

Abstract
The precise role of TGF-β signaling in head and neck squamous cell carcinoma (HNSCC) is not yet fully understood. Here we report generation of an inducible head- and neck-specific knockout mouse model by crossing TGF-β receptor I (Tgfbr1) floxed mice with K14-CreERtam mice. By applying tamoxifen (TM) to oral cavity of the mouse to induce Cre expression, we were able to conditionally delete Tgfbr1 in the mouse head and neck epithelia. Upon tumor induction with 7, 12-dimethylbenzanthracene (DMBA), 45% of Tgfbr1 conditional knockout (cKO) mice (n=42) developed squamous cell carcinomas (SCCs) in the head and neck area starting from 16 weeks after treatment. However, no tumors were observed in the control littermates. A molecular analysis revealed an enhanced proliferation and loss of apoptosis in the basal layer of the head and neck epithelia of Tgfbr1 cKO mice 4 weeks after TM and DMBA treatment. The most notable finding of our study is that the phosphoinositide 3-kinase (PI3K)/Akt pathway was activated in SCCs that developed in the Tgfbr1 cKO mice upon inactivation of TGF-β signaling through Smad2/3 and DMBA treatment. These observations suggest that activation of Smad-independent pathways may contribute cooperatively with inactivation of Smad-dependent pathways to promote head and neck carcinogenesis in these mice. Our results revealed the critical role of the TGF-β signaling pathway and its crosstalk with the PI3K/Akt pathway in suppressing head and neck carcinogenesis.
Keywords: TGF-β, PI3K/Akt, Head and Neck Squamous Cell Carcinoma (HNSCC), Conditional Knockout, Cancer Mouse Model
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the most common types of human cancer (1). Tobacco, alcohol consumption and viral agents are the major risk factors for development of HNSCC. These risk factors together with genetic susceptibility result in the accumulation of multiple genetic and epigenetic alterations in a multistep process of cancer development (2). However, the underlying cellular and molecular mechanisms that contribute to the initiation and progression from normal epithelia to invasive squamous cell carcinoma have not been clearly delineated (3).
There is accumulating evidence which suggests that the TGF-β signal transduction pathway is involved in head and neck carcinogenesis (4, 5). TGF-β is a multifunctional cytokine with diverse biological effects on cellular processes, including cell proliferation, migration, differentiation, and apoptosis. The 3 mammalian TGF-β isoforms, TGF-β1, -β2 and -β3, exert their functions through a cell surface receptor complex composed of type I (TGFBR1) and type II (TGFBR2) serine/threonine kinase receptors. Intracellular signaling is initiated once TGFBR1 has been phosphorylated by TGFBR2, which in turn phosphorylates Smad2 or Smad3. Phosphorylated Smad2 or Smad3 binds to Smad4, and then the complexes translocate from the cytoplasm into the nucleus. This results in the transcriptional activation of TGF-β-responsive genes that mediate the effects of TGF-β at the cellular level. Independent of SMAD proteins, receptor activation also induces other downstream targets, including Ras, RhoA, TAK1, MEKK1, PI3K, and PP2A, to produce the full spectrum of TGF-β responses (6–8).
The effects of TGF-β signaling in carcinogenesis largely depend on the tissue of origin and the tumor type. In most types of human cancer, TGF-β plays a paradoxical role in cancer development by acting as a tumor suppressor in early stages (9), and a tumor promoter in later stages (10, 11). In HNSCC, it is known that TGF-β functions as a potent tumor suppressor (12). However, it is not clear whether TGF-β acts in a pro-oncogenic manner in advanced late-stage HNSCC. The human oral carcinoma cell line, which contained a normal Ras but was growth-inhibited by TGF-β1, led to an increase in cell migration and invasion, and metastasis when transfected with dominant negative TGFBR2 (dn RII) cDNA (13). When TGF-β receptor II (Tgfbr2) was conditionally deleted in mouse head and neck epithelia, 35% of the DMBA-initiated Tgfbr2−/− mice developed jugular lymph node metastasis, suggesting TGF-β may actually in fact suppress metastasis rather than promote it (14).
The correlation between TGF-β receptor-mediated signaling and cancer development has been studied extensively. However, much less attention has been paid to the role of TGFBR1 in carcinogenesis when compared to that of TGFBR2. Although several reports have noted that mutations and polymorphisms of TGFBR1 are associated with HNSCC (15–17), the precise molecular nature of TGFBR1-mediated pro-oncogenic effects is still unknown. In the current study, we conditionally deleted Tgfbr1 in mouse head and neck epithelia using the Cre-LoxP approach to show that deletion of Tgfbr1 alone is not sufficient for spontaneous tumor formation, though it can increase the susceptibility to tumor development initiated by DMBA. The most notable finding of our study is that, in SCCs that developed in the Tgfbr1 cKO mice, the PI3K/Akt pathway, one of the most important Smad-independent receptor-I signaling pathways, was clearly activated in addition to inactivation of the Smad-dependent TGF-β signaling pathway. Our studies identified the critical role of the TGFBR1-mediated signaling pathway and its crosstalk with the PI3K/Akt pathway in suppressing head and neck carcinogenesis. The Tgfbr1 cKO mouse will be a valuable animal model for studying genetic alterations and signaling pathways that play important roles in HNSCC.
Materials and Methods
Generation of Tgfbr1 cKO mice
The Tgfbr1 cKO mice (K14-CreERtam;Tgfbr1f/f) were generated from crosses between Tgfbr1f/f mice (mixed genetic strains of C57BL/6, 129SV/J and FVB/N) (18, 19) and K14-CreERtam mice (genetic strain CD-1) (20). The Tgfbr1 cKO mice and their controls (Tgfbr1f/f, Tgfbr1f/+, and K14-CreERtam;Tgfbr1f/+ mice) were from the same litter and therefore had exactly the same mixed genetic background. The treatment procedures of Tamoxifen and DMBA have been described (20, 14). Additional details are provided in the Supplementary Data.
Histology, immunostaining, and BrdU labeling
Immunohistochemical staining (IHC) and quantifications of IHC slides were performed using a previously published method (21). Intratumoral microvessel density (iMVD) was determined as previously described (22). BrdU labeling and primary antibodies are described in the Supplementary Data.
Western blot analysis
Normal buccal mucosa and tongue from 6 pairs of Tgfbr1f/f and Tgfbr1 cKO mice, together with tumors that developed in DMBA-initiated Tgfbr1 cKO mice, were carefully dissected. A total amount of 40 μg protein from each sample was denatured and then loaded in each lane of NuPAGE 4–12% Bis-Tris precast gel. Additional details are provided in Supplementary Data.
Additional Methods
Information on TUNEL assay, Cre-mediated recombination assessment, quantitative real-time PCR and flow cytometry analysis is detailed in the Supplementary Data.
Statistical analysis
Statistical differences in the levels of mRNA expression between controls and experimental samples were determined using the Student’s t-test.
Results
Inducible deletion of Tgfbr1 in head and neck epithelia is not sufficient for SCC formation in mice
We generated an inducible head- and neck-specific knockout mouse model by crossing Tgfbr1 floxed mice with K14-CreERtam mice. K14 is expressed in proliferating keratinocytes of the basal layer of the epidermis. It is also active in stem cells that regenerate the epidermis, sebaceous glands, hair follicles, and the oral mucosa. Therefore TM treatment causes permanent excision of Tgfbr1 in both epithelia and epidermis of the head and neck region including buccal mucosa, tongue and ears. The Tgfbr1 cKO mice and controls (Tgfbr1f/f) were dissected 10 days after TM treatment. Genomic DNA was extracted from all major organs and tissues. Cre-mediated recombination of the Tgfbr1f/f allele was assessed using a PCR-based assay. Deletions of Tgfbr1 were detected in the buccal mucosa (BM), tongue (Tg), and ear (Er), but not in the esophagus (Es), forestomach (FS), back skin (SK), or any other nonstratified epithelial organs of Tgfbr1 cKO mice (Supplementary Fig. 1). No recombination was detected prior to TM administration. Tgfbr1 mRNA expression was examined by quantitative RT-PCR (qRT-PCR). The expression levels of Tgfbr1 mRNA in Tgfbr1f/f mice were normalized as 1.00 ± 0.23 in the buccal mucosa and 1.00 ± 0.08 in the tongue. The mRNA expression levels were significantly reduced to a mean of 0.65 ± 0.17 in the buccal mucosa (p<0.01) and 0.07 ± 0.05 in SCC of Tgfbr1 cKO mice as well as 0.46 ± 0.05 in the tongue (p<0.001) (Fig. 1A). Using immunostaining, the Tgfbr1 protein level was found to be significantly decreased in the tongue of Tgfbr1 cKO mice, as compared to that of Tgfbr1f/f mice. A similar decrease was also observed in phosphorylated Smad2, an activated mediator of TGF-β signaling (Fig. 1B). However, the expression of both Tgfbr1 and p-Smad2 in the back skin of the same mice remained normal (data not shown). This suggests that, upon oral administration of TM, the deletion of Tgfbr1 and the inactivation of its downstream signaling was localized only in the head and neck epithelia. These results were further confirmed by Western blot (Fig. 1C).
Figure 1.
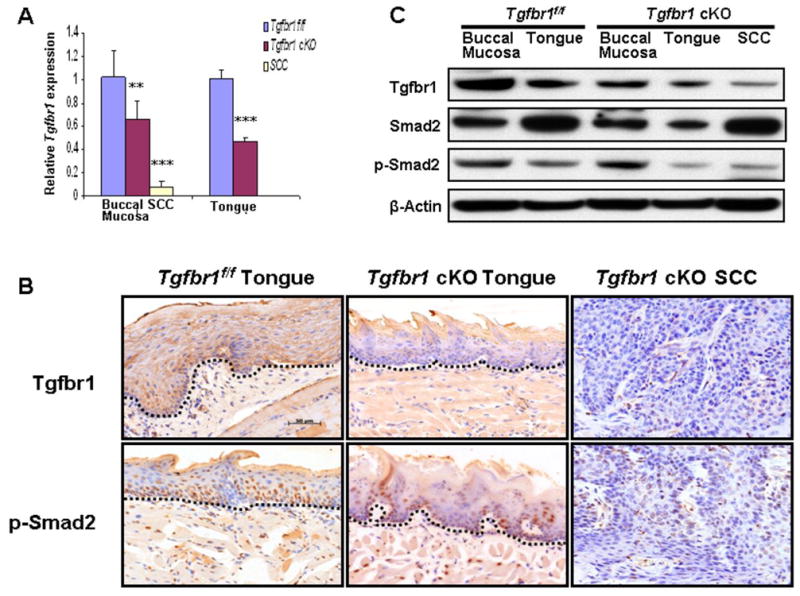
Decreased TGF-β signaling in Tgfbr1 cKO mice. A, Tgfbr1 mRNA significantly reduced in the head and neck epithelia and SCCs of Tgfbr1 cKO mice by qRT-PCR (n=3). **, P<0.01; ***, P<0.001. B, Tgfbr1 and p-Smad2 expression were reduced in the tongue and SCC of Tgfbr1 cKO mice by IHC. The dotted lines delineate the adjacent epithelial compartment. The changes in staining patterns are seen in the epithelium (above the dotted line) in which Tgfbr1 was knocked out. Bar, 50μm. C, Western blot analysis demonstrates that Tgfbr1 and p-Smad2 were reduced in buccal mucosa, tongue and SCCs of Tgfbr1 cKO mice compared to that of Tgfbr1f/f mice.
Out of 31 Tgfbr1 cKO mice, only 3 (9.7%, 3/31) developed spontaneous tumors including 2 SCCs in the periobital region and one in the upper lateral neck. No significant pathological changes in the head and neck region were observed in the remaining Tgfbr1 cKO mice during 1 year of observation. Thus, our results indicate that inactivation of TGF-β signaling alone is not sufficient to promote tumor formation in head and neck epithelia of these mice.
Deletion of Tgfbr1 in the head and neck epithelia together with DMBA initiation induced SCCs in mice
Because spontaneous tumor formation in Tgfbr1 cKO mice was rare, we induced tumors in Tgfbr1 cKO mice by applying a single dose (50 μg per mouse) of DMBA to the mouse oral cavity 10 days after the last TM treatment. DMBA is a commonly used chemical carcinogen, which can induce H-ras mutations in sporadic cells (24). After tumor initiation with DMBA, Tgfbr1 cKO mice started to develop SCCs in the head and neck area as early as 16 weeks, and by 1 year after treatment, 19 out of 42 (45%) Tgfbr1 cKO mice had developed SCCs (Fig. 2B–2E). The sites of tumors that developed in DMBA-treated Tgfbr1 cKO mice included the oral cavity, periorbital region, muzzle area, and skin around the head and neck area (Fig. 2A). 16% (3/19) of mice with tumors had developed metastases in the jugular lymph nodes and/or lungs by the time the mice were dissected (10–12 months after TM and DMBA treatment) (Fig. 2F, 2G). No tumors developed in the heterozygous mice (K14-CreERtam;Tgfbr1f/+, n = 27) or the Tgfbr1 floxed homozygous (Tgfbr1f/f, n = 34) control littermates (also treated with TM and DMBA) during the same time period (Fig. 2H). However, only partial excision of Tgfbr1 in mouse head and neck epithelia were noted by IHC and Western blot, due to relatively low efficiency of the tamoxifen-induced K14-CreERtam mouse line being used in this study (20) (Fig. 1B, 1C).
Figure 2.
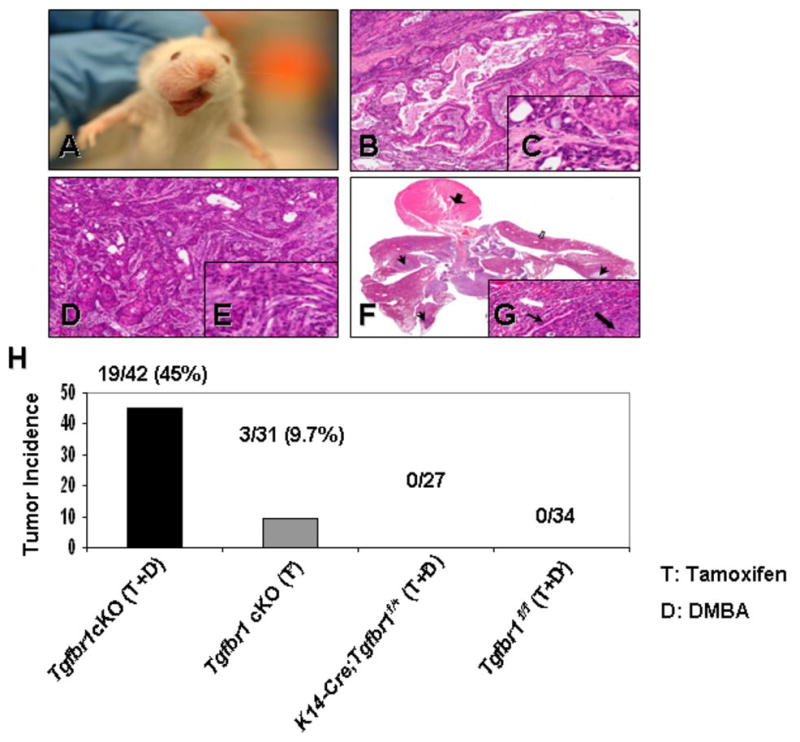
DMBA-initiated Tgfbr1 cKO mice develop HNSCCs. A, Tumor developed in the oral cavity of Tgfbr1 cKO mice. Pathological sections of oral squamous cell carcinoma (B) and infiltrating squamous cell carcinoma (D); (F) Low magnification of the heart (thick black arrow) and lung block. Examples of intrapulmonary metastases are indicated by black arrows; extrapulmonary (lymph nodes) with a white arrow, and non-compromised lung parenchyma by a block white arrow. Inset: the metastasis (black block arrow) is surrounded by compressed lung parenchyma (white block arrow). The arrow indicates a bronchus. The insets in (C), (E) and (G) depict fine details of the malignant cells (magnifications, 20× and 200× for main figure and inset, respectively). H, DMBA-treated Tgfbr1 cKO mice develop SCCs more frequently than Tgfbr1 cKO mice. No tumors were observed in K14-CreERtam;Tgfbr1f/+ or Tgfbr1f/f control littermates during 1 year of observation after TM and DMBA treatment.
Enhanced cell proliferation, inhibition of apoptosis, and down-regulation of cell cycle inhibitors in the head and neck epithelia of Tgfbr1 cKO mice
TGF-β has effects on both cell growth and apoptosis. Four weeks after DMBA treatment, an increased expression of a proliferative marker Ki67 was detected in the basal layer of the tongue of Tgfbr1 cKO mice but not in Tgfbr1f/f mice. A decreased apoptosis was also observed, indicating that the imbalance between cell proliferation and apoptosis occurs early in the head and neck epithelia of Tgfbr1 cKO mice (Fig. 3A). Using BrdU assays, we found a significantly increased number of proliferative cells in Tgfbr1 cKO mice head and neck epithelia and SCCs when compared to those of Tgfbr1f/f mice (Fig. 3B, 3D). However, we did not observe any apoptotic cells in SCCs by TUNEL assays (data not shown). Immunostaining revealed that CDKN1A expression was reduced in tongue and SCCs of Tgfbr1 cKO mice compared to that in Tgfbr1f/f mice. In contrast, c-Myc was overexpressed in tongue of Tgfbr1 cKO mice and its expression was even more remarkable in SCCs (Fig. 3B, 3D). These results were further confirmed by Western blot analysis (Fig 3C). Our results indicate the existence of an imbalance between cell proliferation, differentiation, and apoptosis in SCCs that developed in Tgfbr1 cKO mice, as well as in normal Tgfbr1 cKO mice head and neck epithelia.
Figure 3.
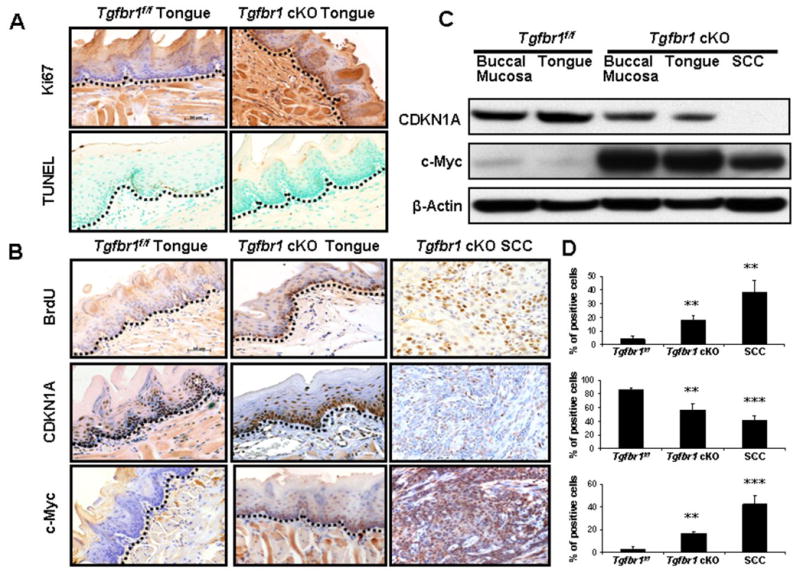
Enhanced growth-promotion and down-regulation of cell cycle inhibitors in Tgfbr1 cKO mice. A, Increased expression of Ki67 and loss of apoptosis in the basal layer of tongue of the Tgfbr1 cKO mice 4 weeks after TM and DMBA treatment. B, A significantly increased number of proliferative cells in tongue and SCCs of Tgfbr1 cKO mice by BrdU assays. CDKN1A expression was reduced in tongue and SCCs of Tgfbr1 cKO mice compared to that in Tgfbr1f/f mice. In contrast, c-Myc was overexpressed in tongue of Tgfbr1 cKO mice and its expression was even more remarkable in SCCs. The results were further confirmed by Western blot (C). D, Percentage of positive cells in tongue or SCCs of Tgfbr1 cKO mice compared with that of Tgfbr1f/f mice. Columns, average of three to five immunostained sections; the dotted lines delineate the adjacent epithelial compartment. Bar, 50 μm. **, P<0.01; ***, P<0.001.
Enhanced paracrine effect of TGF-β on tumor stroma of Tgfbr1 cKO mice
Increased inflammation and angiogenesis have been found in human HNSCCs (25). Deletion of Tgfbr2 in mouse head and neck epithelia resulted in enhanced paracrine effect of TGF-β on tumor stroma (14). To investigate the paracrine effect of TGF-β in tumor progression in the DMBA-treated Tgfbr1 cKO mice, we analyzed the expression level of Cyclooxygenase-2 (Cox-2) (26), Endoglin (CD105) (27), and α-Smooth Muscle Actin (SMA) in tumor stroma (28, 29). We found that Cox-2 expression was absent in normal buccal mucosa and tongue of Tgfbr1f/f mice, as well as in Tgfbr1 cKO mice, but its expression was significantly increased in SCCs, suggesting increased inflammation in tumors (Fig. 4A, 4B). Increased angiogenesis indicated by Endoglin (CD105)-stained microvessels in the stroma surrounding SCCs were also observed (Fig. 4A, 4B). Using immunofluorescent staining, we found that α-SMA, a hallmark of the myofibroblastic phenotype, strongly expressed in the stroma surrounding SCCs, but was not detected in the tongues of Tgfbr1f/f mice (Fig. 4A). To determine whether these enhanced paracrine effects correlate with endogenous TGF-β1 levels in the area surrounding the SCCs, we examined Tgfb1 mRNA expression by qRT-PCR. In comparison to tissues from Tgfbr1f/f mice, the levels of Tgfb1 mRNA expression were increased 2.42 ± 0.31 fold and 27.08 ± 4.42 fold (p<0.01) in DMBA-treated Tgfbr1 cKO mice tongues and SCCs, respectively (Fig. 4D). Immunofluorescent staining indicated significantly increased expression of Tgfb1 located only in the tumor stroma (Fig. 4C).
Figure 4.
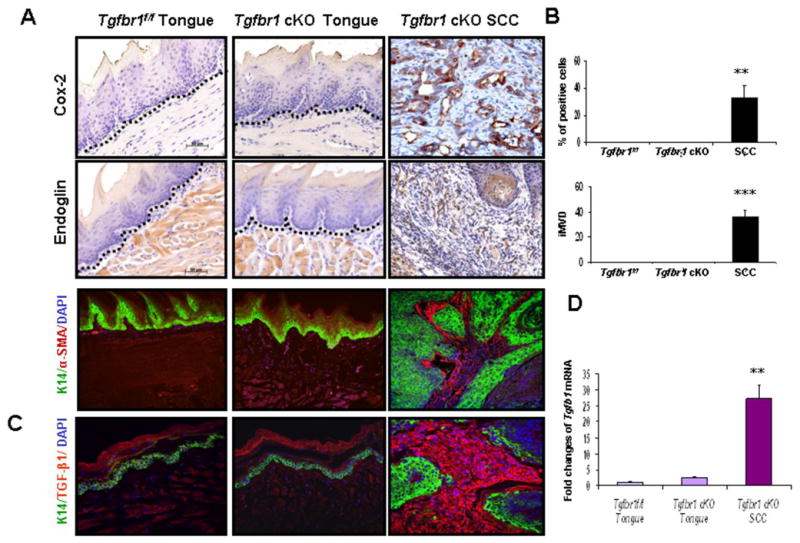
A, Enhanced paracrine effects of TGF-β in Tgfbr1 cKO mice. A, Significantly increased expression of Cox-2 in SCCs as well as overexpression of Endoglin (CD105), α-SMA in the stroma surrounding SCCs of Tgfbr1 cKO mice (magnifications, 200×). No expression was detected in normal tongue of Tgfbr1f/f or Tgfbr1 cKO mice. The dotted lines delineate the adjacent epithelial compartment. Bar, 50 μm. B, Percentage of Cox-2 positive cells and intratumoral microvessel density (iMVD) indicated by Endoglin (CD105)-stained microvessels per 200 × field in tumor stroma of Tgfbr1 cKO mice. Columns, five immunostained sections. **, P<0.01; ***, P<0.001. C, Tgfb1 expression in the tumor stroma by immunofluorescent staining (magnifications, 200×). D, Tgfb1 mRNA expression by qRT-PCR.
Evasion of the immune response is one of the most important features of TGF-β-mediated tumor progression (30, 31). We analyzed the immune status of the Tgfbr1 cKO mice using flow cytometry analysis. Compared with their control littermates, Tgfbr1 cKO mice showed significantly reduced numbers of both CD4+ and CD8+ effector T cells in jugular lymph nodes. In contrast, the regulatory T cells (CD4+CD25+Foxp3+) were increased, indicating active immune suppression in Tgfbr1 cKO mice. Gross changes in inflammation within tumors were noted by H&E staining (Supplementary Fig. 2A, 2B).
Activation of PI3K/Akt signaling in SCCs of Tgfbr1 cKO mice
The PI3K/Akt pathway is important in suppressing apoptosis and in promoting cell growth and proliferation. Hyperactivation of PI3K/Akt in HNSCC is induced either by mutations or by enhanced activity of its upstream activators, including the Ras oncogene or inactivation of PTEN (phosphatase and tensin homolog deleted on chromosome 10) (32). PTEN is a potent tumor suppressor gene and a negative regulator of the PI3K/Akt pathway. Mutations of PTEN have been found in a wide range of human cancers (33). In our study, a significantly increased level of unphosphorylated PTEN, an active form of the protein, was detected in all of the tumors that developed in the DMBA-treated Tgfbr1 cKO mice (Fig. 5B). However, despite the elevated PTEN levels, we observed consistently increased levels of the phosphorylated form of Akt (p-Akt) and its downstream target, the mammalian target of rapamycin (mTOR), in all of the tumors analyzed both by immunostaining and Western blot (Fig. 5A, 5B). These results indicate that in spite of the increased expression of PTEN, the PI3K/Akt pathway was activated in the SCCs that developed in the DMBA-treated Tgfbr1 cKO mice. Our results suggest that Akt activation in the SCCs is independent of effects on PTEN in this mouse model, and that other mechanisms are involved in the activation of this pathway. One of these might be the H-ras mutations caused by DMBA initiation. Indeed, H-ras mutations were detected in 9 out of 17 tumors (53%) at codon 61 in exon 2 of the gene. No K-ras mutations were found in any of these tumors (data not shown). However, the mechanisms underlying the activation of the PI3K/Akt pathway upon TβRI deletion warrant further investigation. A proposed TGF-β signaling alteration that promotes HNSCC in mice through activation of PI3K/Akt pathway is shown in Fig. 6.
Figure 5.
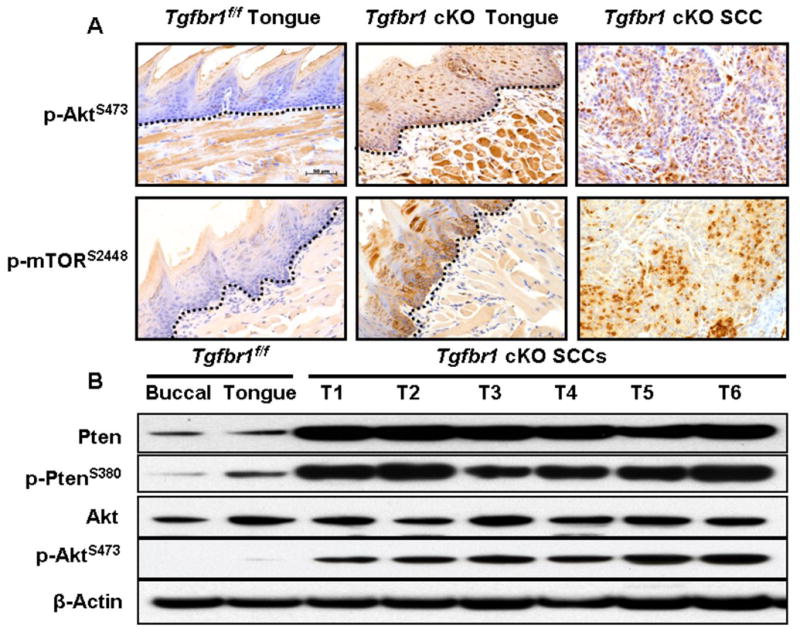
Activation of the PI3K/Akt pathway in Tgfbr1 cKO mice. A, Immunostaining revealed a significantly increased number of positive cells of p-Akt, p-mTOR in the SCCs that developed in Tgfbr1 cKO mice. The dotted lines delineate the adjacent epithelial compartment. Bar, 50 μm. B, A significantly increased level of unphosphorylated PTEN, an active form of the protein, was detected in all SCCs that developed in the DMBA-treated Tgfbr1 cKO mice. However, comparable elevated levels of the phosphorylated form of Akt (p-Akt) were also observed in SCCs by Western blot analysis.
Figure 6.
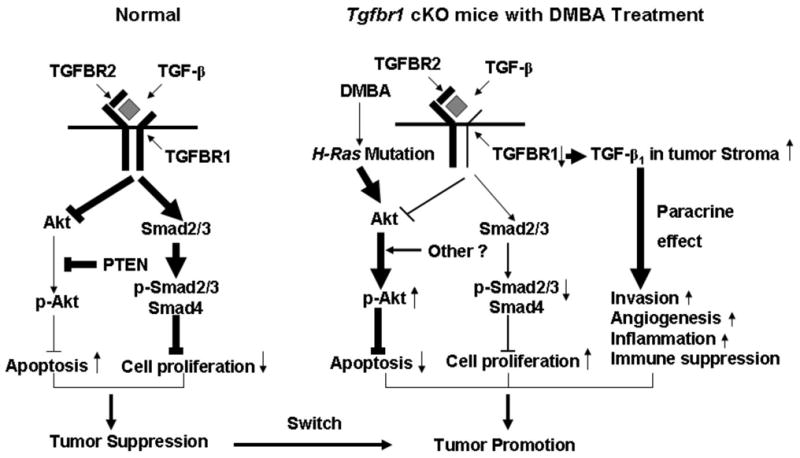
A schematic presentation of the proposed TGF-β signaling alteration that promotes HNSCC in mice. In normal cells, TGF-β inhibits cell proliferation through the Smad-dependent pathway. It also induces apoptosis by repressing the PI3K/Akt pathway results in tumor suppression. Decreased Tgfbr1 expression in Tgfbr1 cKO mice leads to increased cell proliferation and cell survival through PTEN independent activation of the PI3K/Akt pathway. DMBA treatment, which causes H-ras mutation as well as other mechanisms, may also play an important role in Akt activation. Decreased TGFBR1 can also increase TGF-β1 in tumor stroma by as yet unidentified mechanisms, which leads to increased invasion, angiogenesis, and inflammation, as well as immune suppression through the paracrine effect of TGF-β. In summary, inactivation of TGF-β signaling, in the context of ras mutations and aberrant activation of the PI3K/Akt pathway and accompanied by increased paracrine effect of TGF-β, switches TGF-β signaling from tumor suppression in normal cells to tumor promotion in head and neck carcinogenesis of Tgfbr1 cKO mice.
Discussion
TGF-β is a potent growth inhibitor for epithelial cells (34). Inactivating mutations or experimental deletion of components of the TGF-β pathway have been shown to promote tumorigenesis in a variety of organ systems (35, 36). However, the precise role of TGF-β signaling in head and neck carcinogenesis has not been fully understood. As with other organ systems, existing research has been mainly focused on TGFBR2. Inactivation of Tgfbr2 by overexpression of dominant negative receptor constructs or by targeted deletion promotes tumorigenesis in the mammary gland, prostate, pancreas, anogenital region, as well as in the head and neck area (37–40, 14). With one exception (40), inactivation of Tgfbr2 does not result in tumor formation unless cooperating oncogenic lesions are present, suggesting that loss of TGF-β response plays a tumor promoting rather than initiating role (14, 41). Interestingly, mice that harbored an inactivated Tgfbr2 in stromal cells developed intraepithelial neoplasia of the prostate and invasive SCCs in the forestomach. This suggests that alterations in the TGF-β signaling pathway within cells of the tumor microenvironment can also contribute to cancer development and progression (38). Even in cases where the TGF-β pathway is compromised specifically in the epithelium, the effects of this perturbation appear to extend to the stroma. Thus mice with inactivated Tgfbr2 in the mammary epithelium show increased recruitment of F4/80+ cells, increased expression of pro-inflammatory genes, and altered composition of the fibrovascular stroma- all effects that may promote further tumor progression (42). It is clear that perturbations in TGF-β signaling can have far reaching effects throughout the ecosystem of the tumor.
It is important to note that TGFBR2 not only interacts with TGFBR1, but also forms functional complexes with other type I receptors such as ActRI/ALK2, ALK3 or ALK1 (43, 44). Signaling through TGFBR2/Alk1 complexes activates Smad1, Smad5, and Smad8, whereas signaling through the TGFBR2/TGFBR1 complex results in phosphorylation of Smad2 and Smad3. In fact, TGF-β signaling through TGFBR1 and ALK1, in a complex with TGFBR2, showed opposing activities in endothelial cell migration and proliferation (45). Importantly, in epithelial cells TGFBR2 can also directly phosphorylate Par6 without involvement of TGFBR1, and release Par6 from the Par6-TGFBR1 complex. This allows Par6 to trigger the dissolution of tight junctions in the context of epithelial-mesenchymal transitions (46). Therefore, knocking out Tgfbr2 affects not only Smad-mediated TGF-β signaling, but also direct receptor-II–mediated alternative signaling via Par6. Thus knocking out TGFBR1 or TGFBR2 individually could affect downstream signaling differently, leading to distinct biological outcomes.
TGFBR1 forms heterotetrameric complexes with TGFBR2 on the cell surface and is critical for the downstream phosphorylation and activation of the Smads. Mutations and polymorphisms of TGFBR1 have been described: TGFBR1(6A), a 9 bp deletion coding for 3 alanine residues within the 9 alanine repeat region of exon 1, has been particularly associated with HNSCC (15–17). In an earlier study, we showed that 35% of mice with a targeted deletion of Tgfbr1 developed spontaneous SCCs in periorbital and/or perianal regions (19). To specifically study the role of Tgfbr1-mediated signaling in the progression of HNSCCs, we developed a novel inducible knockout mouse model by deleting Tgfbr1 in head and neck epithelia.
Most of our findings on the Tgfbr1 cKO mouse model are consistent with the findings from DMBA-initiated Tgfbr2 cKO mice (14), suggesting that Tgfbr1 functions similarly to Tgfbr2 in the progression of HNSCCs. The lack of spontaneous tumor formation in Tgfbr1 cKO mice, together with the fact that DMBA treatment facilitates tumor development in these mice, suggests that rather than initiation, loss of Tgfbr1 may play a more crucial role in tumor progression in mouse HNSCC. This is also the case for other epithelia, with the sole exception of the anogenital region (40). However, several differences have also been noted in our DMBA-initiated Tgfbr1 cKO mice compared with DMBA-initiated Tgfbr2 cKO mice. For example, none of our DMBA-initiated Tgfbr1 heterozygous mice (K14-CreERtam;Tgfbr1f/+) developed HNSCCs, while about 33% of mice with a heterozygous Tgfbr2 deletion (K5-CrePR1;Tgfbr2f/+) in the head and neck epithelia developed HNSCCs after DMBA initiation. Therefore tumor suppressor activities of TGF-β require a higher threshold level of Tgfbr2 than of Tgfbr1. Furthermore, only 16% of our DMBA-initiated Tgfbr1 cKO mice with tumors developed metastases in jugular lymph nodes and/or lungs by the time the mice were dissected. However, up to 35% of the DMBA-initiated Tgfbr2 cKO mice developed jugular lymph node metastases by 20–39 wks of age. While this difference between the two mouse models may be attributable to differences in mouse genetic background and/or the Cre mouse line being used in the studies, it may also indicate that Tgfbr1 and Tgfbr2 function differently. For example, Tgfbr2 may have more suppressive effects in later stages of cancer development, possibly due to TGFBR1-independent effects.
It is widely believed that TGF-β can affect cancer progression through both autocrine and paracrine effects. Paracrine effects of TGF-β, which are generally tumor promoting, include stimulation of inflammation and angiogenesis, escape from immunosurveillance, and recruitment of myofibroblasts. Autocrine effects of TGF-β in premalignant epithelial cells are tumor suppressive, while more advanced cancer cells with a functional TGF-β receptor complex may exhibit tumor-promoting autocrine effects, due to a convergence of TGF-β signaling with other signaling pathways (47). In the current study, we saw evidence for both types of effect. We found that upon deletion of Tgfbr1 in mouse head and neck epithelia, there is an enhanced cell proliferation and down-regulation of cell cycle inhibitors, due to inactivation of Smad2/3 mediated signaling. An inhibition of apoptosis through activation of the PI3K/Akt pathway in SCCs that developed in Tgfbr1 cKO mice was also observed. These results suggest that in the head and neck epithelia, TGF-β is an early tumor suppressor. In the SCCs that developed in Tgfbr1 cKO mice, we found increased inflammation, angiogenesis, and myofibroblast formation. Similar results have been observed in other mouse models when TGF-β signaling was disrupted (14, 48). Furthermore, elevated levels of endogenous TGF-β1 were detected in tumor stroma of Tgfbr1 cKO mice, as they have been in other studies (14). Therefore, on one hand, the deletion of Tgfbr1 in mouse head and neck epithelia prevents the surrounding increased TGF-β1 from exerting its tumor suppressive effects. On the other hand, the expression of Tgfbr1 in tumor stroma would certainly enhance its tumor promoting function through paracrine effects. Consequently, we believe that the elevated level of TGF-β1 in tumor stroma has direct involvement in the creation of microenvironment for tumor progression (4).
Alternative modes of TGF-β signaling have been categorized (8). Recent work showed that TGF-β induces apoptosis through repression of PI3K/Akt signaling, indicating that there may be negative crosstalk between the TGF-β tumor suppressor and PI3K/Akt pathways (49). The most notable finding of our current study is that in addition to inactivation of the Smad-dependent TGF-β signaling pathway and in spite of increased PTEN levels after deletion of Tgfbr1 in mouse head and neck epithelia and DMBA treatment, the PI3K/Akt pathway is activated in all SCCs that developed in the Tgfbr1 cKO mice. The results from our study indicate that decreased Tgfbr1 expression in Tgfbr1 cKO mice leads to increased cell proliferation and cell survival through PTEN independent activation of PI3K/Akt pathway. This is possibly due to DMBA induced H-ras mutation as well as other unknown mechanisms. These changes accompanied by increased TGF-β1 in tumor stroma, which leads to increased invasion, angiogenesis, inflammation and immune suppression through paracrine effect of TGF-β, switch TGF-β signaling from tumor suppression in normal cells to tumor promotion in head and neck carcinogenesis of Tgfbr1 cKO mice.
In summary, we generated an inducible conditional gene targeting mouse model for head and neck cancer research. We have demonstrated that targeted deletion of Tgfbr1 in the head and neck epithelia is apparently not sufficient for spontaneous tumor formation, but could increase susceptibility to tumor development initiated by DMBA. TGF-β is a major tumor suppressor, and inactivation of TGF-β signaling, in the context of ras mutations and aberrant activation of the PI3K/Akt pathway, may contribute cooperatively to the promotion of head and neck carcinogenesis in these mice. Our results underscore a critical role of the TGF-β signaling pathway and its crosstalk with the PI3K/Akt pathway in suppressing head and neck carcinogenesis. These findings have significant implications for the development of effective therapeutic strategies targeting both the TGF-β and the PI3K/Akt pathways for the treatment of HNSCCs.
Acknowledgments
Grant support: Intramural Research Program, National Institute of Dental and Craniofacial Research, NIH.
We would like to thank Dr Stefan Karlsson for providing us with the Tgfbr1 flox mice, Dr Hynda Kleinman for critical reading of this manuscript, Dr Raj Puri and Dr Andrew Doyle for helpful discussion, and Shelagh Powers for editorial assistance.
Footnotes
Potential conflicts of intrest: None
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Kim MM, Califano JA. Molecular pathology of head-and-neck cancer. Int J Cancer. 2004;112:545–53. doi: 10.1002/ijc.20379. [DOI] [PubMed] [Google Scholar]
- 3.Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5:311–6. doi: 10.1016/s1535-6108(04)00090-x. [DOI] [PubMed] [Google Scholar]
- 4.Lu SL, Reh D, Li AG, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64:4405–10. doi: 10.1158/0008-5472.CAN-04-1032. [DOI] [PubMed] [Google Scholar]
- 5.Qiu W, Schönleben F, Li X, Su GH. Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 2007;245:163–70. doi: 10.1016/j.canlet.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts AB, Wakefield LM. The two faces of transforming growth factor β in carcinogenesis. Proc Natl Acad Sci USA. 2003;100:8621–3. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 8.Massagué J. TGF-β in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor β1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–86. [PubMed] [Google Scholar]
- 10.Piek E, Roberts AB. Suppressor and oncogenic roles of transforming growth factor-beta and its signaling pathways in tumorigenesis. Adv Cancer Res. 2001;83:1–54. doi: 10.1016/s0065-230x(01)83001-3. [DOI] [PubMed] [Google Scholar]
- 11.Tang B, Vu M, Booker T, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–24. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie W, Bharathy S, Kim D, Haffty BG, Rimm DL, Reiss M. Frequent alterations of Smad signaling in human head and neck squamous cell carcinomas: a tissue microarray analysis. Oncol Res. 2003;14:61–73. doi: 10.3727/000000003108748612. [DOI] [PubMed] [Google Scholar]
- 13.Huntley SP, Davies M, Matthews JB, et al. Attenuated type II TGF-beta receptor signaling in human malignant oral keratinocytes induces a less differentiated and more aggressive phenotype that is associated with metastatic dissemination. Int J Cancer. 2004;110:170–6. doi: 10.1002/ijc.20111. [DOI] [PubMed] [Google Scholar]
- 14.Lu SL, Herrington H, Reh D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–42. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen T, Yan W, Wells RG, et al. Novel inactivating mutations of transforming growth factor-beta type I receptor gene in head-and-neck cancer metastases. Int J Cancer. 2001;93:653–61. doi: 10.1002/ijc.1381. [DOI] [PubMed] [Google Scholar]
- 16.Knobloch TJ, Lynch MA, Song H, et al. Analysis of TGF-beta type I receptor for mutations and polymorphisms in head and neck cancers. Mutat Res. 2001;479:131–9. doi: 10.1016/s0027-5107(01)00157-9. [DOI] [PubMed] [Google Scholar]
- 17.Pasche B, Knobloch TJ, Bian Y, et al. Somatic acquisition and signaling of TGFBR1*6A in cancer. JAMA. 2005;294:1634–46. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- 18.Larsson J, Goumans MJ, Sjöstrand LJ, et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663–73. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honjo Y, Bian Y, Kawakami K, et al. TGF-beta receptor I conditional knockout mice develop spontaneous squamous cell carcinoma. Cell Cycle. 2007;6:1360–6. doi: 10.4161/cc.6.11.4268. [DOI] [PubMed] [Google Scholar]
- 20.Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A. 1999;96:8551–6. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czerninski R, Amornphimoltham P, Patel V, Molinolo AA, Gutkind JS. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prev Res. 2009;2:27–36. doi: 10.1158/1940-6207.CAPR-08-0147. [DOI] [PubMed] [Google Scholar]
- 22.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–40. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 24.Kim TW, Chen Q, Shen X, et al. Oral mucosal carcinogenesis in SENCAR mice. Anticancer Res. 2002;22:2733–40. [PubMed] [Google Scholar]
- 25.Chen Z, Malhotra PS, Thomas GR, et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369–79. [PubMed] [Google Scholar]
- 26.Goulart Filho JA, Nonaka CF, da Costa Miguel MC, de Almeida Freitas R, Galvão HC. Immunoexpression of cyclooxygenase-2 and p53 in oral squamous cell carcinoma. Am J Otolaryngol. 2009;30:89–94. doi: 10.1016/j.amjoto.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Wikström P, Lissbrant IF, Stattin P, Egevad L, Bergh A. Endoglin (CD105) is expressed on immature blood vessels and is a marker for survival in prostate cancer. Prostate. 2002;51:268–75. doi: 10.1002/pros.10083. [DOI] [PubMed] [Google Scholar]
- 28.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 29.Lewis MP, Lygoe KA, Nystrom ML, et al. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer. 2004;90:822–32. doi: 10.1038/sj.bjc.6601611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- 31.Kim BG, Li C, Qiao W, et al. Smad4 signaling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441:1015–9. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- 32.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2008 doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 34.Massagué J, Gomis RR. The logic of TGF-β signaling. FEBS Lett. 2006;580:2811–20. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 35.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 36.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 37.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–5. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhowmick NA, Chytil A, Plieth D, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 39.Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313–27. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biswas S, Chytil A, Washington K, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687–92. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- 42.Bierie B, Stover DG, Abel TW, et al. Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res. 2008;68:1809–19. doi: 10.1158/0008-5472.CAN-07-5597. [DOI] [PubMed] [Google Scholar]
- 43.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 44.Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28:6889–902. doi: 10.1128/MCB.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–9. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 47.De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200:429–47. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- 48.Go C, Li P, Wang XJ. Blocking transforming growth factor beta signaling in transgenic epidermis accelerates chemical carcinogenesis: a mechanism associated with increased angiogenesis. Cancer Res. 1999;59:2861–8. [PubMed] [Google Scholar]
- 49.Wang J, Yang L, Yang J, et al. Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 2008;68:3152–60. doi: 10.1158/0008-5472.CAN-07-5348. [DOI] [PubMed] [Google Scholar]