

Abstract
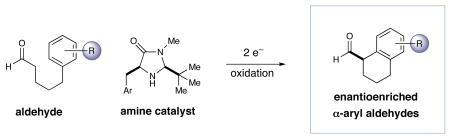
The intramolecular α-arylation of aldehydes has been accomplished using singly occupied molecular orbital (SOMO) catalysis. Selective oxidation of chiral enamines (formed by the condensation of an aldehyde and a secondary amine catalyst) leads to the formation of a 3π-electron radical species. These chiral SOMO-activated radical cations undergo enantioselective reaction with an array of pendent electron-rich aromatics and heterocycles thus efficiently providing cyclic α-aryl aldehyde products (10 examples: ≥70% yield and ≥90% ee). In accordance with our radical mechanism, when there is a choice between arylation at the ortho or para position of anisole substrates, we find that arylation proceeds selectively at the ortho position.
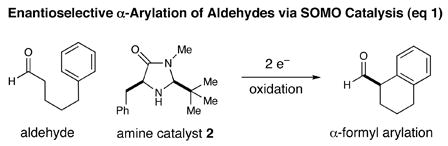
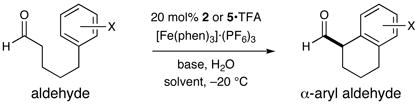
Driven primarily by advances in transition metal chemistry, the catalytic union of enolates with aryl halides has become a mainstay transformation in asymmetric synthesis.1 In particular, the pioneering work of Buchwald and Hartwig has provided a number of enantioselective enolate α-arylations that allow the catalytic formation of α-stereogenic carbonyls that are non-enolizable (e.g. quaternary carbon centers).2–4 Recently, our laboratory introduced a new mode of organocatalytic activation, termed SOMO catalysis, that is founded upon the mechanistic hypothesis that a 3π-radical cation species (e.g. 1) can readily participate in a range of unique asymmetric bond constructions.5 As part of these studies, we documented the first direct and enantioselective α-allylic alkylation,5b α-enolation,5c and α-vinylation5d of aldehydes, three protocols that were not previously known. Continuing this theme, we recently questioned whether the α-arylation of aldehydes might also be accomplished via SOMO catalysis to build and maintain enantioenriched benzylic stereocenters (eq 1). Herein, we describe the enantioselective intramolecular aldehyde α-arylation, a transformation that allows the highly efficient production of enolizable α-formyl-α-aryl substrates without post-reaction epimerization or quaternary carbon formation.6 Moreover, this protocol is highly ortho-selective using 1,3-disubstituted aryl rings, a finding that is consistent with a radical based (open shell) arylation mechanism.7
 |
(1) |
 |
(2) |
Design Plan
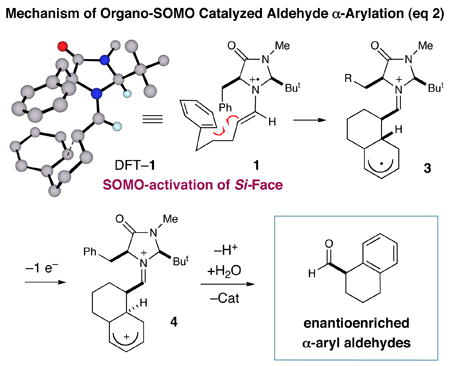
In accord with our previous SOMO-catalysis studies,5 we reasoned that exposure of an aryl-tethered aldehyde to amine catalyst 2 in the presence of a suitable oxidant would render the radical-cation 1 (DFT–1). Rapid and enantioselective addition of the pendent aryl ring system to the 3π-electron species would then provide a bicyclic radical cation 3 that upon further oxidation would generate a dienyl cation 4 (eq 2). At this stage, we presumed that rearomatization via proton loss would be facile while hydrolysis of the iminium species would reconstitute the catalyst and deliver the desired formyl α-arylation product. Given the predilection of electron-rich aryl rings to undergo ortho-selective addition to radical cations,7 we presumed that high levels of regiochemical control should be possible if a traditional SOMO catalysis mechanism is operative. Moreover, we presumed that the activated radical DFT–1 would position the 3π-electron system away from the bulky tert-butyl group, while the benzyl group on the imidazolidinone framework effectively shields the Re-face leaving the Si-face open to enantioselective arylation.
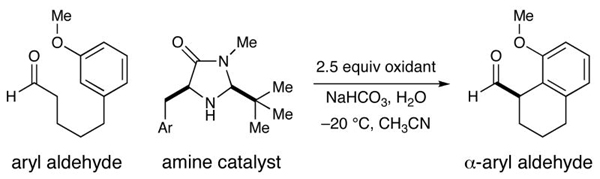
Our intramolecular aldehyde α-arylation was first evaluated using 3-anisole pentanaldehyde, catalyst 2 or 5, and a selection of oxidants (Table 1). Initial experiments with ceric ammonium nitrate (CAN) revealed that the proposed transformation was possible, however reaction efficiency and enantioselectivity levels were found to be moderate. Not satisfied with the general utility of these reaction conditions, we sought to find a superior catalyst/oxidant combination. As revealed in Table 1, use of [Fe(phen)3]·(PF6)3 as a single electron oxidant allowed for a significant increase in enantiocontrol while application of a recently designed imidazolidinone catalyst 5 enabled optimal reaction efficiency and a further increase in stereocontrol (with 20 mol % catalyst). High-throughput experiments using a robotic platform (see Supporting Information) revealed that tightly controlled quantities of additives such as pivalic acid and water could also be employed to boost efficiency (entries 3–6). Given the notable selectivities achieved with both amines 2 and 5, we selected both catalysts for further exploration.
Table 1.
Development of Organo-SOMO Aldehyde α-Arylation.
 | ||||
|---|---|---|---|---|
| entry | oxidant | catalyst (Ar) % | yielda | % eeb |
| 1 | CAN | Ph, 2 (30 mol %) | 63 | 84 |
| 2 | [Fe(phen)3]·(PF6)3 | Ph, 2 (30 mol %) | 60 | 92 |
| 3c | [Fe(phen)3]· PF6)3 | Ph, 2 (30 mol %) | 85 | 92 |
| 4c | [Fe(phen)3]·(PF6)3 | Ph, 2 (20 mol %) | 61 | 90 |
| 5c | [Fe(phen)3]·(PF6)3 | 1-Naphth, 5 (20 mol %) | 70 | 97 |
| 6c,d | [Fe(phen)3]·(PF6)3 | 1-Naphth, 5 (20 mol %) | 80 | 98 |
Isolated yields of the corresponding alcohol.
Enantiomeric excess determined by chiral HPLC.
30 mol % pivalic acid.
50 mol % H2O.
As highlighted in Table 2, a wide array of aryl and heteroaryl ring systems readily participate as SOMOphiles in this intramolecular α-aldehyde coupling. For example, anisole, naphthylene, indole, pyrrole, thiophene and furan ring systems all function well9 in this new enantioselective ring forming reaction. Moreover, aldehydes that incorporate nitrogen, oxygen or carbonyl containing tethers worked well in this protocol (entries 2, 4, and 10, catalyst 5: 93–98% ee). As is clearly shown in Table 2, the imidazolidinone catalyst 5 is far superior to amine 2 with respect to enantioselectivity and cyclization efficiency. It is also important to note that these electron-rich aryl ring systems are completely tolerant to the mild oxidative conditions employed in this study. Furthermore, substrates that have the potential for aromatic substitution at two regiochemical positions (ortho or para) were in fact highly selective for ortho-functionalization (entries 1–5, ortho-substitution only).10 Such regioselectivity provides further evidence that a radical-mediated addition pathway is operative, as proposed in equation 2. It should be noted that a cationic Friedel-Crafts mechanism was recently proposed for a similar arylation reaction using our imidazolidinone catalyst 2;8 a mechanistic interpretation based on the exclusive observation of para-selective products. However, the authors have subsequently issued a correction8b that several of their adducts were in fact formed with ortho-selectivity (Table 3, entries 1 and 3). As such, given (i) our previous mechanistic studies into SOMO-activation, wherein an open-shell pathway was definitively shown using a radical-clock probe,5a and (ii) the observation of only ortho-selective products using 1,3-disubstituted aryl rings, we presume that a radical cyclization is operative for the substrates described in this manuscript.9, 10
Table 2.
Scope of the Organo-SOMO Intramolecular α-Arylation.
| |||||||
|---|---|---|---|---|---|---|---|
| entrya | Product | cat: yield,b eec | entrya | product | cat: yield,b eec | ||
| 1 | 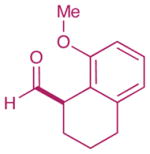 |
2: 61% yield 92% ee 5: 80% yield 98% ee |
2 | 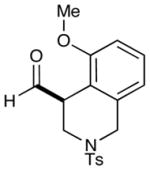 |
2: 84% yield 74% ee 5: 86% yield 95% ee |
||
| 3 | 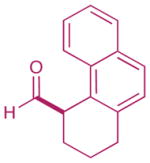 |
2: 83% yield 93% ee 5: 73% yield 96% ee |
4 | 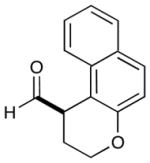 |
2: 72% yield 92% ee 5: 70% yield 95% ee |
||
| 5 | 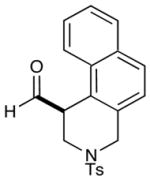 |
2: 70% yield 97% ee 5: 71% yield 96% ee |
6 | 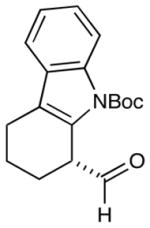 |
2: 86% yield 92% ee 5: 84% yield 96% ee |
||
| 7 | 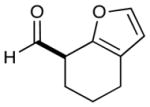 |
2: 89% yield 58% ee 5: 96% yield 90% ee |
8 | 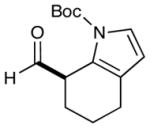 |
2: 84% yield 91% ee 5: 82% yield 96% ee |
||
| 9 | 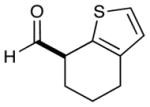 |
2: 86% yield 76% ee 5: 96% yield 94% ee |
10 | 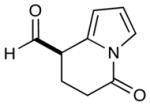 |
2: 72% yield 81% ee 5: 72% yield 93% ee |
||
Conditions, entries 1–2: [Fe(phen)3]·(PF6)3, NaHCO3, HOPiv, −20 °C MeCN; entries 3–5: [Fe(phen)3]·(PF6)3, Na2HPO4, −30 °C acetone; entries 6–10: CAN, NaHCO3, NaO2CCF3, −30 °C acetone.
Isolated yields after NaBH4 reduction.
Determined by chiral HPLC of corresponding alcohols.
 |
(4) |
 |
(5) |
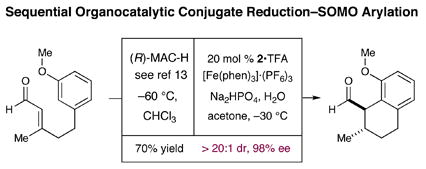
We have been able to combine our novel SOMO a-arylation reaction into a two-step organocatalytic sequence that first involves a Hantzsch ester hydride (HEH) reduction11 of an anisole tethered α,β-unsaturated aldehyde followed by the protocol described herein. As shown in equation 4, this sequence provides the resulting bicyclic ring system as a single (anti) diastereomer in 70% yield and 98% ee.
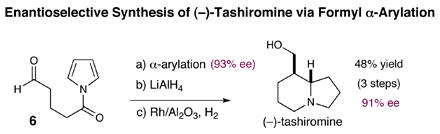
We have also been able to apply this new carbonyl α-arylation technology towards the total synthesis of (−)-tashiromine.12 As outlined in equation 5, exposure of the pyrrole amide tethered aldehyde 6 to this new catalytic procedure provides the desired [6,5]-bicyclic ring system in 72% yield and in 93% ee. The resulting amide was then reduced in the presence of AlCl3 with LiAlH4 in 83% yield prior to exhaustive pyrrole hydrogenation using catalytic Rh/Al2O3 and HOCH(CF3)2 to provide (−)-tashiromine in 81% yield and 4:1 diastereoselectivity.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM078201-01-01) and kind gifts from Merck. JCC thanks NSERC for a postdoctoral fellowship.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided (x pages, PDF).
References
- 1.a) Muratake H, Nakai H. Tetrahedron Lett. 1999;40:2355. [Google Scholar]; b) García-Fortanet J, Buchwald SL. Angew Chem Int Ed. 2008;47:8108. doi: 10.1002/anie.200803809. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vo GD, Hartwig JF. Angew Chem Int Ed. 2008;47:2127. doi: 10.1002/anie.200705357. [DOI] [PubMed] [Google Scholar]
- 2.a) Liao X, Weng Z, Hartwig JF. J Am Chem Soc. 2008;130:195. doi: 10.1021/ja074453g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) García-Fortanet J, Buchwald SL. Angew Chem Int Ed. 2008;47:8108. doi: 10.1002/anie.200803809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.This racemization has been exploited for dynamic kinetic resolutions: Xie JH, Zhou ZT, Kong WL, Zhou QL. J Am Chem Soc. 2007;129:1868. doi: 10.1021/ja0680109.
- 4.With quinone Michael acceptors: Alemán J, Cabrera S, Maerten E, Overgaard J, Jørgensen KA. Angew Chem Int Ed. 2007;46:5520. doi: 10.1002/anie.200701207.
- 5.a) Beeson TD, Mastracchio A, Hong J-B, Ashton K, MacMillan DWC. Science. 2007;316:582. [PubMed] [Google Scholar]; b) Jang H-Y, Hong J-B, MacMillan DWC. J Am Chem Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]; c) Kim H, MacMillan DWC. J Am Chem Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]; d) Graham TH, Jones CM, Jui NT, MacMillan DWC. J Am Chem Soc. 2008;130:16494. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Details of this work were described at the ACS Centennial Symposium MacMillan, David W. C. Abstracts of Papers, 236th ACS National Meeting, Philadelphia, PA, United States, August 17–21, 2008 (2008), ORGN-545. CODEN: 69KXQ2 AN 2008:954577 CAPLUS
- 7.For ortho-selective radical additions to anisole see: Tiecco M, Testaferri L. In: Reactive Intermediates. Abramovitch RA, editor. Vol. 3. Plenum Press; New York: 1983. p. 61.
- 8.Nicolaou KC, Reingruber R, Sarlah D, Bräse S. J Am Chem Soc. 2009;131:2086. doi: 10.1021/ja809405c.(b) A correction for ref 8a was submitted on the same day as the original version of this manuscript (which also noted the mischaracterizations of ref 8a): J. Am. Chem. Soc. 2009, 131, 6640. While the authors of 8a and 8b did not provide experimental verification of the reassigned structures, our submission did so, see Supporting Information.
- 9.It should be noted that Nicolaou and co-workers observed para-selectivity with highly electron-rich 1,3,4-trisubstituted aryl rings (see ref 8a).
- 10.An open shell, radical pathway is also consistent with the successful addition of highly electron-deficient styrenes to radical cations such as 1 (see ref 5d).
- 11.a) Ouellet SG, Walji A, MacMillan DWC. Acc Chem Res. 2007;40:1327. doi: 10.1021/ar7001864. [DOI] [PubMed] [Google Scholar]; b) Ouellet SG, Tuttle JB, MacMillan DWC. J Am Chem Soc. 2005;127:32. doi: 10.1021/ja043834g. [DOI] [PubMed] [Google Scholar]
- 12.Banwell MG, Beck DAS, Smith JA. Org Biomol Chem. 2004;2:157. doi: 10.1039/b312552a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.