

Summary
Haemophagocytosis (hemophagocytosis) is the phenomenon of activated macrophage consumption of red and white blood cells, including professional phagocytes and lymphocytes. It can occur in patients with severe cases of intracellular microbial infection, including avian influenza, leishmaniasis, tuberculosis and typhoid fever. While well-known to physicians since at least the mid-1800s, haemophagocytosis has been little studied due to a paucity of tractable animal and cell culture models. Recently, haemophagocytosis has been described in a mouse model of typhoid fever, and it was noted that the infectious agent, Salmonella enterica, resides within haemophagocytic macrophages in mice. In addition, a cell culture model for haemophagocytosis revealed that S. enterica preferentially replicate in haemophagocytic macrophages. This review describes how, at the molecular and cellular levels, S. enterica may promote and take advantage of haemophagocytosis to establish long-term systemic infections in mammals. The role, relevance and possible molecular mechanisms of haemophagocytosis are discussed within the context of other microbial infections and of genetic deficiencies in which haemophagocytosis occurs and is associated with morbidity.
Introduction
A recent explosion of data reveals the exquisite molecular sophistication of host pathogen interactions (Monack et al., 2004; Fink and Cookson, 2007; Wick, 2007; García-Del Portillo et al., 2008; Haraga et al., 2008). Here we describe how an intracellular pathogen, Salmonella enterica, may survive long term in a mammalian host. The Gram-negative bacteria S. enterica is the causative agent of typhoid fever. As early as the mid-1800s, haemophagocytic macrophages, which are macrophages that have consumed red and white blood cells, were observed in the tissues and blood of recently deceased typhoid fever patients (Mallory, 1898; Fisman, 2000). Haemophagocytosis, defined as the phagocytosis of haematopoietic cells by activated macrophages, is associated with diverse and medically important infectious diseases, including typhoid fever, tuberculosis, leishmaniasis and influenza (Fisman, 2000; Janka, 2007; La Gruta et al., 2007). The role and relevance of haemophagocytosis are discussed within the context of systemic salmonellae and other microbial infections in which the phenomenon occurs.
Natural S. enterica infections and laboratory mice
Salmonella enterica subspecies cause a variety of natural infections in a wide range of host animals. Colonization can be limited to the gastrointestinal (GI) tract, resulting in enteritis, as with subspecies Typhimurium in humans. Alternatively, infection can become systemic with colonization of the liver, spleen and mesenteric lymph nodes (Tsolis et al., 1999). Subspecies Typhi or Paratyphi A, B and C cause human typhoid fever, an acute systemic infection associated with significant morbidity and mortality in populations lacking access to treated water. Approximately 5% of people in endemic regions are asymptomatic chronic carriers of the pathogen. Carriers are a public health concern because they shed the pathogen in the environment over the course of decades, resulting in the infection of naïve hosts (Parry et al., 2002).
Wild mice naturally become infected with S. enterica serotype Typhimurium (hereafter referred to as Salmonella) and develop systemic disease with colonization of the liver, spleen and mesenteric lymph nodes (Tsolis et al., 1999). Inbred laboratory mice inoculated with Salmonella have been used to model typhoid fever. There are two basic classes of mouse systemic infection models, those for studying acute disease and those for examining chronic infection and transmission (Monack et al., 2004). Acute disease is studied in mice defective for innate immunity. Generally, Slc11a1G169A homozygous mice, such as the Balb/C and C57Bl6 strains, are used. Slc11a1 (previously known as Nramp1) is an anti-porter of divalent metals and protons (Techau et al., 2007) in the endocytic membranes of neutrophils, macrophages and dendritic cells. Slc11a1G169A is a recessive loss of function allele that is pleiotropic and affects the production of reactive oxygen and nitrogen species, iron regulation and antigen presentation to T-cells (Gruenheid et al., 1997; Miller and Britigan, 1997; Canonne-Hergaux et al., 2002; Cellier et al., 2007; Stober et al., 2007). Slc11a1G169A mice are extremely susceptible to Salmonella infection and die of organ failure within a week (Vidal et al., 1995). These mice are also highly susceptible to leishmaniae and mycobacteriae infection (Huynh and Andrews, 2008). Exactly why Slc11a1 is needed to limit growth of intravacuolar pathogens is unclear, but it is likely a complex process.
Chronic systemic infections are modelled in mice that are wild type for Slc11a1. The disease course in these mice is analogous to that of human typhoid fever, as the animals suffer acute infection but generally recover even in the absence of antibiotic treatment. This model enables study of the transition from acute to chronic infection, maintenance of chronic infection and bacterial transmission to naïve animals (Monack et al., 2004; Nix et al., 2007; Lawley et al., 2008). Wild-type mice infected with Salmonella also provide a tractable system in which haemophagocytosis is studied (Nix et al., 2007).
A brief overview of systemic Salmonella infection
We describe a possible course of systemic infection in wild-type mice upon oral inoculation. Emphasis is placed on events that occur after the bacteria breach the intestinal barrier and become systemic, with the aim of integrating the phenomenon of haemophagocytosis into the existing literature.
From ingestion to deep tissue phagocytes
Salmonella infections begin with the ingestion of contaminated food or water (Ohl and Miller, 2001). Enteric bacteria, including salmonellae, encounter diverse elements of innate immunity in the GI tract, and use multiple molecular mechanisms to withstand mucosal immunity (Kagnoff, 2006; Pamer, 2007). To establish systemic infection, salmonellae must breach the epithelial wall of the GI tract. Once in the mouse small intestine, Salmonella can be ingested by Peyer’s Patch M-cells and transcytosed to underlying phagocytes (Jones et al., 1995). Alternatively, Salmonella may be ingested by dendritic cells, which return to systemic circulation carrying the bacteria (Vazquez-Torres et al., 1999; Rescigno et al., 2001). Both pathways culminate with the bacteria inside of a professional phagocyte, be it a macrophage, dendritic cell or neutrophil. Professional phagocytes, including neutrophils (Beauvillain et al., 2007), rapidly ingest microbes and travel to local lymph nodes to present microbial antigens to T-cells, thereby activating adaptive immunity. Salmonella appears to use phagocytes to gain access to the lymph nodes, spleen and liver, where the bacteria establish long-term infections.
Irrespective of dose or infection route, more than 80% of Salmonella within tissues are inside of professional phagocytes (Richter-Dahlfors et al., 1997; Salcedo et al., 2001; Monack et al., 2004). An intracellular location may allow bacteria to avoid immune system components (i.e. complement and antibodies), replicate or manipulate the host immune response. Macrophages are the cell type in which Salmonella are consistently found in both wild type and Slc11a1G169A mice (Fig. 1A) (Richter-Dahlfors et al., 1997; Salcedo et al., 2001; Sheppard et al., 2003; Monack et al., 2004; Nix et al., 2007). Salmonella mutants that cannot survive within tissue culture macrophages cannot survive within mice, indicating that bacterial survival within macrophages is fundamental for systemic colonization of the host (Buchmeier and Heffron, 1989). The bacteria have also been observed within neutrophils and dendritic cells. While dendritic cells are important for the early stages of infection, their role in later stages is unclear (Rydström and Wick, 2007; Wick, 2007). The role of neutrophils in systemic infection is also not well understood. Neutrophils ingest and eliminate Salmonella in Slc11a1G169A mice after oral infection (Rydström and Wick, 2007), but allow engulfed Salmonella to replicate after intraperitoneal inoculation with a large dose (Geddes et al., 2007). Whether Salmonella can replicate within neutrophils in more natural situations, such as in wild-type mice where the host immune system is not overwhelmed and the animal survives infection, is unknown.
Fig. 1.
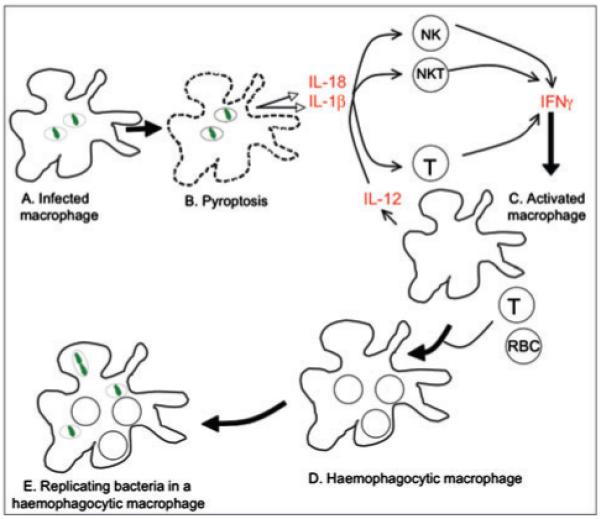
Model for the development of haemophagocytosis in S. enterica-infected mice. A.S. enterica (green) resides within macrophage vacuoles. B. The bacteria kill the macrophage via pyroptosis, releasing the inflammatory cytokines IL-18 and IL-1β. Early in infection, IL-18 and IL-1β stimulate NK and NKT cells to produce IFNγ. After the onset of adaptive immunity, T-cells (CD4 and CD8) also produce IFNγ. C. Positive feedback loops, including IL-12 stimulation of NK, NKT and T-cells, help maintain IFNγ production. D. IFNγ stimulates macrophages to phagocytose non-adherent cells. E. Haemophagocytic macrophages may provide S. enterica with a survival niche. It is unknown whether S. enterica reside within vacuoles in haemophagocytic macrophages, as drawn.
Salmonella kill phagocytes via pyropotosis, a cell death process that promotes IFNγ production
Even though Salmonella reside within phagocytes, the bacteria can kill macrophages and dendritic cells (Brennan and Cookson, 2000; van der Velden et al., 2003; Fink and Cookson, 2007) via a process that promotes host IFNγ production (Fink and Cookson, 2007). Phagocyte killing can be carried out by either of two type 3 secretion systems (T3SS), each of which manipulate host cell processes by delivering multiple bacterial proteins (effectors) into and/or across host cell membranes (Haraga et al., 2008). Both Salmonella T3SS are required for systemic chronic infection (Lawley et al., 2006) and both kill phagocytes via a specialized programmed cell death pathway called pyroptosis. Pyroptosis differs from the better-known process of apoptosis in at least two important ways. First, the molecular pathways involved in pyroptosis and apoptosis are distinct. Second pyroptosis, unlike apoptosis, causes tissue inflammation (Fink and Cookson, 2007). Pyroptosis requires host caspase-1 (Hersh et al., 1999; Brennan and Cookson, 2000; Jesenberger et al., 2000), which triggers the activation of the inflammatory transcription factor NF-κB (Lamkanfi et al., 2004), and also cleaves and activates the inflammatory cytokines IL-18 and IL-1β (Fantuzzi and Dinarello, 1999; Hersh et al., 1999). During pyroptosis, the host cell membrane breaks down (Brennan and Cookson, 2000), releasing the active IL-18 and IL-1β (Fig. 1B) (Fink and Cookson, 2007). Mice lacking caspase-1, IL-18 or IL-1β have increased susceptibility to Salmonella. IL-18 and IL-1β may help limit Salmonella replication early during infection at least in part by promoting IFNγ production (Lara-Tejero et al., 2006; Raupach et al., 2006). IL-18 and IL-1β stimulate natural killer (NK) cells and natural killer T (NKT) cells to secrete IFNγ within a few days of oral Salmonella inoculation of mice (Kirby et al., 2002; Berntman et al., 2005). IL-18 also contributes to CD4 T-cell IFNγ production during Salmonella infection (Srinivasan et al., 2007). Moreover, exogenous delivery of recombinant IL-18 protects mice from lethal doses of Salmonella in an IFNγ-dependent manner (Mastroeni et al., 1999). These observations collectively indicate that early during infection, pyroptosis may help the host control Salmonella by promoting IFNγ production.
IFNγ stimulates the formation of haemophagocytic macrophages, which may provide Salmonella with a survival niche in mice
IFNγ is a key cytokine for systemic Salmonella infection. It is released not only during pyroptosis but also from granules within splenic macrophages and neutrophils upon Salmonella infection of mice (Kirby et al., 2002). IFNγ stimulates macrophages to make IL-12, which in turn activates NK, NKT, CD4 T-cells and CD8 T-cells to secrete more IFNγ (Fig. 1C) (Berg and Forman, 2006). IFNγ is important for limiting Salmonella replication both during the first week of infection and in chronically infected mice (Nauciel and Espinasse-Maes, 1992; Monack et al., 2004). IFNγ-/- mice succumb to infection within days (Mastroeni et al., 1999), and treatment of chronically infected mice with neutralizing anti-IFNγ antibodies causes relapse into acute infection (Monack et al., 2004). High IFNγ levels in Salmonella-infected mice could contribute to haemophagocytic macrophage development (Fig. 1D). Macrophages within the liver of Salmonella-infected mice are haemophagocytic, as they contain multiple nuclei, many of which are surrounded by actin rings and colocalize with markers for lymphocytes or neutrophils. These observations in mice are consistent with cell culture experiments in which classically activated [e.g. IFNγ and lipopolysaccharide (LPS)-stimulated] macrophages incubated with non-adherent cell types, such as lymphocytes, become haemophagocytic (Nix et al., 2007). These data support the hypothesis that IFNγ contributes to the formation of haemophagocytic macrophages in Salmonella-infected mice.
Haemophagocytic macrophages may be important for the establishment and maintenance of chronic Salmonella infections because they can provide the bacteria with a survival niche. Salmonella have been observed within haemophagocytic macrophages in the liver and spleen of mice at 1, 3 and 8 weeks post infection, and cell culture haemophagocytic macrophages are permissive for Salmonella replication (Nix et al., 2007). In contrast, classically activated non-haemophagocytic macrophages, which have phagocytosed nothing or polystyrene beads, kill the bacteria (Vazquez-Torres et al., 2000; Mosser, 2003; Nix et al., 2007). Thus, haemophagocytic macrophages may enable Salmonella to chronically infect mice (Fig. 1E).
IFNγ, haemophagocytosis and human disease
Haemophagocytic cells in the blood and bone marrow are common in typhoid fever patients and have been referred to as ‘Typhoidal cells’ (Macias, 1975; Shin et al., 1994). However, the phenomenon is by no means unique to typhoid fever. Haemophagocytosis is associated with a variety of genetic lesions and diverse intracellular microbial pathogens, including viruses, mycobacteriae, spirochaetes, fungi and protozoal parasites (Fisman, 2000; Janka, 2007). For example, haemophagocytosis is a classic, although uncommon, feature of tuberculosis (Claessens et al., 2006), and a formally recognized characteristic of severe human influenza (La Gruta et al., 2007).
Haemophagocytosis is a clinical feature of haemophagocytic lymphohistiocytosis (HLH), a syndrome characterized by the uncontrolled activation and proliferation of macrophages and T-cells. Additional symptoms of HLH include fever, cytopenias, hepatosplenomegaly and high serum ferritin and cytokines (e.g. TNFα and IFNγ) (Grom, 2004; Janka, 2007). HLH is frequently associated with infections caused by intracellular microbial pathogens. Administered therapies vary between patients but generally include one or more of the following: corticosteroids (e.g. methlyprednisolone), cell cycle inhibitors (e.g. cyclosporin A), TNF inhibitors (e.g. etanercept) and blood transfusions (Fisman, 2000; Grom, 2004). Unfortunately, significant numbers of patients respond poorly to therapy and die of organ failure (Janka, 2007). HLH clearly represents an extreme situation in which haemophagocytosis occurs. An understanding of the mechanisms that cause haemophagocytosis could suggest novel therapies for HLH patients.
A standard clinical feature of patients with haemophagocytosis is high IFNγ levels. This, and data from Salmonella mouse and tissue culture models, suggest that IFNγ contributes to the phenomenon of haemophagocytosis. One pathway by which too much IFNγ can be produced involves NK or CD8 T-cells, which normally use perforin to create a pore in the target cell membrane through which apoptosiscausing proteins, granzymes, are delivered. Mutations that impair perforin-mediated target cell killing, including defects in perforin, granzymes or granule exocytosis, are associated with haemophagocytosis in humans. The idea is that NK and CD8 T-cells recognize but cannot kill target cells and remain in a partially activated state in which they continue to produce IFNγ, causing excessive macrophage activation. The mechanism(s) responsible for normal IFNγ downregulation after target cell killing is unknown (Grom, 2003; Janka, 2007). Experiments with perforin knockout mice support this model. Normally, perforin-/- mice appear healthy, but infection with lymphocytic choriomeningitic virus (LCMV) results in haemophagocytosis. LCMV is an RNA virus that causes natural, non-cytopathic infections in wild mice. Perforin-/- mice infected with LCMV develop haemophagocytosis and high serum cytokine levels, including IFNγ, within 10-12 days. Depletion of CD8 T-cells or treatment with anti-IFNγ antibodies decreases serum IFNγ levels and reduces associated cytopenias caused by macrophage ingestion of red and white blood cells. In contrast, treatment with neutralizing antibodies to a panel of cytokines (TNFα, M-CSF, GM-CSF, IL-10, IL-12 and IL-18) or depletion of NK or CD4 T-cells has no effect. These data suggest that a lack of perforin leads to over-production of INFγ by CD8 T-cells, and that high INFγ levels can promote haemophagocytosis (Jordan et al., 2004).
INFγ may also contribute to haemophagocytosis observed in cases of severe influenza caused by the highly pathogenic avian H5N1 viruses (La Gruta et al., 2007). When cultured dendritic cells present a particular fragment of the influenza hemagglutinin (H5) protein to human CD8 T-cells, survival of both the dendritic cells and the T-cells increases, effectively prolonging their interaction. Moreover, perforin protein levels within the T-cells decline, and IFNγ secretion increases (Hsieh and Chang, 2006). This suggests that presentation of select influenza antigens to CD8 T-cells can result in increased INFγ production and contribute to haemophagocytosis.
In the case of Typhoid fever, bacterially induced macrophage pyroptosis could contribute to increasing host IFNγ levels. High-tissue IFNγ could then be maintained throughout chronic infection by feedback mechanisms that would not necessarily require continuous pyroptosis. For instance, positive feedback occurs between macrophages producing IL-12 and NK or T-cells producing IFNγ. Similarly, splenocytes exposed to IFNγ produce IL-18, which further increases IFNγ production (Mastroeni et al., 1999). IFNγ stimulation of macrophage haemophagocytosis could then provide Salmonella with a host cell in which the bacteria can survive and replicate.
Questions
Is haemophagocytosis always detrimental to the host, or could it be beneficial at low levels?
The fact that haemophagocytosis appears to be a common host response to intracellular pathogens suggests that it may reflect a normal host process that is co-opted or gets out of control. For instance, liver and splenic macrophages normally phagocytose and remove senescent red blood cells (RBCs), and infection-associated haemophagocytosis could be an amplification and extension of this process to include phagocytosis of leucocytes. Whether the haemophagocytic macrophages that accumulate during infection descend from tissue macrophages or are recruited as monocytes from the blood is not yet known. Another idea is that modest numbers of haemophagocytic cells generated by localized IFNγ production may benefit the host by controlling or clearing intracellular pathogens. For instance, in the early stages of Salmonella infection, haemophagocytic macrophages could shift the balance from acute to chronic disease, benefiting the host because chronic disease is asymptomatic. Excessive haemophagocytosis may result from high systemic IFNγ levels, which, in extreme cases, can be fatal, as RBC depletion reduces oxygen delivery to tissues. A need to have an appropriate level and distribution of IFNγ may be conceptually analogous to the situation of TNFα, which is essential locally for containing microbes, but causes lethal shock when released systemically in large amounts.
How are haemophagocytic and non-haemophagocytic macrophages different?
In vivo, haemophagocytic macrophages are distinguished from non-haemophagocytic macrophages by the observation that the former contain haematopoietic cells (Janka, 2007), a characteristic that reveals little about potential functional differences. Why tissue culture haemophagocytic macrophages enable Salmonella to replicate is also unknown, but more than one mechanism may be involved. First, RBC uptake by macrophages could protect bacteria from damaging reactive oxygen species (ROS), as oxidation of haemoglobin may decrease the amount of ROS available for killing microbes (Hand and King-Thompson, 1983). This is consistent with cell culture experiments in which macrophages that phagocytose RBCs do not efficiently kill Salmonella or Staphylococcus aureus during the first hour of infection (Gill et al., 1966; Hand and King-Thompson, 1983). Second, iron accumulation in macrophages could also interfere with macrophage killing of microbes. Iron overload in mice increases susceptibility to microbes, including salmonellae (Jones et al., 1977; Sawatzki et al., 1983). Humans and mice with natural or experimentally induced RBC accumulation in liver macrophages also have increased susceptibility to bacterial infections (Kaye and Hook, 1963; Roy et al., 2007). Thus, excess iron may promote microbe survival by functioning as a cofactor for microbial enzymes, by decreasing macrophage TNFα, NO and ROS production (Collins et al., 2002; Nairz et al., 2007), or by affecting T-cell activation (Mencacci et al., 1997).
Salmonella can also replicate in macrophages that have phagocytosed lymphocytes instead of RBCs. Specifically, macrophages stimulated with IFNγ and LPS [such that they are classically activated (Mosser, 2003)] can phagocytose live or dead lymphocytes, but only those with live lymphocytes allow Salmonella to replicate (Nix et al., 2007). How macrophages become haemophagocytic, and how live lymphocytes change macrophages are not known. It is possible that IFNγ and LPS stimulation can alter macrophages such that they can phagocytose live cells, thus becoming haemophagocytic. This could involve, for instance, new receptors on macrophages or loss of inhibitory ligands. Phagocytosis of live lymphocytes may change how macrophages respond to pathogens. This could be mediated through receptors on live cells that normally prevent uptake by macrophages, such as CD31 and CD47. CD31 is an immunoglobulin family member that promotes live cell detachment from macrophages and thereby prevents uptake (Brown et al., 2002). CD47 is an integrin-associated protein that binds SIRPα/SHPS-1 on macrophages. Phosphorylation of SIRPα leads to activation of SHP-1 (Src homology-containing tyrosine phosphatase-1), which blocks antibody- and complement-mediated phagocytosis (Oldenborg, 2004; Gardai et al., 2006). Thus, signals from lymphocytes received by haemophagocytic macrophages during or after engulfment may render macrophages unable to kill Salmonella.
What enables microbes to replicate within haemophagocytic macrophages?
An important issue is whether haemophagocytic macrophages are generally permissive for microbial replication or only permit certain microbes to replicate. If only Salmonella can replicate in haemophagocytic macrophages, there must be Salmonella-specific genes that allow survival. If most or all microbes can replicate in haemophagocytic macrophages, this would suggest that microbes have common strategies for long-term survival in animals. Further studies are needed to establish whether other intracellular pathogens reside in vivo in haemophagocytic macrophages.
Can markers of haemophagocytic cells be identified?
One current limitation to the study of haemophagocytosis is the lack of molecular markers for haemophagocytic cells. In the clinic, haemophagocytic blood or bone marrow cells are identified by standard microscopic analysis of blood or bone marrow cells (Shin et al., 1994). In the laboratory, haemophagocytic cells within solid tissue can be identified with confocal microscopy using a combination of fluorescent probes to macrophages and other cell types. As many different non-adherent cell types have been observed within haemophagocytic macrophages (Fisman, 2000), the simultaneous use of many different markers would be required to quantify tissue haemophagocytic macrophages. In addition, it is important to distinguish haemophagocytic macrophages from cells that have undergone nuclear division or cell-cell fusion. It is at least formally possible that a macrophage could be haemophagocytic, undergo division of its endogenous nucleus and also fuse with other cells. Thus, specific molecular markers of haemophagocytic cells are needed.
Conclusions
Haemophagocytosis is a poorly understood phenomenon associated with intracellular infections and genetic lesions. The recent development of mouse models of infection-associated haemophagocytosis (Jordan et al., 2004; Nix et al., 2007) and of cell culture models for haemophagocytic cells (Nix et al., 2007) will facilitate a molecular and cellular understanding of the development of haemophagocytosis in infectious disease. This may lead to more effective therapies for patients with severe haemophagocytosis.
Acknowledgements
We thank Kim Erickson, Greg Plano and Josh Myatt for critically reading the manuscript. Due to space constraints, many important studies were not directly referenced but are instead described within cited reviews. Supported by NIH R01-AI072492 and R21-AI076682 to CSD.
References
- Beauvillain C, Delneste Y, Scotet M, Peres A, Gascan H, Guermonprez P, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood. 2007;110:2965–2973. doi: 10.1182/blood-2006-12-063826. [DOI] [PubMed] [Google Scholar]
- Berg RE, Forman J. The role of CD8 T cells in innate immunity and in antigen non-specific protection. Curr Opin Immunol. 2006;18:338–343. doi: 10.1016/j.coi.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Berntman E, Rolf J, Johansson C, Anderson P, Cardell SL. The role of CD1d-restricted NK T lymphocytes in the immune response to oral infection with Salmonella typhimurium. Eur J Immunol. 2005;35:2100–2109. doi: 10.1002/eji.200425846. [DOI] [PubMed] [Google Scholar]
- Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418:200–203. doi: 10.1038/nature00811. [DOI] [PubMed] [Google Scholar]
- Buchmeier NA, Heffron F. Intracellular survival of wild-type Salmonella typhimurium and macrophage-sensitive mutants in diverse populations of macrophages. Infect Immun. 1989;57:1–7. doi: 10.1128/iai.57.1.1-7.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canonne-Hergaux F, Calafat J, Richer E, Cellier M, Grinstein S, Borregaard N, Gros P. Expres- sion and subcellular localization of NRAMP1 in human neutrophil granules. Blood. 2002;100:268–275. doi: 10.1182/blood.v100.1.268. [DOI] [PubMed] [Google Scholar]
- Cellier MF, Courville P, Campion C. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infection. 2007;9:1662–1670. doi: 10.1016/j.micinf.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Claessens Y, Pene F, Tulliez M, Cariou A, Chiche J. Life-threatening haemophagocytic syndrome related to mycobacterium tuberculosis. Eur J Emerg Med. 2006;13:172–174. doi: 10.1097/01.mej.0000190275.85107.55. [DOI] [PubMed] [Google Scholar]
- Collins HL, Kaufmann SHE, Schaible UE. Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst. J Immunol. 2002;168:3458–3463. doi: 10.4049/jimmunol.168.7.3458. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1 beta: two cytokine substrates for ICE (caspase-1) J Clin Immunol. 1999;19:1–11. doi: 10.1023/a:1020506300324. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- Fisman DN. Haemophagocytic syndromes and infection. Emerg Infect Dis. 2000;6:601–608. doi: 10.3201/eid0606.000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Del Portillo F, Núñez-Hernández C, Eisman B, Ramos-Vivas J. Growth control in the Salmonella-containing vacuole. Curr Opin Microbiol. 2008;11:46–52. doi: 10.1016/j.mib.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol. 2006;79:896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- Geddes K, Cruz F, Heffron F. Analysis of cells targeted by salmonella type III secretion in vivo. PLoS Pathog. 2007;3:e196. doi: 10.1371/journal.ppat.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill FA, Kaye D, Hook EW. The influence of erythrophagocytosis on the interaction of macrophages and salmonella in vitro. J Exp Med. 1966;124:173–183. doi: 10.1084/jem.124.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grom AA. Macrophage activation syndrome and reactive haemophagocytic lymphohistiocytosis: the same entities? Curr Opin Rheumatol. 2003;15:587–590. doi: 10.1097/00002281-200309000-00011. [DOI] [PubMed] [Google Scholar]
- Grom AA. Natural killer cell dysfunction: a common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and haemophagocytic lymphohistiocytosis? Arthritis Rheum. 2004;50:689–698. doi: 10.1002/art.20198. [DOI] [PubMed] [Google Scholar]
- Gruenheid S, Pinner E, Desjardins M, Gros P. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med. 1997;185:717–730. doi: 10.1084/jem.185.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand WL, King-Thompson NL. Effect of erythrocyte ingestion on macrophage antibacterial function. Infect Immun. 1983;40:917–923. doi: 10.1128/iai.40.3.917-923.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol. 2008;6:53–66. doi: 10.1038/nrmicro1788. [DOI] [PubMed] [Google Scholar]
- Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA. 1999;96:2396–2401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh S, Chang S. Insufficient perforin expression in CD8+ T cells in response to hemagglutinin from avian influenza (H5N1) virus. J Immunol. 2006;176:4530–4533. doi: 10.4049/jimmunol.176.8.4530. [DOI] [PubMed] [Google Scholar]
- Huynh C, Andrews NW. Iron acquisition within host cells and the pathogenicity of Leishmania. Cell Microbiol. 2008;10:293–300. doi: 10.1111/j.1462-5822.2007.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janka GE. Haemophagocytic syndromes. Blood Rev. 2007;21:245–253. doi: 10.1016/j.blre.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Jesenberger V, Procyk KJ, Yuan J, Reipert S, Baccarini M. Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis. J Exp Med. 2000;192:1035–1046. doi: 10.1084/jem.192.7.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B, Pascopella L, Falkow S. Entry of microbes into the host: using M cells to break the mucosal barrier. Curr Opin Immunol. 1995;7:474–478. doi: 10.1016/0952-7915(95)80091-3. [DOI] [PubMed] [Google Scholar]
- Jones RL, Peterson CM, Grady RW, Kumbaraci T, Cerami A, Graziano JH. Effects of iron chelators and iron overload on Salmonella infection. Nature. 1977;267:63–65. doi: 10.1038/267063a0. [DOI] [PubMed] [Google Scholar]
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of haemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- Kagnoff MF. Microbial-epithelial cell crosstalk during inflammation: the host response. Ann N Y Acad Sci. 2006;1072:313–320. doi: 10.1196/annals.1326.038. [DOI] [PubMed] [Google Scholar]
- Kaye D, Hook EW. The influence of hemolysis or blood loss on susceptibility to infection. J Immunol. 1963;91:65–75. [PubMed] [Google Scholar]
- Kirby AC, Yrlid U, Wick MJ. The innate immune response differs in primary and secondary Salmonella infection. J Immunol. 2002;169:4450–4459. doi: 10.4049/jimmunol.169.8.4450. [DOI] [PubMed] [Google Scholar]
- La Gruta NL, Kedzierska K, Stambas J, Doherty PC. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol. 2007;85:85–92. doi: 10.1038/sj.icb.7100026. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Kalai M, Saelens X, Declercq W, Vandenabeele P. Caspase-1 activates nuclear factor of the kappa-enhancer in B cells independently of its enzymatic activity. J Biol Chem. 2004;279:24785–24793. doi: 10.1074/jbc.M400985200. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Chan K, Thompson LJ, Kim CC, Govoni GR, Monack DM. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2006;2:e11. doi: 10.1371/journal.ppat.0020011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Bouley DM, Hoy YE, Gerke C, Relman DA, Monack DM. Host transmission of Salmonella enterica serovar Typhimurium is controlled by virulence factors and indigenous intestinal microbiota. Infect Immun. 2008;76:403–416. doi: 10.1128/IAI.01189-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias EG. Letter: typhoidal cells. Lancet. 1975;2:927–928. doi: 10.1016/s0140-6736(75)92165-0. [DOI] [PubMed] [Google Scholar]
- Mallory FB. A histological study of typhoid fever. J Exp Med. 1898;3:611–638. [PMC free article] [PubMed] [Google Scholar]
- Mastroeni P, Clare S, Khan S, Harrison JA, Hormaeche CE, Okamura H, et al. Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulent Salmonella typhimurium. Infect Immun. 1999;67:478–483. doi: 10.1128/iai.67.2.478-483.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencacci A, Cenci E, Boelaert JR, Bucci P, Mosci P, Fè d’Ostiani C, et al. Iron overload alters innate and T helper cell responses to Candida albicans in mice. J Infect Dis. 1997;175:1467–1476. doi: 10.1086/516481. [DOI] [PubMed] [Google Scholar]
- Miller RA, Britigan BE. Role of oxidants in microbial pathophysiology. Clin Microbiol Rev. 1997;10:1–18. doi: 10.1128/cmr.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Bouley DM, Falkow S. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J Exp Med. 2004;199:231–241. doi: 10.1084/jem.20031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Mueller A, Falkow S. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol. 2004;2:747–765. doi: 10.1038/nrmicro955. [DOI] [PubMed] [Google Scholar]
- Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Nairz M, Theurl I, Ludwiczek S, Theurl M, Mair SM, Fritsche G, Weiss G. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007;9:2126–2140. doi: 10.1111/j.1462-5822.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- Nauciel C, Espinasse-Maes F. Role of gamma interferon and tumor necrosis factor alpha in resistance to Salmonella typhimurium infection. Infect Immun. 1992;60:450–454. doi: 10.1128/iai.60.2.450-454.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix RN, Altschuler SE, Henson PM, Detweiler CS. Haemophagocytic macrophages harbor Salmonella enterica during persistent infection. PLoS Pathog. 2007;3:e193. doi: 10.1371/journal.ppat.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohl ME, Miller SI. Salmonella: a model for bacterial pathogenesis. Annu Rev Med. 2001;52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- Oldenborg P. Role of CD47 in erythroid cells and in autoimmunity. Leuk Lymphoma. 2004;45:1319–1327. doi: 10.1080/1042819042000201989. [DOI] [PubMed] [Google Scholar]
- Pamer EG. Immune responses to commensal and environmental microbes. Nat Immunol. 2007;8:1173–1178. doi: 10.1038/ni1526. [DOI] [PubMed] [Google Scholar]
- Parry CM, Hien TT, Dougan G, White NJ, Farrar JJ. Typhoid fever. N Engl J Med. 2002;347:1770–1782. doi: 10.1056/NEJMra020201. [DOI] [PubMed] [Google Scholar]
- Raupach B, Peuschel S, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescigno M, Rotta G, Valzasina B, Ricciardi-Castagnoli P. Dendritic cells shuttle microbes across gut epithelial monolayers. Immunobiology. 2001;204:572–581. doi: 10.1078/0171-2985-00094. [DOI] [PubMed] [Google Scholar]
- Richter-Dahlfors A, Buchan AM, Finlay BB. Murine salmonellosis studied by confocal microscopy: Salmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J Exp Med. 1997;186:569–580. doi: 10.1084/jem.186.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Riendeau N, Bédard C, Hélie P, Min-Oo G, Turcotte K, et al. Pyruvate kinase deficiency confers susceptibility to Salmonella typhimurium infection in mice. J Exp Med. 2007;204:2949–2961. doi: 10.1084/jem.20062606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydström A, Wick MJ. Monocyte recruitment, activation, and function in the gut-associated lymphoid tissue during oral Salmonella infection. J Immunol. 2007;178:5789–5801. doi: 10.4049/jimmunol.178.9.5789. [DOI] [PubMed] [Google Scholar]
- Salcedo SP, Noursadeghi M, Cohen J, Holden DW. Intracellular replication of Salmonella typhimurium strains in specific subsets of splenic macrophages in vivo. Cell Microbiol. 2001;3:587–597. doi: 10.1046/j.1462-5822.2001.00137.x. [DOI] [PubMed] [Google Scholar]
- Sawatzki G, Hoffmann FA, Kubanek B. Acute iron overload in mice: pathogenesis of Salmonella typhimurium infection. Infect Immun. 1983;39:659–665. doi: 10.1128/iai.39.2.659-665.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard M, Webb C, Heath F, Mallows V, Emilianus R, Maskell D, Mastroeni P. Dynamics of bacterial growth and distribution within the liver during Salmonella infection. Cell Microbiol. 2003;5:593–600. doi: 10.1046/j.1462-5822.2003.00296.x. [DOI] [PubMed] [Google Scholar]
- Shin BM, Paik IK, Cho HI. Bone marrow pathology of culture proven typhoid fever. J Korean Med Sci. 1994;9:57–63. doi: 10.3346/jkms.1994.9.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A, Salazar-Gonzalez R, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in salmonella-infected mice is dependent on IL-18. J Immunol. 2007;178:6342–6349. doi: 10.4049/jimmunol.178.10.6342. [DOI] [PubMed] [Google Scholar]
- Stober CB, Brode S, White JK, Popoff J, Blackwell JM. Slc11a1, formerly Nramp1, is expressed in dendritic cells and influences major histocompatibility complex class II expression and antigen-presenting cell function. Infect Immun. 2007;75:5059–5067. doi: 10.1128/IAI.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techau ME, Valdez-Taubas J, Popoff J, Francis R, Seaman M, Blackwell JM. Evolution of differences in transport function in Slc11a family members. J Biol Chem. 2007;282:35646–35656. doi: 10.1074/jbc.M707057200. [DOI] [PubMed] [Google Scholar]
- Tsolis RM, Kingsley RA, Townsend SM, Ficht TA, Adams LG, Bäumler AJ. Of mice, calves, and men. Comparison of the mouse typhoid model with other Salmonella infections. Adv Exp Med Biol. 1999;473:261–274. [PubMed] [Google Scholar]
- Vazquez-Torres A, Jones-Carson J, Bäumler AJ, Falkow S, Valdivia R, Brown W, et al. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804–808. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- Vazquez-Torres A, Jones-Carson J, Mastroeni P, Ischiropoulos H, Fang FC. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J Exp Med. 2000;192:227–236. doi: 10.1084/jem.192.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velden AWM, Velasquez M, Starnbach MN. Salmonella rapidly kill dendritic cells via a caspase-1-dependent mechanism. J Immunol. 2003;171:6742–6749. doi: 10.4049/jimmunol.171.12.6742. [DOI] [PubMed] [Google Scholar]
- Vidal S, Tremblay ML, Govoni G, Gauthier S, Sebastiani G, Malo D, et al. The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J Exp Med. 1995;182:655–666. doi: 10.1084/jem.182.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick MJ. Monocyte and dendritic cell recruitment and activation during oral Salmonella infection. Immunol Lett. 2007;112:68–74. doi: 10.1016/j.imlet.2007.07.007. [DOI] [PubMed] [Google Scholar]