiol. 2000;71(3):347–350. doi: 10.1562/0031-8655(2000)071<0347:sotota>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 75.Devoiselle JM, Soulié-Bégu S, Mordon S, Desmettre T, Maillols HA. Preliminary Study of the In Vivo Behaviour of an Emulsion Formulation of Indocyanine Green. Lasers Med Sci. 1998;13:279–282. doi: 10.1007/s101030050008. [DOI] [PubMed] [Google Scholar]
- 76.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable Long-Circulating Polymeric Nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 77.Torchilin VP, Trubetskoy VS. Which polymers can make nanoparticulate drug carriers long-circulating? Adv Drug Delivery Rev. 1995;16:141–155. [Google Scholar]
- 78.Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 79.Stolnick S, Illum L, Davis SS. Long circulating microparticulate drug carriers. Adv Drug Delivery Rev. 1995;16(1995):195–214. [Google Scholar]
- 80.Rossin R, Muro S, Welch MJ, Muzykantov VR, Schuster DP. In Vivo Imaging of 64Cu-Labeled Polymer Nanoparticles Targeted to the Lung Endothelium. J Nucl Med. 2008;49(103111):103. doi: 10.2967/jnumed.107.045302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.