

Abstract
Zipper-interacting protein kinase (ZIP kinase) has been thought to be involved in apoptosis and the C-terinal leucine zipper motif is important for its function. Recent studies have revealed that ZIP kinase also plays a role in regulating myosin phosphorylation. Here we found novel ZIP kinase isoform in which the C-terminal non-kinase domain containing a leucine zipper is eliminated (hZIPK-S). hZIPK-S binds to myosin phosphatase targeting subunit 1(MYPT1) similar to the long isoform (hZIPK-L). In addition, we found that hZIPK-S as well as hZIPK-L binds to myosin. These results indicate that a leucine zipper is not critical for the binding of ZIP kinase to MYPT1 and myosin. Consistently, hZIPK-S localized with stress-fibers where they co-localized with myosin. The residues 278-311, the C-terminal side of the kinase domain common to the both isoforms, is involved in the binding to MYPT1, while the myosin binding domain is within the kinase domain. These results suggest that the newly found hZIPK-S as well as the long isoform plays an important role in the regulation of myosin phosphorylation.
Keywords: ZIP kinase, myosin, myosin light chain phosphatase, smooth muscle, myosin light chain phosphorylation, autophosphorylation
Introduction
Zipper-interacting protein kinase (ZIP) kinase, also termed death-associated protein kinase like kinase (DLK), was originally identified by two different groups and classified as a subfamily of the death-associated protein kinase (DAPK) [1, 2]. ZIP kinase is a serine/threonine-specific protein kinase that contains a high homology to the kinase domain of the DAPK at the N-terminus and leucine zipper domain at the C-terminus [1, 2]. Previous studies suggested that ZIP kinase is oligomerized through its C-terminal leucine zipper structure and it has been thought that the oligomerization is critical for the activation of the enzyme [1]. It has been thought that murine ZIP kinase is involved in apoptosis since overexpression of active ZIP kinase but not its inactive form induces apoptosis. Consistent with the proposed role of ZIP kinase in apoptosis, it has been shown that murine ZIP kinase predominantly localizes in the nucleus [3]. On the other hand, it was found that human ZIP kinase is predominantly localized in cytosol [4], suggesting a physiological role of ZIP kinase through the phosphorylation of its cytoplasmic substrates.
Recently, it was found that ZIP kinase phosphorylates the regulatory light chain (MLC20) of smooth muscle myosin [5, 6]. This finding raised a possibility that ZIP kinase plays a role in cell contractile activity since the function of smooth muscle and non-muscle myosin is regulated by the phosphorylation of its MLC20 [7-9].
The site responsible for the activation of the myosin II motor function had been identified to be Ser19 of MLC20. On the other hand, it is known that MLC20 is also phosphorylated at Thr18 in addition to Ser19 [10, 11], which stabilizes the formation of myosin filaments and the myosin phosphorylated at both sites tends to form large thick filaments [12, 13].
While the physiological significance of Thr18 phosphorylation is obscure, Thr18 phosphorylation occurs in vivo and it has been reported that di-phosphorylation of MLC20 at Thr 18 and Ser19 occurs in smooth muscles induced by external stimuli [14, 15] and in non-muscle cells in conjunction with the cellular shape change and exocytosis [16-18]. However, physiological relevance and regulation of di-phosphorylation of MLC20 has been puzzling because MLCK, which has been thought to be the predominant kinase responsible for MLC20 phosphorylation, phosphorylates the Thr18 at a much slower rate than that for Ser19 of MLC20 [10]. Interestingly, it was found that ZIP kinase phosphorylates Ser19 and Thr18 of MLC20 at the same rate constant, suggesting that this kinase is responsible for the production of di-phosphorylated myosin in smooth muscle and non-muscle cells [6]. Actually, it was found that ZIP kinase is primarily responsible for MLC20 phosphorylation in motile mammalian cells [18], although a recent study suggested that integrin linked kinase is rather responsible for MLC20 phosphorylation in the Triton-X100 permeabilized rat caudal artery fiber using a protein kinase inhibitor, AV25, as a probe [19].
On the other hand, MacDonald et al. [20] reported that a low molecular mass protein kinase (32kDa) purified from bovine bladder smooth muscle tissue cross-reacted with the anti-ZIP kinase antibody, and termed ZIP kinase like kinase. ZIP kinase like kinase has an affinity for MYPT1, a regulatory subunit of myosin light chain phosphatase (MLCP), and phosphorylated MYPT1 at Thr641, the site inhibiting MLCP activity originally found as a Rho kinase specific site [21, 22], but subsequently was found to be the site of multiple-kinases [20, 23, 24]. However, the origin and identity of this kinase is unknown. Although it could be a degradation product of ZIP kinase during biochemical manipulations, these results raise a hypothesis that there are multiple ZIP kinase isoforms present and they function to regulate MLC20 phosphorylation level in cells.
In the present study, we cloned a novel ZIP kinase isoform from human bladder smooth muscle (hZIPK-S). It contained the entire catalytic domain and retained a protein kinase activity accordingly, but lacked the leucine zipper domain. We also demonstrate smooth muscle specific expression of human ZIP kinases in human bladder tissue. We found that hZIPK-S bound directly to myosin in addition to MYPT1, and localized at stress fibers in ARPE19 cells where myosin and MYPT1 are present. Our results are consistent with the notion that hZIPK isoforms are responsible for MLCK independent regulation of myosin phosphorylation.
Materials and methods
Cloning of ZIP kinase isoforms
In order to clone potential splicing variants of human ZIP-kinase, 1 μg of total RNA from human bladder (Ambion Inc., Austin, TX) was reverse transcribed using SuperScriptII reverse transcriptase (Invitrogen Life technologies, Carlsbad, CA) and 3′-RACE adapter primer [5′-GCG AGC ACA GAA TTA ATA CGA CTC ACT ATA GG (T)12 VN-3′]. 1/20 volume of reverse transcription reaction was subjected to two rounds of polymerase chain reaction (PCR). Primer pairs used for the 1st round PCR were Exon2 specific forward primer [5′-CGGCGT TCA CTA CCT GCA CTC TAA G-3′] and “Outer” adapter reverse primer [5′-GCG AGC ACA GAA TTA ATA CGA CT -3′]. Primer pairs used for the 2nd round PCR were Exon3 specific forward primer [5′-TCG GCA TCG CGC ACA AGA TC-3′] and “Inner” adapter primer [5′-CGC GGA TCC GAA TTA ATA CGA CTC ACT ATA GG]. PCR was conducted using Pfx DNA polymerase (Invitrogen lifetechnologies, Carlsbad, CA). The thermal cycle profile was 94°C 30sec, 60°C 30sec, 68°C 2min for 30 cycles. The PCR product was run on 1% agarose gel and each band was gel purified. The purified PCR products were subcloned into pCR2.1-TOPO cloning vector (Invitrogen Lifetechnologies, Carlsbad, CA).
Production of cDNA constructs
All the constructs used in this study were produced by PCR using gene specific composite primers containing appropriate restriction enzyme sites. The PCR products were digested with appropriate restriction enzymes and in-frame ligated with expression vectors (derivatives of pFastBac or pEGFP-C1). Fidelity of the entire coding regions of all the expression vectors was confirmed by automatic DNA sequencing performed by the Nucleic Acid Facility at the University of Massachusetts Medical School. Mammalian expression vector with N-terminal myc tag: pMyc, was generated by replacing the EGFP sequence from pEGFP-C1 (BD Bioscience Clonetech, Palo Alto, CA) with custom myc-tag linker [Forward, 5′-CTA GCG GTC GCC ACC ATG GCA TCA ATG CAG AAG CTG ATC TCA GAG GAG GAC CTG CTT T-3′, Reverse, 5′-CCG GAA AGC AGG TCC TCC TCT GAG ATC AGC TTC TGC ATT GAT GCC ATG GTG GCG ACC G-3′] using NheI and BspE1 sites. Primers used to generate GST fusion proteins were as follows: Forward primer for all pFastBac clones [5′-GAA GGC GGA ATT CCC GCC ATG TCC ACG TTC AGG-3′], Reverse primers for pFastBac-GST∷hZIPK-L [5′-CGC TCG AGC TAG CGC AGC CCG CAC TCC ACG CC-3′], pFastBac-GST∷hZIP-S [5′-GGT ACC TCG AGC TAG GCG TCC ACA GGC GCG ACG AT-3′]. pFastBac-GST∷hZIPK-K was generated by site directed mutagenesis of hZIPK-S at I278, replacing to stop codon using forward primer [5′-CAT TCC TGG ATT AAG GCG TAG CGG CGG CGG AAC GTG CGT G-3′], and reverse primer [5′-CAC GCA CGT TCC GCC GCC GCT ACG CCT TAA TCC AGG AAT G-3′]. Inserts for the myc-tag expression vector was prepared from pFastBac vector. To generate S110A, T112A and D139N mutants, site-directed mutagenesis was conducted by the PCR mutagenesis method using pFastBacGST∷hZIPK-L and -KD as templates and high fidelity DNA dependent DNA polymerase, Pfu (Invitrogen). hZIPK-KD construct was produced by introducing a stop codon at the 3′ side of 277th codon (Ala277) using PCR mutagenesis method. To express hZIPK-S without GST, the hZIPK-S cDNA was inserted to pFastBac1 baculovirus transfer vector. The expressed hZIPK-S with no exogenous amino acids was subjected to western blot to compare the molecular masses of ZIP kinase isoforms expressed in bladder.
Preparation of recombinant proteins
Expression and purification of recombinant MYPT1 and ZIPKs were done using the Baculovirus expression system as described previously [6]. Smooth muscle myosin was isolated from bovine bladder as described previously [25]. MLC20 was prepared as described previously [26]. Actin was prepared from rabbit skeletal muscle according to Spudich and Watt [27].
Western blotting of ZIP kinase
Whole tissue lysates of normal adult human bladder were purchased from BioChain Institute, Inc (Hayward, CA) or ProSci Inc. (Poway, CA). NIH3T3 fibroblast cells were maintained in DME containing 10% newborn calf serum. Immunoblotting was done as described using a Nitrocellulose membrane [6, 28]. In brief, the samples were loaded on a 7-20% polyacrylamide gradient slab gel containing 0.1 % SDS and electrophoresis was carried out according to Laemli (1970). The proteins separated by SDS-PAGE were transblotted to a Nitrocellulose membrane (BioRad Laboratories). The trans-blotted membrane was incubated with polyclonal anti ZIP kinase antibodies [6] in 5% milk-tris buffered saline (TBS) at 4°C for overnight. After washing with TBS 0.05% Tween20, the membrane was incubated with the second antibody conjugated with horseradish peroxidase (BioRad) in 5% milk-TBS for 1 hr at room temperature, then washed with TBS containing 0.05% Tween20 several times. The membrane was stained using the enhanced cheniluminescence detection method.
Determination of protein kinase activity and autophosphorylation of ZIPK
Phosphorylation of the isolated smooth muscle myosin, MLC20 or recombinant MYPT1 was carried out as described previously [6]. Briefly, various amounts of substrates and hZIPK (4 or 20μg/ml) were incubated at 25 °C in the reaction buffer containing 30mM Tris-HCl, pH7.5, 1mM MgCl2, 100mM NaCl, 1mM EGTA, 1mM DTT, 1μM microcystin LR, 0.2mM [γ-32P] ATP (PerkinElmer Life Sciences, Boston, MA), and then the reaction was terminated by heat denaturation. Aliquots were subjected to 7.5-20% SDS-PAGE and then subjected to autoradiography. The phosphorylated protein bands were excised and the radioactivity was determined by a liquid scintillation counter as described previously [6].
To detect the autophosphorylation of hZIPK, hZIPK was incubated with 0.2 mM [γ-32P]ATP in the reaction buffer described above various times. The samples were subjected to SDS-PAGE followed by autoradiography.
Cell culture, transfection, immunocytochemistry
ARPE-19 human retinal pigmented epithelial cells that show clear stress fiber and cortical actin structure were used and maintained with 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium containing 1.2 g/L sodium bicarbonate, 2.5 mM L-glutamine, 15 mM HEPES, 10% fetal bovine serum, antibiotics, and antimycotics as a growth medium. Transient transfection was carried out using FuGENE6 (Roche Applied Science, Indianapolis, IL). ARPE-19 cells were passed into a 12-well plate with a cover glass 24 hours before transfection, and a mixture of 0.5μg plasmid DNA and 3μl FuGENE6 was overlayed onto the cells after changing the medium. After 15-16 hrs, the cells were subjected to immunocytochemistry. The cells were fixed in 4% paraformaldehyde, 2 mM MgCl2, 1 mM EGTA in PBS for 10 min at 15 hours post-transfection, incubated with 0.1% TritonX-100 in PBS for 10min, and then blocked with 3% BSA in PBS for 30 min. Primary Abs were diluted in blocking buffer and incubated at 4°C for 16 hours. Primary Abs and dilutions used in this study were as follows: Mouse monoclonal ant-myc Ab (1:500, clone 9E10, Santa Cruz Biotechnology, Santa Cruz, CA), or rabbit polyclonal anti-diphosphorylated myosin light chain (pTS) Ab (1:1000, [18]. Secondary Abs labeled with FITC or Cy5 (Jackson ImmunoResearch Lab. Inc., West Grove, PA) were applied, and incubated for 60 min at an ambient temperature. Actin structure was visualized with Alexa Fluor 546 Phalloidin (Molecular Probes, Eugene, OR). Samples were observed using Leica DM IRBE confocal microscopy equipped with TCS SP2 scanner and images were acquired using Leica confocal software (Leica Microsystems, Bannockburm, IL).
GST-pull-down assay
GST fusion proteins were incubated with Glutathion Sepharose 4B (Amersham Biosciences, Piscataway, NJ) on ice for 2hr in PBS containing 50mM TrisHCl pH7.5, 150mM NaCl, 2mM EDTA, 2mM EGTA 0.1% Triton X-100, 5mM DTT, 2mM PMSF, 0.2 mM Nα-p-tosyl-L-lysine chloromethyl ketone (TLCK), 0.2 mM N-tosyl-L-phenylalanine chloromethyl ketone (TPCK), and 10μg/ml Leupeptin and then washed with PBS. Then the recombinant MYPT1, isolated smooth muscle myosin, or F-actin was mixed with GST-fusion proteins in buffer A (50mM TrisHCl pH7.5, 150mM NaCl, 1mM EGTA, 2mM MgCl2, 0.5% NP-40, and 0.5mM PMSF) at 4°C for 1 hour with rotation, followed by the wash with buffer A for three times to remove unbound proteins. The bound proteins were dissolved in SDS sample loading buffer and separated by 7.5-20% polyacrylamide gradient slab gel. The samples were then subjected to Western blot using rabbit polyclonal anti-MYPT1 Ab (1:2000), mouse monoclonal anti-myosin Ab (clone mm8, 1:500) [29], rabbit polyclonal anti-pan-Actin Ab (Cytoskeleton, Denver, CO), or mouse monoclonal anti-GST Ab (1:1000, clone B-14, Santa Cruz Biotechnology, Santa Cruz, CA). The signals were detected by horseradish peroxidase conjugated secondary Ab and chemiluminescence (Pierce Biotechnology, Rockford, IL).
To examine the effect of autophosphorylation of ZIPK on the binding to myosin and MYPT1, Glutathion Sepharose 4B bounded GST tagged hZIPK was incubated with ATP at 25°C for 5 min for maximum phosphorylation as described above. After the autophosphorylation, free ATP was removed by washing the beads with PBS twice and then the GST-hZIPK was subjected to GST-pull-down assay.
Results
Molecular cloning of ZIPK isoform from human bladder
ZIP kinase contains a conserved kinase domain at the N-terminal half of the molecule. Therefore, we performed a 3′-RACE using human bladder total RNA as a template to obtain ZIP kinase isoforms because the putative ZIP kinase isoforms may contain a 3′ untranslated region different from that of known ZIP kinase (see MATERIALS AND METHODS). We found a cDNA fragment whose molecular mass is much smaller than the known human ZIP kinase (hZIPK-L) (Fig. 1A). This cDNA fragment was subcloned into pCR2.1-TOPO vector and sequenced. It was found in the short ZIP kinase isoform that the originally assigned exon 8 was internally spliced out to yield a C-terminal domain truncated variant (Fig. 1B, C). The new human ZIP kinase isoform, hZIPK-S, contains the entire kinase domain, truncated at nucleotide 978 of Accession number AB022341, and lacks leucine zipper domain. This eliminates the original termination codon and the part of the original 3′ untranslated region was utilized to produce a unique ten amino acid residues with a novel termination codon at nucleotide 1972, and utilizes the original polyA signal at nucleotide 2035 (Fig.1 B). The calculated molecular mass of hZIPL-S is 37.0kDa. The result suggests that mRNA encoding hZIPK is present in bladder tissue.
Fig. 1.
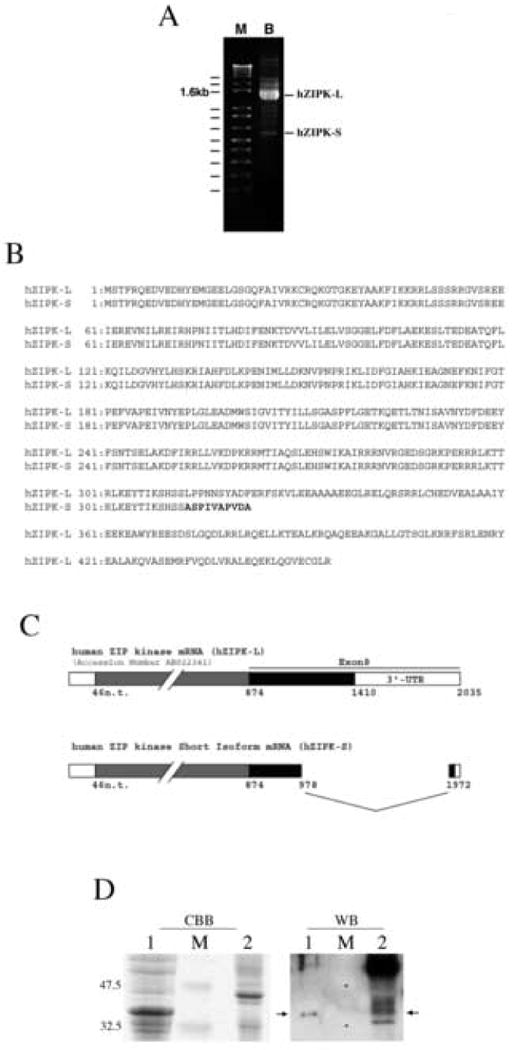
Cloning of novel ZIP kinase isoform. A. 3′-RACE of ZIP kinase isoforms using human bladder RNA. 3′-RACE was performed as described in MATERIALS AND METHODS. B. Amino acid sequence alignment of hZIPK-L and hZIPK-S. Bold letters show the unique sequence of the short isoform. C. Schematic representation of hZIPK-S exon structure, and the nucleotide sequence of hZIPK-S. Sequence analysis of hZIPK isoforms from human bladder revealed that internal splicing of Exon 8 resulted in C-terminal truncation (hZIPK-S). hZIPK-S contains entire kinase domain, truncated at nucleotide 978 of Accession number AB022341, and lacks the leucine zipper domain. The 3′ end of the 3′ untranslated region was translated to produce a novel termination codon at nucleotide 1972, and utilizes original poly A signal at nucleotide 2035. D. Coomassie brilliant blue (CBB) staining and Western blot (WB) of hZIPK-S and human bladder using anti-ZIPK antibodies. Lane 1, total cell lysates of sf9 cells expressing hZIPK-S; lane 2, total tissue homogenates of human bladder. Lane M is the molecular mass markers. The molecular masses (kDa) are indicated on the left. For western blot, the total sf9 cell lysates were diluted by 200 times. The arrows indicate hZIPK-S.
A low molecular mass protein kinase recognized by anti-ZIP kinase antibodies has been purified and termed ZIP kinase like kinase (19). Consistently, there were smaller molecular mass protein bands detected in bladder tissue by Western blot and one of the low molecular mass band coincided with the recombinant ZIPK-S (Fig. 1 D). Small molecular mass bands recognized by anti-ZIP kinase antibodies were also detected previously in addition to the 55 kDa band [18]. The results support the notion that there is a small molecular mass ZIP kinase isoform, although it may not be identical to the previously reported ZIP kinase like kinase.
Enzymatic characterization of hZIPK isoforms
It was shown previously that ZIP kinase can phosphorylate myosin and MYPT1 [6, 20]. We examined the kinase activity of the hZIPK-S against myosin and MYPT1 and compared its activity with the long isoform (hZIPK-L). The rate of myosin phosphorylation of hZIPK-S was significantly faster than that of ZIPK-L, while the rate of MYPT1 phosphorylation of hZIPK-S was slower than hZIPK-L (Fig. 2D). The rate constant was measured as a function of the substrate concentration and the kinetic parameters were obtained for hZIPK isoforms. Vmax and Km for myosin phosphorylation were 248 nmol/min/mg and 12 μM for the short isoforms and 120 nmol/min/mg and 6.2 μM for the long isoforms, respectively. Similarly, Vmax and Km for MLC20 phosphorylation were 1.3 μmol/min/mg and 73 μM for the short isoforms and 271 nmol/min/mg and 10.4 μM for the long isoforms, respectively. It should be noted that myosin is a much better substrate than MYPT1 for hZIPK-S and the rate of phosphorylation was 10 fold higher for myosin (Fig. 2C).
Fig. 2.
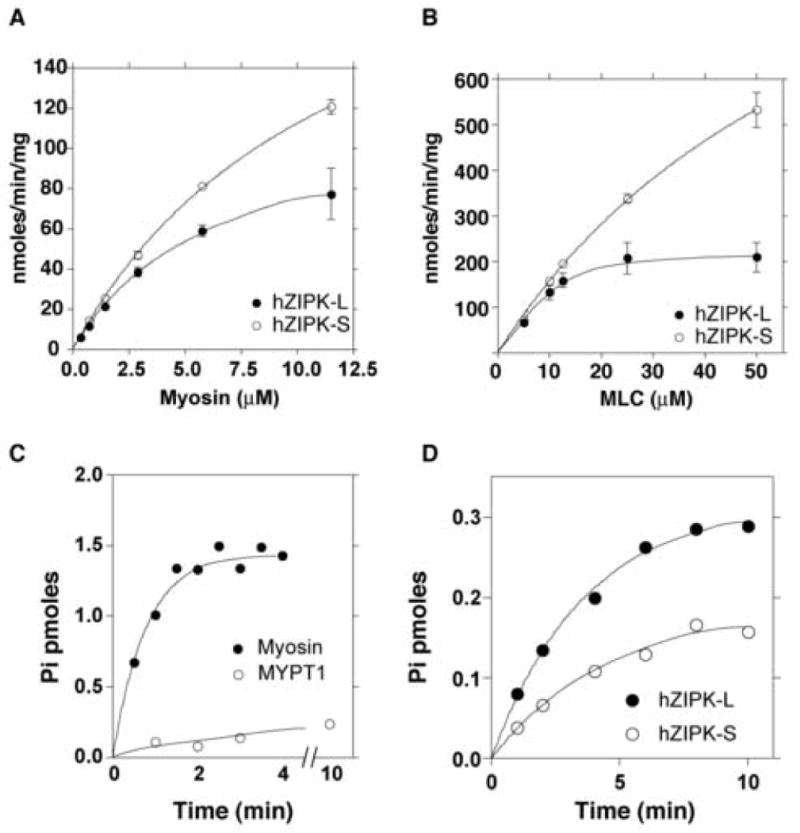
Enzymatic characterization of hZIPK-L and S. A. Phosphorylation of myosin by hZIPK isoforms as a function of myosin concentration. Open symbols and closed symbols are hZIPK-S and hZIPK-L, respectively. Bars represent standard error of three independent experiments. It should be noted that phosphorylation of myosin was not observed in the absence of hZIPK indicating that there was practically no contaminated kinases in myosin preparation used. B. Phosphorylation of isolated MLC20 by hZIPK isoforms as a function of MLC20 concentration. Open circle, hZIPK-S; Closed circle, hZIPK-L. Bars represent standard error of three independent experiments. C. Phosphorylation of MYPT1 and myosin by hZIPK-S. 20 μg/ml of substrates were used. Open circle, MYPT1; Closed circles, myosin. D. Phosphorylation of MYPT1 by hZIPK isoforms. Open symbols and closed symbols are hZIPK-S and hZIPK-L, respectively. It should be noted that the contaminated kinase activity is not recognized in myosin preparation used. Assay conditions are as described in MATERIALS AND METHODS.
Both hZIPK isoforms underwent autophosphorylation, and the autophosphorylation was achieved within 1 min at 25°C (Fig. 3A). Phosphoamino acid analysis revealed that both threonine and serine were phosphorylated for both isoforms (not shown). The extent of phosphorylation was the same for the two isoforms, suggesting that the C-terminal domain unique to the long isoform does not include the major sites for autophosphorylation. This view is consistent with the previous report that the multiple autophosphorylation sites are not located at the C-terminal domain of hZIPK-L [30].
Fig. 3.
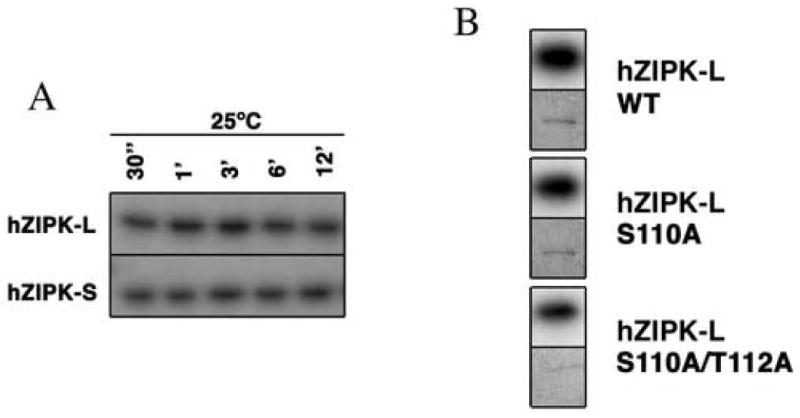
Autophosphorylation of hZIPK isoforms. A. Time course of autophosphorylation of hZIPK isoforms. SDS-PAGE of the autophosphorylated hZIPK isoforms were done as described in MATERIALS AND METHODS. B. Autophosphorylation of S110A and T112A mutants of hZIPK-L. Autophosphorylation was done at 25°C for 15 min. Upper and lower panels are autoradiography and CBB staining, respectively.
It was reported previously that ZIP-like kinase autophosphorylated at the serine and threonine residues [20]. The autophosphorylation sites of ZIP-like kinase were identified and the sequence around the phosphorylation sites was highly homologous to hZIPK isoforms. The sites are corresponding to Thr112 and Ser110 of hZIPK isoforms, therefore, we examined whether these residues are the autophosphorylation sites of hZIPK isoforms. As shown in Fig. 3B, both S110A and T112A/S110A hZIPK-L were autophosphorylated and the mutation of these sites did not much reduce the autophosphorylation, suggesting that these sites are not the predominant autophosphorylation sites of hZIPK isoforms. We also produced a hZIPK mutant in which the 45 amino acid residues from the C-terminal end of hZIPK-S were deleted (hZIPK kinase domain, hZIPK-KD). Interestingly, the truncation completely abolished the autophosphorylation of hZIPK-KD, while the kinase activity was not markedly affected (Fig. 4A). The results suggest that the C-terminal 45 residues of the short isoform contain the autophosphorylation sites.
Fig. 4.
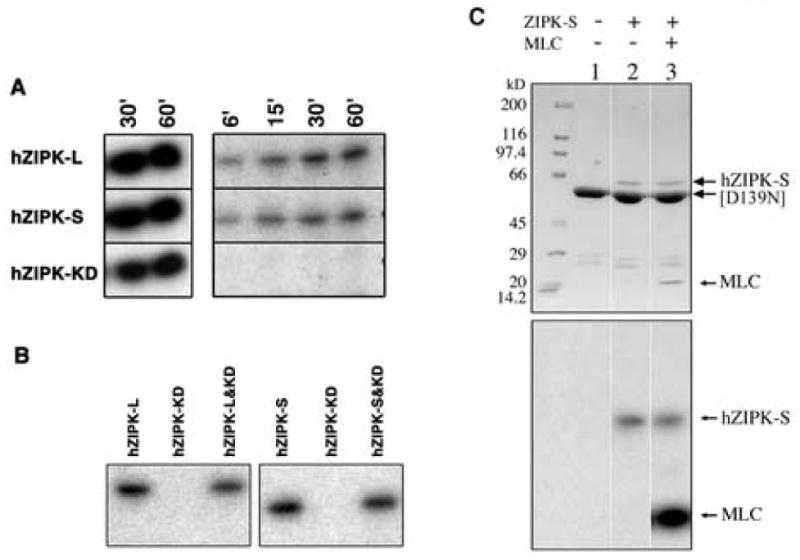
Autphosphorylation sites within the non-kinase domain of hZIPK-S. A. Autophosphorylation of hZIPK isoforms and hZIPL-KD. Autophosphorylation was done as described in MATERIALS AND METHODS. Left panel shows the autoradiogram of MLC20 phosphorylation by hZIPK. Right panel shows the autoradiogram of hZIPK autophosphorylation. B. Failure of phosphorylation of hZIPK-KD by hZIPK isoforms. Left panel: hZIPK-L (first lane), hZIPK-KD (second lane), or hZIPK-L + hZIPK-KD (third lane) were phosphorylated with 0.2 mM [γ32P]-ATP at 25°C for 15 min in the reaction buffer. Right panel: hZIPK-S (first lane), hZIPK-KD (second lane), or hZIPK-S + hZIPK-KD (third lane). Note that only hZIPK-L or-S but not hZIPK-KD was phosphorylated. C. Failure of phosphorylation of D139N hZIPK-KD by hZIPK-S. The hZIPK-KD (D139N) mutant was incubated with hZIPK-S in the presence of 0.2 mM [γ-32P] ATP at 25°C for 15 min. Upper panels show Coomassie Brilliant Blue (CBB) staining, and lower panels show autoradiography. Note that hZIPK-S phosphorylated MLC20 but not [D139N] hZIPK-KD.
To further evaluate this notion, we mixed hZIPK-KD with hZIPK-L or hZIPK-S and measured the phosphorylation of hZIPK-KD. Neither hZIPK-L nor hZIPK-S phosphorylated hZIPK-KD (Fig. 4B). The results clearly indicate that the phosphorylation sites are within 278-311 at the C-terminal end of the short isoform that is present in both isoforms. Furthermore, we produced the mutant ZIPK-KD, in which the invariant Asp in the catalytic loop is substituted by Asn, thus inactivating the kinase activity ([D139N]ZIPK-KD). As shown in Fig. 4C, ZIPK-S did not phosphorylate [D139N]ZIPK-KD, while ZIPK-S is autophosphorylated. The result is consistent with the result in Fig. 3B, and showed that the catalytic domain of the expressed huZIPK cannot be autophosphorylated. However, the above experiments cannot exclude the possibility that the hZIPK-KD is pre-autophosphorylated and the incubation with [γ-32P]ATP does not incorporate the radioactivity into the kinase domain of hZIPK (see DISCUSSION).
Binding of hZIPK isoforms to MYPT1 and myosin
Since hZIPK-S phosphorylates MLC20 and MYPT1, we examined whether hZIPK-S binds to MYPT1 and myosin. Isolated GST-hZIPK-S was incubated with myosin or MYPT1 then centrifuged with glutathione-coated beads at low speed. After the beads were washed, the precipitated beads were subjected to SDS-PAGE followed by Western blot (Fig. 5). MYPT1 was co-precipitated with hZIPK-S. Myosin was also co-precipitated with hZIPK-S. The result suggests that ZIPK-S directly binds to both MYPT1 and myosin (Fig. 5A). To determine the region of hZIPK-S responsible for the binding to MYPT1 and myosin, the binding of hZIPK-KD to these proteins was examined. Interestingly, hZIPK-KD binds to myosin but not MYPT1 (Fig. 5A). The results suggest that residues 278-311 are involved in the binding to MYPT1 while myosin binds to the kinase domain. We also examined the binding of hZIPK to actin by GST pull-down assay. The proteins bound to the beads as well as the unbound fraction were determined by SDS-PAGE. The entire amount of actin was recovered in the supernatant and we could not detect actin in the pellets (not shown). Furthermore, we examined whether autophosphorylation influence the binding of hZIPK-S to myosin or MYPT1. The autophosphorylation did not markedly change the binding of hZIPK-S to MYPT1 and myosin (Fig. 5B). The signal changes were quantified by three independent experiments and found that the amount of the bound phosphorylated hZIPK-S was 80% of the unphosphorylated one for both myosin II and MYPT1. On the other hand, the phosphorylation of myosin and MYPT1 significantly affected the interaction with hZIPK-S. Myosin and MYPT1 were phosphorylated by hZIPK-S and, then subjected to the GST-pull-down assay. As shown in Fig. 5C, the binding of hZIPK-S to both myosin and MYPT1 was markedly attenuated by phosphorylation.
Fig. 5.
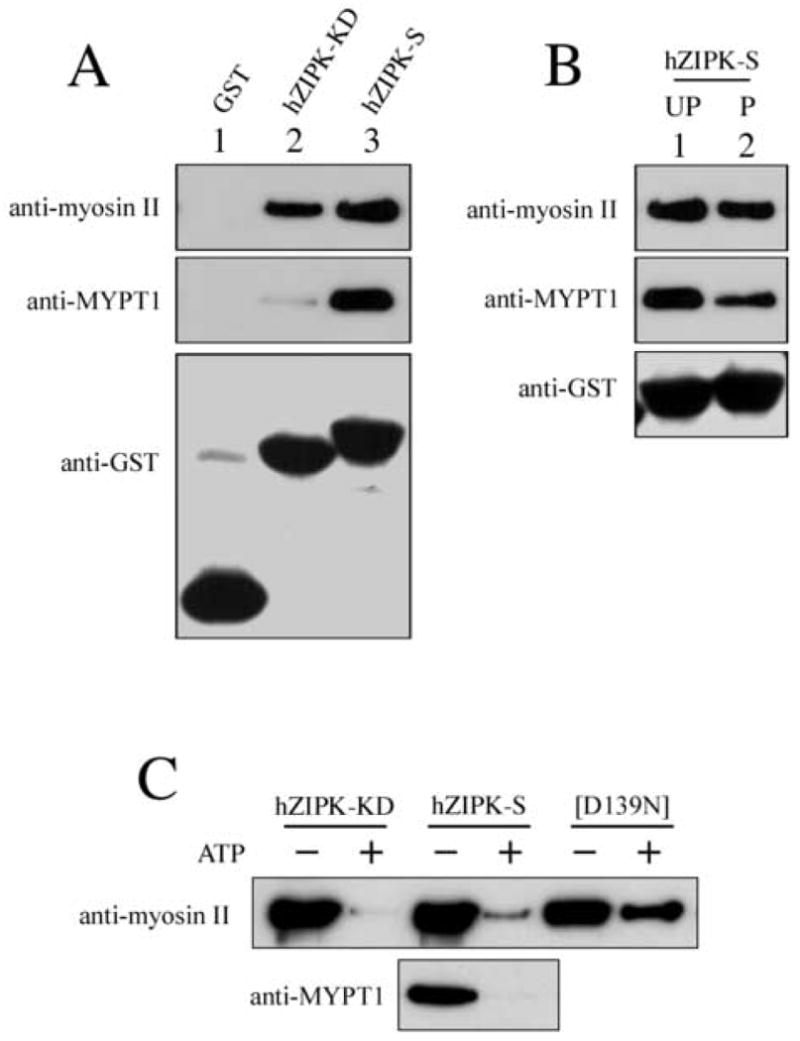
Interaction of hZIPK isoforms with MYPT1 and myosin. A. Binding of hZIPK isoforms to myosin and MYPT1. GST tagged hZIPK isoforms were incubated with myosin or MYPT1, and then subjected to GST-pull-down assay. The proteins precipitated with GST-Sepharose were subjected to SDS-PAGE followed by Western blot by using specific Abs (anti-myosin II Ab, anti-MYPT1 Ab and anti-GST Ab, respectively). B. Effect of autophosphorylation on the binding of hZIPK-S to myosin and MYPT1. Phosphorylated and non-phosphorylated GST tagged hZIPK-S were incubated with myosin or MYPT1 and then subjected to GST-pull-down assay. UP, unphosphorylated; P, phosphorylated. C. Effect of substrate phosphorylation on the binding of hZIPK-S to myosin and MYPT1. hZIPK-KD, hZIPK-S and [D139N] hZIPK-KD were incubated with either myosin II or MYPT1 in the presence of ATP at 25°C for 30 min for phosphorylation. The proteins were also incubated in the absence of ATP as a control. The samples were then subjected to the GST-pull-down assay.
Intracellular localization of hZIPK isoforms
Figure 6 shows the localization of hZIPK isoforms in ARPE19 cells. Both isoforms of hZIPK showed filamentous localization, which colocalizes actin stress fibers as revealed by the double staining of Phalloidine (Fig. 6A,B, panel a and b). On the other hand, the majority of the kinase domain of hZIPK (hZIPK-KD) showed diffuse localization although we found some cells showing filamentous localization (not shown). It should be noted that hZIPK isoforms are also present in the cytosolic pool and nucleus and there were cells showing the cytosolic localization with no significant filamentous localization of hZIPK isoforms. Fig. 6 also shows the co-localization of hZIPK isoforms with the phosphorylated myosin probed by the di-phosphorylated MLC20 specific Ab [18]. Phosphorylated myosin showed stress-fiber localization consistent with the previous study [18] and was highly colocalized with hZIPK isoforms (Fig. 6A,B, panel d). To more precisely evaluate the colocalization, fluorescence distribution of the transverse section of the images of hZIPK, actin and di-phosphorylated myosin II was compared. The distribution of the GFP signal well coincided with those of actin and di-phosphorylated myosin II signals (Fig. 6 A,B panel e). It should be noted that hZIPK-L was present predominantly in cytosol while hZIPK-S was present in both cytosol and nucleus. The results were consistent with the finding that both hZIPK isoforms binds to myosin in vitro (Fig. 5).
Fig. 6.
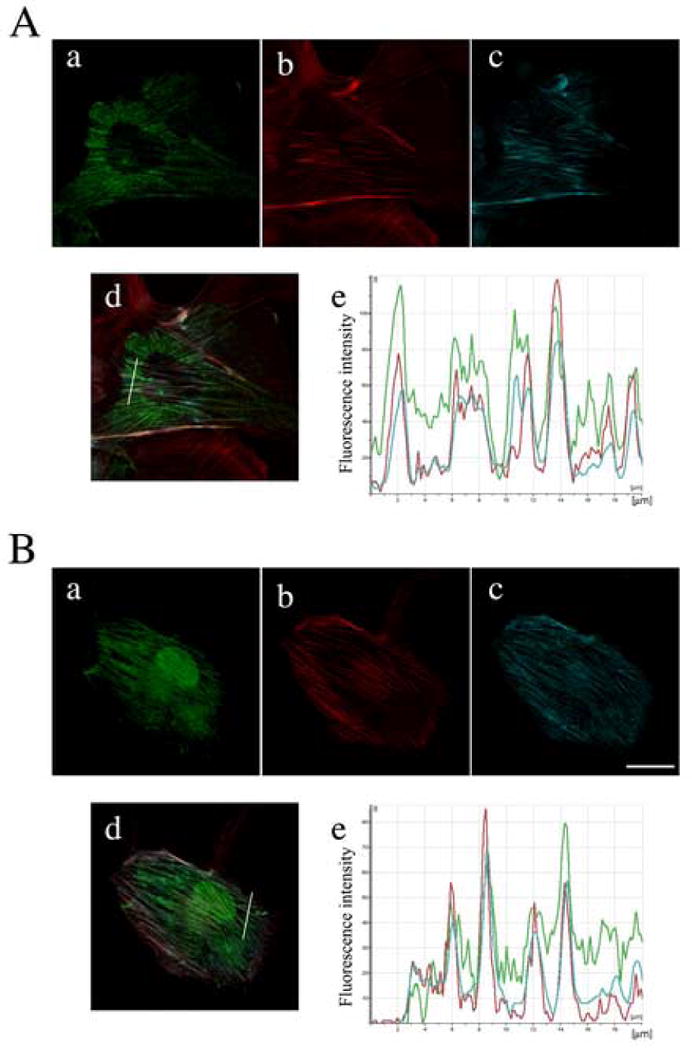
Co-localization of hZIPK isoforms with phosphorylated myosin II in ARPE19 cells. The cells were transiently transfected with various myc-tagged hZIPK constructs as indicated. Fifteen hours post transfection, cells were fixed and stained with anti-myc Ab. Cells were also stained with tetramethyl-rhodamine labeled phalloidine to visualize actin structure, and anti-pTS Ab to stain the di-phosphorylated myosin as described in MATERIALS AND METHODS. A, hZIPK-L; B, hZIPK-S. a, myc-tagged hZIPK; b, actin; c, di-phosphorylated myosin; d, merged images of a, b and c; e, Fluorescence intensity distribution of the transverse section of the image ‘d’ shown by white lines. Green trace, hZIPK signals; red trace, F-actin; blue trace, di-phosphorylated myosin. Bars represent 20 μm.
Discussion
We report here a novel ZIP kinase isoform (hZIPK-S) in which a majority of originally assigned exon 8, encoding the C-terminal non-kinase domain, is spliced out. This type of splicing has been known as intron retention [31, 32]. A similar type of the splicing variants are found for myosin IXb [33](Kambara and Ikebe, unpublished observation). Previously, MacDonald et al. [20] reported that ZIP-like kinase purified from cow bladder had an apparent molecular mass of 32 kDa that was much smaller than the reported mass of ZIP kinase, although one cannot eliminate the possibility that the 32 kDa peptide is produced by proteolytic degradation of ZIP Kinase during biochemical purification from cow bladder tissues. The calculated molecular mass of hZIPK-S found in the present study is 37kDa, and an apparent molecular mass estimated with the mobility of SDS-PAGE is 34 kDa. Based upon the similarity of the molecular mass, previously reported ZIP-like kinase may be the short isoform of human ZIP kinase (hZIPK-S) found in the present study. However, the partial amino acid sequence of ZIP-like kinase determined by Edman degradation of the three isolated peptides showed that seven amino acid residues out of the thirty four determined residues were different from those of kinase domain of ZIP kinase. On the other hand, the kinase domain of hZIPK-S was completely identical to that of the long isoform (hZIPK-L). The amino acid sequence of the kinase domain of ZIP kinase is highly conserved among the mammalian species [1] and the region of the three peptides, determined for ZIP-like kinase, is completely identical among the ZIP kinase from various species. Therefore, it is unlikely that the different sequence of bovine ZIP-like kinase from ZIP kinases is due to the species difference. Therefore, ZIP-like kinase might be different from the short isoform of ZIP kinase (hZIPK-S) identified in the present study, although one cannot eliminate the possibility that the difference in the sequence is due to the amino acid sequencing error of the ZIP-like kinase.
It was reported recently that there were no spliced variants of the ZIP kinase in vascular smooth muscle [34]. Our result clearly demonstrated that there is a short isoform present in smooth muscle tissues that is produced due to the splicing at the exon 8. As shown in Fig.1, the original 3′ untranslated region is almost completely deleted in hZIPK-S and the presence of this isoform could only be found using the 3′ RACE technique. The sequence used for the minus strand PCR primers in the previous study are at the regions upstream of the 5′ region of the exon 8 and these primers do not yield the PCR product containing hZIPK-S unique sequence. Therefore, it is reasonable to assume that the previous study could not detect the present short ZIP kinase isoform (ZIPK-S)[34].
We found that hZIPK-S can bind to MYPT1 similar to hZIPK-L. The result indicates that the C-terminal domain of the long isoform including the leucine zipper is not critical to the binding of ZIP kinase to MYPT1. We found that the kinase domain of hZIPK (Met1-Ala277, hZIPK-KD) has no binding activity to MYPT1, suggesting that the amino acids, Ile278-Ser312, at least in part responsible for the MYPT1 binding site of ZIP kinase. On the other hand, we found that both the hZIPK-S and hZIPK-KD bind to myosin. This result suggests that the myosin binding site is different from that of the MYPT1 binding site.
Initially nuclear localization of rodent ZIP kinase was reported in association with cell death inducing activity [3]. Subsequently, human ZIP kinase was cloned from Hela cells [5], which also induces cell death in Hela cells and showed essentially cytoplasmic localization. Our data supports that, unlike rodent ZIP kinases, human ZIP kinase resides in cytoplasm and may play a role other than inducing cell death in the cytoplasm. Interestingly, the kinase activity of the ZIPK-S was significantly higher for myosin but not for MYPT1. Furthermore, our biochemical analysis revealed that the ZIPK-S has different kinase properties from the ZIPK-L, suggesting that the C-terminal domain including the leucine zipper of ZIP kinase is important for its substrate specificity. Consistently, it was reported that the leucine zipper of human ZIP kinase (ZIPK-L) affects its enzymatic activity and cellular localization [30].
It was previously shown that ZIP kinase localizes at the stress fiber in mammalian motile cells where a significant level of di-phosphorylated MLC20 is localized (18). In the present study, we found the localization of the newly found hZIPK-S isoform at the stress fiber in addition to the previously found hZIPK-L isoform. Furthermore, we found that hZIPK isoforms were co-localized with myosin at the stress fiber. These results are consistent with the finding that hZIPK isoforms bind to myosin in vitro. Based on these findings, it is thought that hZIPK binds to myosin at the stress fibers where hZIPK isoforms phosphorylates myosin to stabilize the thick filament formation. On the other hand, it has been shown that MYPT1 in part localized at the stress fiber [35]. Therefore, it is plausible that hZIPK also phosporylates the inhibitory site of MYPT1 at the stress fiber thus down-regulating the MLCP activity, and attenuates the dephosphorylation of myosin II at the stress fiber.
It was shown previously that ZIP kinase undergoes autophosphorylation, yet the effect of autophosphorylation on the function has been obscure. We examined the effect of autophosphorylation on hZIPK-S function. Of interest is whether the autophosphorylation of hZIPK-S alters the binding to MYPT1 because the region overlaps the segment responsible for the binding of hZIPK-S to MYPT1. However, we could not find a significant effect of autophosphorylation on the binding to MYPT1. On the other hand, we found a significant decrease in the binding of hZIPK-S to the targeting proteins, i.e., myosin and MYPT1, when they were phosphorylated. The result suggests that ZIP kinase leaves away from the targeting proteins as soon as it finishes phosphorylation. Therefore, it is thought that the localization of hZIPK isoforms at the stress fiber predominantly reflects the binding to the unphosphorylated myosin present in the stress fiber. Because it is reasonable to assume that the number of the ZIP kinase molecules is much less than those of myosin II and MYPT1, the low affinity of ZIP kinase for the phosphorylated target proteins enables it to efficiently phopshorylate the unphosphorylated target proteins.
Our results indicate that the major phosphorylation sites of ZIP kinase are centered within Ile278-Ser312, which is included in the short isoform found in the present study, but not in the catalytic core. Graves et al. [30] reported that human ZIP kinase expressed in HEK293 cells is autophosphorylated at Thr180 and Thr225, and Thr265 in the catalytic core revealed by 32P incorporation into the corresponding peptides. They claimed that the phosphorylation was necessary for the kinase activity of ZIP kinase because the mutation at Thr180, Thr225, or Thr265 abolished the kinase activity. The apparent discrepancy of the results is unclear. However, since the reported three residues are in the kinase activation T loop, the substrate binding groove, and the kinase subdomain X, respectively [30], it is likely that the mutation itself disturbs the structure of the kinase thus inactivating the enzyme activity. Supporting this notion, T180D and T225D mutation caused nearly complete inactivation of the kinase rather than mimicking phosphorylation (activation) [30]. Interestingly, the kinase dead ZIP kinase was also phosphorylated at S311 and T265 that was argued to be phosphorylated by endogenous ZIP kinase associated with the dead kinase [30], therefore, the observed 32P incorporation to Thr180, Thr225 and Thr265 might be catalyzed by the associated kinases.
Quite recently, it was shown that the kinase domain of the recombinant mouse ZIP kinase expressed in E. coli can be autophosphorylated at Thr265 and this plays an important role in its activity [36]. The apparent discrepancy from our data is likely due to the difference in the expression systems. As is known, many protein kinases are phosphorylated right after the protein synthesis which is important for the activation of the kinase activity in eukaryote, while such an apparatus is lacking in prokaryote. Therefore, it is reasonable to assume that the expressed huZIPK in sf9 cells is pre-phosphorylated by endogenous kinases due to post-translational modification. Supporting this notion, it is shown that ZIP kinase can be phosphorylated by endogenous kinases associated with ZIP kinase in mammalian cultured cells [30, 37]. These results suggest that ZIP kinase is phosphorylated at the activation loop by post translational modification thus producing an active kinase and the additional sites located at the C-terminal side of the catalytic domain are phosphorylated either by autophosphorylation or by other kinases. At present, the significance of the regulatory phosphorylation on the function of ZIP kinase is obscure and the understanding of the regulation of ZIP kinase requires further study.
Acknowledgments
This work was supported by NIH grants AR41653 and HL073050 (M.I.) and the American Heart Association grant 0535419T (S.K.).
Abbreviations
- ZIPK
Zipper-interacting protein kinase
- DAPK
death-associated protein kinase
- DLK
death-associated protein kinase like kinase
- MLCK
myosin light chain kinase
- MLC20
myosin regulatory light chain
- MLCP
myosin light chain phosphatase
- MYPT1
regulatory subunit of myosin light chain phosphatase
- GST
glutathion-S-transferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, Matsumoto M, Takeda K, Sanjo H, Akira S. Mol Cell Biol. 1998;18:1642–1651. doi: 10.1128/mcb.18.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kogel D, Plottner O, Landsberg G, Christian S, Scheidtmann KH. Oncogene. 1998;17:2645–2654. doi: 10.1038/sj.onc.1202204. [DOI] [PubMed] [Google Scholar]
- 3.Kogel D, Bierbaum H, Preuss U, Scheidtmann KH. Oncogene. 1999;18:7212–7218. doi: 10.1038/sj.onc.1203169. [DOI] [PubMed] [Google Scholar]
- 4.Murata-Hori M, Fukuta Y, Ueda K, Iwasaki T, Hosoya H. Oncogene. 2001;20:8175–8183. doi: 10.1038/sj.onc.1205055. [DOI] [PubMed] [Google Scholar]
- 5.Murata-Hori M, Suizu F, Iwasaki T, Kikuchi A, Hosoya H. FEBS Lett. 1999;451:81–84. doi: 10.1016/s0014-5793(99)00550-5. [DOI] [PubMed] [Google Scholar]
- 6.Niiro N, Ikebe M. J Biol Chem. 2001;276:29567–29574. doi: 10.1074/jbc.M102753200. [DOI] [PubMed] [Google Scholar]
- 7.Hartshorne DJ. In: Physiology of the Gastrointestinal Tract. Johnson LR, editor. New York: Raven; 1987. pp. 423–482. [Google Scholar]
- 8.Kamm KE, Stull JT. Annu Rev Physiol. 1989;51:299–313. doi: 10.1146/annurev.ph.51.030189.001503. [DOI] [PubMed] [Google Scholar]
- 9.Tan JL, Ravid S, Spudich JA. Annu Rev Biochem. 1992;61:721–759. doi: 10.1146/annurev.bi.61.070192.003445. [DOI] [PubMed] [Google Scholar]
- 10.Ikebe M, Hartshorne DJ. J Biol Chem. 1985;260:10027–10031. [PubMed] [Google Scholar]
- 11.Ikebe M, Hartshorne DJ, Elzinga M. J Biol Chem. 1986;261:36–39. [PubMed] [Google Scholar]
- 12.Ikebe M, Reardon S. Biochemistry. 1990;29:2713–2720. doi: 10.1021/bi00463a014. [DOI] [PubMed] [Google Scholar]
- 13.Kamisoyama H, Araki Y, Ikebe M. Biochemistry. 1994;33:840–847. doi: 10.1021/bi00169a027. [DOI] [PubMed] [Google Scholar]
- 14.Colburn JC, Michnoff CH, Hsu LC, Slaughter CA, Kamm KE, Stull JT. J Biol Chem. 1988;263:19166–19173. [PubMed] [Google Scholar]
- 15.Singer HA. Am J Physiol. 1990;259:C631–639. doi: 10.1152/ajpcell.1990.259.4.C631. [DOI] [PubMed] [Google Scholar]
- 16.Itoh K, Hara T, Yamada F, Shibata N. Biochim Biophys Acta. 1992;1136:52–56. doi: 10.1016/0167-4889(92)90084-o. [DOI] [PubMed] [Google Scholar]
- 17.Choi OH, Adelstein RS, Beaven MA. J Biol Chem. 1994;269:536–541. [PubMed] [Google Scholar]
- 18.Komatsu S, Ikebe M. J Cell Biol. 2004;165:243–254. doi: 10.1083/jcb.200309056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson DP, Sutherland C, Borman MA, Deng JT, Macdonald JA, Walsh MP. Biochem J. 2005;392:641–648. doi: 10.1042/BJ20051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacDonald JA, Borman MA, Muranyi A, Somlyo AV, Hartshorne DJ, Haystead TA. Proc Natl Acad Sci USA. 2001;98:2419–2424. doi: 10.1073/pnas.041331498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 22.Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, Nakano T. J Biol Chem. 1999;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- 23.Kiss E, Muranyi A, Csortos C, Gergely P, Ito M, Hartshorne DJ, Erdodi F. Biochem J. 2002;365:79–87. doi: 10.1042/BJ20011295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muranyi A, MacDonald JA, Deng JT, Wilson DP, Haystead TA, Walsh MP, Erdodi F, Kiss E, Wu Y, Hartshorne DJ. Biochem J. 2002;366:211–216. doi: 10.1042/BJ20020401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikebe M, Hartshorne DJ. J Biol Chem. 1985;260:13146–13153. [PubMed] [Google Scholar]
- 26.Ikebe M, Reardon S, Schwonek JP, Sanders CR, 2nd, Ikebe R. J Biol Chem. 1994;269:28165–28172. [PubMed] [Google Scholar]
- 27.Spudich JA, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 28.Yano K, Araki Y, Hales SJ, Tanaka M, Ikebe M. Biochemistry. 1993;32:12054–12061. doi: 10.1021/bi00096a016. [DOI] [PubMed] [Google Scholar]
- 29.Ikebe M, Komatsu S, Woodhead JL, Mabuchi K, Ikebe R, Saito J, Craig R, Higashihara M. J Biol Chem. 2001;276:30293–30300. doi: 10.1074/jbc.M101969200. [DOI] [PubMed] [Google Scholar]
- 30.Graves PR, Winkfield KM, Haystead TA. J Biol Chem. 2005;280:9363–9374. doi: 10.1074/jbc.M412538200. [DOI] [PubMed] [Google Scholar]
- 31.Black DL. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 32.Andreadis A, Gallego ME, Nadal-Ginard B. Annu Rev Cell Biol. 1987;3:207–242. doi: 10.1146/annurev.cb.03.110187.001231. [DOI] [PubMed] [Google Scholar]
- 33.Grewal PK, Jones AM, Maconochie M, Lemmers RJ, Frants RR, Hewitt JE. Gene. 1999;240:389–398. doi: 10.1016/s0378-1119(99)00459-x. [DOI] [PubMed] [Google Scholar]
- 34.Endo A, Surks HK, Mochizuki S, Mochizuki N, Mendelsohn ME. J Biol Chem. 2004;279:42055–42061. doi: 10.1074/jbc.M403676200. [DOI] [PubMed] [Google Scholar]
- 35.Murata K, Hirano K, Villa-Moruzzi E, Hartshorne DJ, Brautigan DL. Mol Biol Cell. 1997;8:663–673. doi: 10.1091/mbc.8.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato N, Kamada N, Muromoto R, Kawai T, Sugiyama K, Watanabe T, Imoto S, Sekine Y, Ohbayashi N, Ishida M, Akira S, Matsuda T. Immunol Lett. 2006;103:127–134. doi: 10.1016/j.imlet.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 37.Shani G, Marash L, Gozuacik D, Bialik S, Teitelbaum L, Shohat G, Kimchi A. Mol Cell Biol. 2004;24:8611–8626. doi: 10.1128/MCB.24.19.8611-8626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]