

Abstract
Aicardi-Goutières syndrome (AGS) is a genetically determined encephalopathy demonstrating phenotypic overlap both with the sequelae of congenital infection and with systemic lupus erythematosus (SLE). Recent molecular advances have revealed that AGS can be caused by mutations in any one of five genes, most commonly on a recessive basis but occasionally as a dominant trait. Like AGS, SLE is associated with a perturbation of type I interferon metabolism. Interestingly then, heterozygous mutations in the AGS1 gene TREX1 underlie a cutaneous subtype of SLE-called familial chilblain lupus, and mutations in TREX1 represent the single most common cause of monogenic SLE identified to date. Evidence is emerging to show that the nucleases defective in AGS are involved in removing endogenously produced nucleic acid (NA) species, and that a failure of this removal results in activation of the immune system. This hypothesis explains the phenotypic overlap of AGS with congenital infection and some aspects of SLE, where an equivalent type I interferon-mediated innate immune response is triggered by viral and self NAs, respectively. The combined efforts of clinicians, geneticists, immunologists and cell biologists are producing rapid progress in the understanding of AGS and overlapping autoimmune disorders. These studies provide important insights into the pathogenesis of SLE and beg urgent questions about the development and use of immunosuppressive therapies in AGS and related phenotypes.
INTRODUCTION
A disturbance of interferon alpha (IFN-α) homeostasis is central to the pathogenesis of the prototypic autoimmune disorder systemic lupus erythematosus (SLE) (1–3). As in lupus, perturbation of IFN-α metabolism is a major pathogenic feature of the inflammatory encephalopathy Aicardi-Goutières syndrome (AGS) (4). In keeping with this, some children with AGS develop an early-onset form of SLE (5–8); heterozygous mutations in the AGS1 gene TREX1 underlie a cutaneous subtype of SLE-called familial chilblain lupus (FCL) (9); and, remarkably, ∼2% of SLE patients harbour pathogenic mutations in TREX1 (10). Rare but high-penetrant causes of lupus are important to identify because they provide immediate insights into pathogenesis.
TREX1 deficiency results in the intracellular accumulation of DNA and a type I interferon response accrues from the activation of a Toll-like receptor (TLR)-independent pathway (11,12). Distinct from defects in central and peripheral lymphocyte tolerance or activation of non-cell-autonomous innate immune recognition through TLRs, these studies define a new mechanism for the initiation of autoimmunity by interferon-stimulatory nucleic acid (NA).
In this report, we provide a selected overview of the important clinical and molecular features of AGS and related phenotypes, and discuss recent data linking disordered NA metabolism with autoimmunity.
MOLECULAR BASIS OF AGS AND RELATED PHENOTYPES
In its classic presentation, AGS can be considered as a Mendelian mimic of the sequelae of in utero viral infection (13). Severe neurological dysfunction becomes apparent in infancy, and manifests as progressive microcephaly, spasticity, psychomotor retardation and, in ∼40% of cases, death in early childhood (14). Intracranial calcification, white matter destruction and brain atrophy are evident on neuroimaging, similar to the radiological findings seen in congenital infections, while outside the nervous system, thrombocytopenia, hepatosplenomegaly, elevated hepatic transaminases and intermittent fevers also erroneously suggest an infective process (15).
AGS is a genetically heterogeneous disorder caused by mutations in any of the genes encoding the 3′→5′ exonuclease TREX1 (AGS1) (16), the three non-allelic components of the RNASEH2 endonuclease complex (AGS2, 3 and 4) (17) and the uncharacterized SAMHD1 protein (AGS5) (18) (Table 1). Evidence of further genetic heterogeneity exists.
Table 1.
Genes so far identified to be associated with the AGS phenotype
| Gene | Locus | Protein | % of families with mutations |
|---|---|---|---|
| AGS1 | 3p21 | TREX1/DNaseIII | 35 |
| AGS2 | 13q14 | RNASEH2B | 45 |
| AGS3 | 11q13 | RNASEH2C | 15 |
| AGS4 | 19p13 | RNASEH2A | <5 |
| AGS5 | 20q11 | SAMHD1/DCIP | ∼10 |
AGS-RELATED GENES AND PROTEINS
TREX1
TREX1 (DNase III) represents the major 3′→5′ DNA exonuclease activity measured in mammalian cells (19). It was originally thought that TREX1 may serve to excise mismatched dNTPs during lagging strand synthesis or gap-filling during base excision repair (20). However, TREX1 null mice did not show an increased spontaneous mutation frequency (21). Instead, like AGS patients, TREX1 knock-out mice exhibit an inflammatory phenotype although, in contrast to AGS patients, they do not demonstrate neurological involvement but show an inflammatory cardiomyopathy (21). There is evidence to suggest that TREX1 associates, perhaps through its polyproline II helix (22,23), with the SET complex, a caspase-independent cell death pathway also including the DNA binding protein HMGB2 and the endonucleases APE1 and NM23-H1, where TREX1 might rapidly degrade 3′ ends of nicked dsDNA (24). The significance of this possibility in relation to human disease is unclear.
The majority of recessive AGS-causing TREX1 mutations are predicted as null alleles (with the recurrent missense transition c.341G>A involving the arginine residue at 114 crucial for protein dimerization and so likely also abrogating protein function) (14). Following the identification of AGS-causing biallelic mutations in TREX1 in 2006, heterozygous TREX1 mutations were described in a cutaneous form of SLE-called FCL (9,25,26), in rare cases of dominantly inherited AGS (9) and in patients with an adult-onset cerebro-retinal microangiopathy associated with Raynaud's phenomenon [retinal vasculopathy with cerebral leukodystrophy (RVCL)] (27). Furthermore, in light of the phenotypic and biochemical overlap of AGS with lupus, heterozygous TREX1 mutations were searched for and found in ∼2% of a cohort of individuals with SLE (10) (Table 2).
Table 2.
Summary of recognized phenotypes associated with TREX1 mutations
| Phenotype | AGS | RVCL | FCL | SLE |
|---|---|---|---|---|
| Inheritance | AR (rare AD cases) | AD | AD | Rare monogenic forms |
| Genes | TREX1(9,16), RNASEH2A/B/C (17), SAMHD1 (18) | TREX1 (27) | TREX1 (9,25,26) | Monogenic: TREX1 (10), DNASE1 (28), complement deficiency (29) |
| Onset | Prenatal—usually <12 months | 30–50 years | Childhood | Usually 15–40 years |
| Mortality | 40% <10 years of age | 5–20% 10 year mortality (from onset) | Non-lethal | 5–20% 10 year mortality |
| Neurological involvement | Severe intellectual and physical disability | Strokes, seizures, migraine, cognitive decline | None | Neuro-lupus: strokes, seizures, psychosis, cognitive decline |
References shown in brackets.
The pathogenicity of, and phenotypic variability associated with, heterozygous TREX1 mutations is explained, at least partly, by the residues involved and the position of the mutation (Fig. 1).
Figure 1.

Schematic of (selected) disease-associated TREX1 mutations. Mutations: black, recessive AGS; purple, dominant AGS; green, FCL; blue, SLE; red, RVCL. Exo1, 2, 3 domains; PII, polyproline II domain; TM, transmembrane domain.
In particular, all RVCL-, and most lupus-, associated mutations cluster in the C-terminus, responsible for tethering the protein in the endoplasmic reticulum (10,27). Thus, C-terminus RVCL and lupus frameshift mutations have been shown to result in cellular mislocalization rather than a loss of enzymatic activity per se (although disturbed localization might effectively equate with an absence of enzymatic activity in the right place). Meanwhile, the heterozygous mutations at D18 and D200 (published in association with dominant FCL and de novo dominant AGS respectively) involve key catalytic residues which, in vitro, have been shown to possess significantly reduced exonuclease activity against double-stranded DNA (dsDNA) and to inhibit wild-type protein activity (30). It is of note that some AGS patients have C-terminus frameshift mutations, and there is considerable overlap of the missense mutations reported in lupus patients (including the R114H mutation identified on more than 60 AGS-associated disease alleles) with those seen in AGS (14). These observations suggest that some AGS patients and their heterozygous parents may be at risk of developing RVCL and SLE.
RNASEH2
Ribonucleases H (RNASEH) are endonucleases that cleave the RNA of RNA/DNA hybrids in a sequence non-specific manner (31). Eukaryotes have two types of RNASEH (H1/I and H2/II) showing distinct enzymatic and site-specific activity with RNASEH2 enzymes able to recognize single ribonulceotides embedded in DNA duplexes (32,33). RNASEH2 is the major source of cellular ribonuclease activity in eukaryotes (33,34). Eukaryotic RNASEH2 is composed of three different proteins, the catalytic subunit (2A), and two further subunits (2B, 2C) (encoded by AGS4, AGS2 and AGS3, respectively) that have no prokaryotic counterparts and as yet unknown functions but that are necessary for catalysis (17,35,36).
In contrast to TREX1, the overwhelming majority of AGS2, 3 and 4 mutations are hypomorphic, and it is of note that biallelic null mutations in any of these genes have never been observed (14), suggesting that such a state may be lethal or result in presently unrecognized phenotypes. In keeping with this, recent work demonstrates that most RNASEH2 complex mutations so far studied do not alter recorded nuclease activity (36,37). The failure to note differences in activity in vitro suggests that changes in assembly, stability or localization of the complex might be important in producing a disease phenotype.
The ability of RNASEH2 to recognize and cleave a single ribonucleotide in a DNA duplex suggested a possible role for the enzyme in DNA repair where DNA polymerases might mistakenly incorporate a ribo- rather than deoxyribnulceotide (38). Other proposed functions are in the suppression of R-loops transiently formed during transcription (31,39), and the hydrolysis of Okazaki fragment RNA primers (40). Recent data indicate that RNASEH2B may interact with proliferating cell nuclear antigen (PCNA), via a PCNA-interacting peptide at its C-terminus (36), a protein essential for eukaryotic Okazaki fragment processing during lagging strand synthesis.
SAMHD1
This summer, mutations in the AGS5 gene encoding SAMHD1 were reported to cause AGS (18). As for TREX1, mutations in SAMHD1 include biallelic null alleles as well as missense mutations. The functions of the 626 amino acid protein SAMHD1 are currently unknown. SAMHD1 was originally identified in a human dendritic cell cDNA library as an orthologue of the mouse IFN-γ-induced gene Mg11 (41), hence the alternative name dendritic cell-derived IFN-γ-induced protein (DCIP) (NB. Mg11 was cloned by Lafuse et al. (42) although the cited reference discusses an unrelated gene Mg21). Other evidence also implicates SAMHD1 in immunity since it is upregulated in response to viral infections (43–45) and may have a role in mediating TNF-α proinflammatory responses (46). The SAMHD1 name derives from the presence of a sterile alpha motif (SAM) and a HD domain in tandem, an arrangement which is apparently unique amongst human proteins. SAMs are 65–70 residues in length and can serve as protein-interaction modules mediating interactions with other SAM domain and non-SAM domain-containing proteins (47). Additionally, the SAM domains of S. cerevisiae Vts1p and its D. melanogaster homolog Smaug bind an RNA stem-pentaloop hairpin in a sequence non-specific manner (48). The HD domain, characterized by a doublet of divalent-cation-coordinating His and Asp residues, is found in a diverse superfamily of enzymes with predicted or known phosphohydrolase activity (49). It is noteworthy that nucleotides are the substrates of five HD-domain enzymes characterized to date (50), while a sixth, YhaM, has a known exonuclease activity (51).
PATHOGENESIS
Two recent high-profile papers on TREX1 have provided crucial insights into the pathogenesis of AGS. In 2007, Yang et al. (11), using TREX1−/− mouse embryonic fibroblasts and AGS-derived patient fibroblasts, corroborated previous data to show that TREX1 predominantly localizes to the cytoplasm. They also demonstrated a relocation of the protein to BrdU positive foci in the nucleus during S phase and presented data suggesting that TREX1 null cells exhibit defective G1/S transition and chronic ATM-dependent checkpoint protein activation. Remarkably, TREX1 null cells were shown to accumulate 60–65 nucleotide long single-stranded DNA (ssDNA) species proposed to derive from cells in S phase. Taken together, these data were interpreted to indicate that TREX1 acts on ssDNA polynucleotides, generated from the processing of replication intermediates, to attenuate checkpoint signalling and prevent pathological immune activation. Considering a possible common substrate on which both TREX1 and the RNASEH2 complex might act, the authors invoked a model involving the ‘folding back’ of short flaps of DNA with an attached RNA primer, produced by the resolution of Okazaki fragments during lagging strand synthesis.
Having previously defined the IFN-stimulatory DNA (ISD) response, a cytosolic antiviral pathway that detects DNA, in 2008 Stetson et al. (12) showed that TREX1 is an essential negative regulator of the ISD response. Using a series of mouse crosses to dissect the pathway linking TREX1 deficiency to lethal autoimmunity, they demonstrated a TLR-independent pathway signalling through the transcription factor IRF3 (Table 3). As expected from human studies, an intact type I IFN response was necessary to develop the disease phenotype. Of considerable interest, by crossing the TREX1 null mouse with a knock-out for the DNA recombinase RAG2 required for generating functional lymphocytes, they also showed that the inflammatory pathology was dependent on antibody production (Table 3). Controversially, unlike Yang et al., Stetson et al. (12) found no evidence for the activation of DNA damage checkpoint signalling, which they suggested might be an artefact related to cell line immortalization. Rather, although they also demonstrated an accumulation of ssDNA in TREX1 null cells, they suggested that such DNA derived from endogenous retroelements otherwise metabolized by functional TREX1.
Table 3.
Summary of mouse cross data presented by Stetson et al. (12)
| Mouse model | Phenotype and IFN status |
|---|---|
| TREX1 null | Inflammatory cardiomyopathy: high IFN |
| TREX1/IRF3 DKO | ‘Cured’: low IFN |
| TREX1/IFNαR1 DKO | ‘Cured’: low IFN |
| TREX1/RAG2 DKO | ‘Cured’: high IFN |
DKO, double knock-out; IRF3, interferon regulatory factor 3; IFNαR1, type I IFN-receptor.
Nucleic acid metabolism in AGS
As TREX1 and RNASEH2 are nucleases, it was previously hypothesized that these proteins might remove ‘waste’ NAs, and that a failure of this process could result in immune activation (17,52). The work of both Yang et al. (11) and Stetson et al. (12) shows that TREX1 deficiency does indeed lead to the intracellular accumulation of DNA, and the data generated by Stetson et al. further demonstrate activation of the immune system by these accumulated NA (12). Distinct from defects in central and peripheral lymphocyte tolerance or activation of non-cell-autonomous innate immune recognition through TLRs, these studies define a novel cell-autonomous, TLR-independent mechanism for the initiation of autoimmunity by IFN-stimulatory NA (Fig. 2).
Figure 2.

Model of disease pathogenesis in deficiency of TREX1, RNASEH2 and SAMHD1 activity. NA (ssDNA in TREX1 deficiency putatively derived from endogenous retroelements (12) and/or Okazaki fragments (11); possibly RNA:DNA hybrids in absence of RNASEH2 (17): unknown in SAMHD1 deficiency (18)) accumulate and are recognized by as yet undefined sensors leading to the TLR-independent induction of type I IFN via the transcription factor IRF3. At least in TREX1 deficiency, functional lymphocytes are necessary to propagate the disease phenotype (12).
Nucleic acids and autoimmunity
The innate immune system detects viral infections, induces antiviral effectors that neutralize the spread of infection and activates antigen-specific adaptive responses (53). Type I interferons play an important role in the coordination of this response. In many cases, the presence of virus is detected by receptors that recognize viral NA. TLRs are transmembrane proteins localized at the cell membrane or in endoplasmic compartments of specialized immune cells. TLR-3, -7/8 and -9 recognize viral double-stranded (ds) RNA, single-stranded (ss) RNA or DNA, respectively, that are delivered to endosomes during the infection process. Other receptors are more broadly expressed and almost all cell types can mount an IFN response to cytosolic NA. RIG-I, MDA5 and LGP-2 are helicase proteins that constitute a family of receptors sensing infection with RNA viruses, including influenza A virus and hepatitis C virus, amongst others (54). This cell-autonomous response involves signalling through the adaptor protein IPS-1. Cytosolic DNA triggers IFN induction via an IPS1-independent pathway and DAI has been implicated as a possible receptor (55,56). Other pathways for the recognition of cytosolic NA exist as well. For example, a multi-protein complex termed the inflammasome triggers processing of pro-IL-1b and pro-IL-18 (57). The mature forms of these cytokines are potent proinflammatory modulators. Recently, a cytoplasmic sensor coupling DNA recognition to the inflammasome has been identified (58–61). Further, viral RNA too has been suggested to trigger the inflammasome (62).
The existence of NA sensors raises an important question of self/non-self discrimination; that is, how do sequence-independent sensors avoid recognition of self-DNA/RNA? Separation of NA from putative receptors, differential modification of endogenous vis-à-vis exogenous NA and disposal of self-NA are all important in this regard (Table 4).
Table 4.
Mechanisms which might be involved in avoiding an immune reaction against self NA
However, such mechanisms are imperfect and it is becoming absolutely clear that the metabolism of endogenous NA is a central theme in the pathogenesis of autoimmunity (Table 5).
Table 5.
Examples of perturbation of NA metabolism in autoimmune phenotypes
| Molecule | Phenotype (reference) |
|---|---|
| DNase I | Systemic lupus erythematosus (28) |
| DNase II | Rheumatoid arthritis-like (65) |
| TREX1 (DNase III) | Systemic lupus erythematosus (10) |
| FEN1 (DNase IV) | Systemic lupus erythematosus-like (66) |
| Yaa (TLR7) | Systemic lupus erythematosus (67) |
| HMGB1/RAGE | Systemic lupus erythematosus (68) |
| LL37 | Psoriasis (69) |
| P202 (IfI202) | Systemic lupus erythematosus (61) |
| MDA5 (IFIH1) | Type I diabetes mellitus (70) |
Since a hallmark of SLE is the production of antibodies directed against RNA and DNA, the finding of defective NA metabolism in lupus, where NAs can act as both antigen and adjuvant, is unsurprising. Moreover, considering the importance of NA in inducing a type I IFN response, the observation of a disturbance of IFN-α homeostasis as central to the pathogenesis of SLE is also credible. Taken together, these recent studies also offer an elegant mechanistic explanation for the phenotypic overlap of AGS with SLE and congenital infection. That is, in the absence of TREX1, RNASEH2 or SAMHD1 activity, endogenous NAs accumulate and are sensed as ‘non-self’, leading to the induction of an IFN-α-mediated immune response.
SUMMARY
Broadly speaking, two clinical presentations of AGS can be delineated; an early-onset neonatal form highly reminiscent of congenital infection seen particularly with TREX1 mutations, and a later-onset presentation, sometimes occurring after several months of normal development and occasionally associated with remarkably preserved neurological function, most frequently due to RNASEH2B mutations (14). Interestingly, whichever presentation, little disease progression seems to occur beyond the initial encephalopathic period. These observations are important because they indicate that the treatment in the early stages of the disease should result in attenuation of the associated inflammation and consequent tissue damage. By defining the precise pathways linking NA accumulation to activation of the immune response we believe that it will be possible to develop treatments, for example antagonists of IRF3 or anti-IFN antibodies, to interrupt the AGS disease process at one or more points (Fig. 3). It is expected that these therapies will be relevant to the treatment of FCL and subtypes of lupus. Exciting precedents exist for such an approach in other immune-mediated inflammatory diseases (71).
Figure 3.
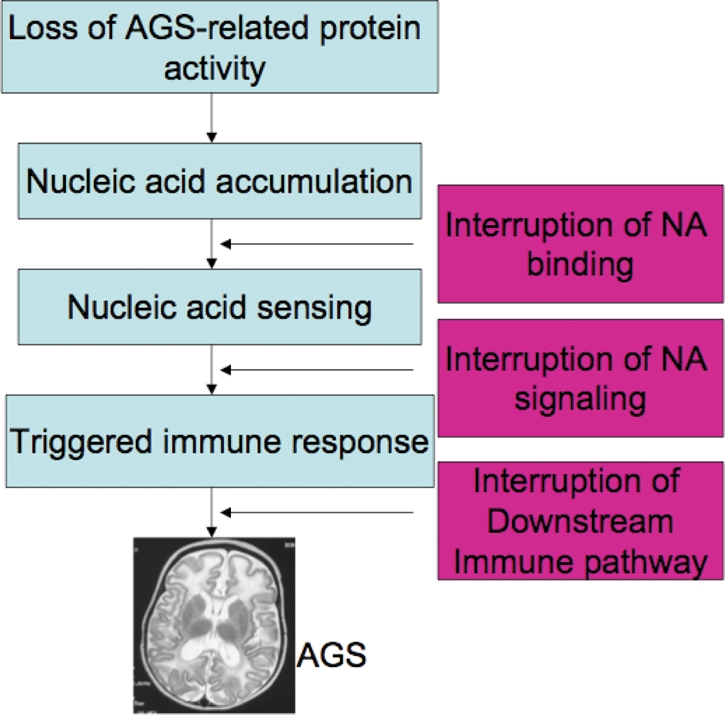
Stages in the pathogenesis of AGS which might be amenable to targeted interruption.
FUNDING
This work was supported by BDF Newlife, the Royal Society, the Wellcome Trust and the Manchester NIHR Biomedical Research Centre. J.R. is a recipient of FEBS and HFSP long term fellowships.
ACKNOWLEDGEMENTS
We sincerely thank the participating families for the use of genetic samples and clinical information, all collaborating clinicians and colleagues for helpful discussions—most particularly Gillian Rice, Hannah Gornall, David Bonthron, Caetano Reis e Sousa, Dan Stetson, Tomas Lindahl, Debbie Barnes and Fred Perrino.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Stetson D.B., Medzhitov R. Antiviral defense: interferons and beyond. J. Exp. Med. 2006;203:1837–1841. doi: 10.1084/jem.20061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crow M.K. Type I interferon in systemic lupus erythematosus. Curr. Top Microbiol. Immunol. 2007;316:359–386. doi: 10.1007/978-3-540-71329-6_17. [DOI] [PubMed] [Google Scholar]
- 3.Ronnblom L., Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dussaix E., Lebon P., Ponsot G., Huault G., Tardieu M. Intrathecal synthesis of different alpha-interferons in patients with various neurological diseases. Acta Neurol. Scand. 1985;71:504–509. doi: 10.1111/j.1600-0404.1985.tb03235.x. [DOI] [PubMed] [Google Scholar]
- 5.Aicardi J., Goutieres F. Systemic lupus erythematosus or Aicardi-Goutieres syndrome? Neuropediatrics. 2000;31:113. doi: 10.1055/s-2000-7533. [DOI] [PubMed] [Google Scholar]
- 6.Dale R.C., Tang S.P., Heckmatt J.Z., Tatnall F.M. Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics. 2000;31:155–158. doi: 10.1055/s-2000-7492. [DOI] [PubMed] [Google Scholar]
- 7.De Laet C., Goyens P., Christophe C., Ferster A., Mascart F., Dan B. Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutieres syndrome. Neuropediatrics. 2005;36:399–402. doi: 10.1055/s-2005-873058. [DOI] [PubMed] [Google Scholar]
- 8.Rasmussen M., Skullerud K., Bakke S.J., Lebon P., Jahnsen F.L. Cerebral thrombotic microangiopathy and antiphospholipid antibodies in Aicardi-Goutieres syndrome—report of two sisters. Neuropediatrics. 2005;36:40–44. doi: 10.1055/s-2004-830532. [DOI] [PubMed] [Google Scholar]
- 9.Rice G., Newman W.G., Dean J., Patrick T., Parmar R., Flintoff K., Robins P., Harvey S., Hollis T., O'Hara A., et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee-Kirsch M.A., Gong M., Chowdhury D., Senenko L., Engel K., Lee Y.A., de Silva U., Bailey S.L., Witte T., Vyse T.J., et al. Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 11.Yang Y.G., Lindahl T., Barnes D.E. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 12.Stetson D.B., Ko J.S., Heidmann T., Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crow Y.J., Livingston J.H. Aicardi-Goutieres syndrome: an important Mendelian mimic of congenital infection. Dev. Med. Child Neurol. 2008;50:410–416. doi: 10.1111/j.1469-8749.2008.02062.x. [DOI] [PubMed] [Google Scholar]
- 14.Rice G., Patrick T., Parmar R., Taylor C.F., Aeby A., Aicardi J., Artuch R., Montalto S.A., Bacino C.A., Barroso B., et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchis A., Cervero L., Bataller A., Tortajada J.L., Huguet J., Crow Y.J., Ali M., Higuet L.J., Martinez-Frias M.L. Genetic syndromes mimic congenital infections. J. Pediatr. 2005;146:701–705. doi: 10.1016/j.jpeds.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 16.Crow Y.J., Hayward B.E., Parmar R., Robins P., Leitch A., Ali M., Black D.N., van Bokhoven H., Brunner H.G., Hamel B.C., et al. Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 17.Crow Y.J., Leitch A., Hayward B.E., Garner A., Parmar R., Griffith E., Ali M., Semple C., Aicardi J., Babul-Hirji R., et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat. Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 18.Rice G.I., Bond J., Asipu A., Brunette R., Manfield I.W., Carr I.M., Fuller J.C., Jackson R.M., Lamb T., Briggs T.A., et al. Mutations in Aicardi-Goutières syndrome implicate SAMHD1 as a novel regulator of the innate immune response. Nat. Genet. 2009 doi: 10.1038/ng.373. epub ahead of print June 14, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazur D.J., Perrino F.W. Structure and expression of the TREX1 and TREX2 3′ –>5′ exonuclease genes. J. Biol. Chem. 2001;276:14718–14727. doi: 10.1074/jbc.M010051200. [DOI] [PubMed] [Google Scholar]
- 20.Hoss M., Robins P., Naven T.J., Pappin D.J., Sgouros J., Lindahl T. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J. 1999;18:3868–3875. doi: 10.1093/emboj/18.13.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morita M., Stamp G., Robins P., Dulic A., Rosewell I., Hrivnak G., Daly G., Lindahl T., Barnes D.E. Gene-targeted mice lacking the Trex1 (DNase III) 3′–>5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brucet M., Querol-Audi J., Serra M., Ramirez-Espain X., Bertlik K., Ruiz L., Lloberas J., Macias M.J., Fita I., Celada A. Structure of the dimeric exonuclease TREX1 in complex with DNA displays a proline-rich binding site for WW Domains. J. Biol. Chem. 2007;282:14547–14557. doi: 10.1074/jbc.M700236200. [DOI] [PubMed] [Google Scholar]
- 23.de Silva U., Choudhury S., Bailey S.L., Harvey S., Perrino F.W., Hollis T. The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a polyproline II helix for protein partnering. J. Biol. Chem. 2007;282:10537–10543. doi: 10.1074/jbc.M700039200. [DOI] [PubMed] [Google Scholar]
- 24.Chowdhury D., Beresford P.J., Zhu P., Zhang D., Sung J.S., Demple B., Perrino F.W., Lieberman J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell. 2006;23:133–142. doi: 10.1016/j.molcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Lee-Kirsch M.A., Gong M., Schulz H., Ruschendorf F., Stein A., Pfeiffer C., Ballarini A., Gahr M., Hubner N., Linne M. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am. J. Hum. Genet. 2006;79:731–737. doi: 10.1086/507848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee-Kirsch M.A., Chowdhury D., Harvey S., Gong M., Senenko L., Engel K., Pfeiffer C., Hollis T., Gahr M., Perrino F.W., et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. 2007;85:531–537. doi: 10.1007/s00109-007-0199-9. [DOI] [PubMed] [Google Scholar]
- 27.Richards A., van den Maagdenberg A.M., Jen J.C., Kavanagh D., Bertram P., Spitzer D., Liszewski M.K., Barilla-Labarca M.L., Terwindt G.M., Kasai Y., et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- 28.Yasutomo K., Horiuchi T., Kagami S., Tsukamoto H., Hashimura C., Urushihara M., Kuroda Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001;28:313–314. doi: 10.1038/91070. [DOI] [PubMed] [Google Scholar]
- 29.Truedsson L., Bengtsson A.A., Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40:560–566. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- 30.Lehtinen D.A., Harvey S., Mulcahy M.J., Hollis T., Perrino F.W. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J. Biol. Chem. 2008;283:31649–31656. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerritelli S.M., Crouch R.J. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frank P., Braunshofer-Reiter C., Poltl A., Holzmann K. Cloning, subcellular localization and functional expression of human RNase HII. Biol. Chem. 1998;379:1407–1412. doi: 10.1515/bchm.1998.379.12.1407. [DOI] [PubMed] [Google Scholar]
- 33.Frank P., Braunshofer-Reiter C., Wintersberger U., Grimm R., Busen W. Cloning of the cDNA encoding the large subunit of human RNase HI, a homologue of the prokaryotic RNase HII. Proc. Natl Acad. Sci. USA. 1998;95:12872–12877. doi: 10.1073/pnas.95.22.12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eder P.S., Walder J.A. Ribonuclease H from K562 human erythroleukemia cells. Purification, characterization, and substrate specificity. J. Biol. Chem. 1991;266:6472–6479. [PubMed] [Google Scholar]
- 35.Jeong H.S., Backlund P.S., Chen H.C., Karavanov A.A., Crouch R.J. RNase H2 of Saccharomyces cerevisiae is a complex of three proteins. Nucleic Acids Res. 2004;32:407–414. doi: 10.1093/nar/gkh209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chon H., Vassilev A., DePamphilis M.L., Zhao Y., Zhang J., Burgers P.M., Crouch R.J., Cerritelli S.M. Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucleic Acids Res. 2009;37:96–110. doi: 10.1093/nar/gkn913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perrino F.W., Harvey S., Shaban N.M., Hollis T. RNaseH2 mutants that cause Aicardi-Goutieres syndrome are active nucleases. J. Mol. Med. 2009;87:25–30. doi: 10.1007/s00109-008-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eder P.S., Walder R.Y., Walder J.A. Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie. 1993;75:123–126. doi: 10.1016/0300-9084(93)90033-o. [DOI] [PubMed] [Google Scholar]
- 39.Drolet M., Phoenix P., Menzel R., Masse E., Liu L.F., Crouch R.J. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl Acad. Sci. USA. 1995;92:3526–3530. doi: 10.1073/pnas.92.8.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murante R.S., Henricksen L.A., Bambara R.A. Junction ribonuclease: an activity in Okazaki fragment processing. Proc. Natl Acad. Sci. USA. 1998;95:2244–2249. doi: 10.1073/pnas.95.5.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li N., Zhang W., Cao X. Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol. Lett. 2000;74:221–224. doi: 10.1016/s0165-2478(00)00276-5. [DOI] [PubMed] [Google Scholar]
- 42.Lafuse W.P., Brown D., Castle L., Zwilling B.S. Cloning and characterization of a novel cDNA that is IFN-gamma-induced in mouse peritoneal macrophages and encodes a putative GTP-binding protein. J. Leukoc. Biol. 1995;57:477–483. doi: 10.1002/jlb.57.3.477. [DOI] [PubMed] [Google Scholar]
- 43.Hartman Z.C., Kiang A., Everett R.S., Serra D., Yang X.Y., Clay T.M., Amalfitano A. Adenovirus infection triggers a rapid, MyD88-regulated transcriptome response critical to acute-phase and adaptive immune responses in vivo. J. Virol. 2007;81:1796–1812. doi: 10.1128/JVI.01936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prehaud C., Megret F., Lafage M., Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao D., Peng D., Li L., Zhang Q., Zhang C. Inhibition of G1P3 expression found in the differential display study on respiratory syncytial virus infection. Virol. J. 2008;5:114. doi: 10.1186/1743-422X-5-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao W., Bao Z., Cheng C., Mok Y.K., Wong W.S. Dendritic cell-derived interferon-gamma-induced protein mediates tumor necrosis factor-alpha stimulation of human lung fibroblasts. Proteomics. 2008;8:2640–2650. doi: 10.1002/pmic.200700954. [DOI] [PubMed] [Google Scholar]
- 47.Qiao F., Bowie J.U. The many faces of SAM. Sci. STKE. 2005;2005:re7. doi: 10.1126/stke.2862005re7. [DOI] [PubMed] [Google Scholar]
- 48.Oberstrass F.C., Lee A., Stefl R., Janis M., Chanfreau G., Allain F.H. Shape-specific recognition in the structure of the Vts1p SAM domain with RNA. Nat. Struct. Mol. Biol. 2006;13:160–167. doi: 10.1038/nsmb1038. [DOI] [PubMed] [Google Scholar]
- 49.Aravind L., Koonin E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998;23:469–472. doi: 10.1016/s0968-0004(98)01293-6. [DOI] [PubMed] [Google Scholar]
- 50.Zimmerman M.D., Proudfoot M., Yakunin A., Minor W. Structural insight into the mechanism of substrate specificity and catalytic activity of an HD-domain phosphohydrolase: the 5′-deoxyribonucleotidase YfbR from Escherichia coli. J. Mol. Biol. 2008;378:215–226. doi: 10.1016/j.jmb.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oussenko I.A., Sanchez R., Bechhofer D.H. Bacillus subtilis YhaM, a member of a new family of 3′-to-5′ exonucleases in gram-positive bacteria. J. Bacteriol. 2002;184:6250–6259. doi: 10.1128/JB.184.22.6250-6259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alarcon-Riquelme M.E. Nucleic acid by-products and chronic inflammation. Nat. Genet. 2006;38:866–867. doi: 10.1038/ng0806-866. [DOI] [PubMed] [Google Scholar]
- 53.Pichlmair A., Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 54.Yoneyama M., Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 55.Stetson D.B., Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 56.Takaoka A., Wang Z., Choi M.K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 57.Franchi L., Eigenbrod T., Munoz-Planillo R., Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burckstummer T., Baumann C., Bluml S., Dixit E., Durnberger G., Jahn H., Planyavsky M., Bilban M., Colinge J., Bennett K.L., et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 59.Fernandes-Alnemri T., Yu J.W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., Latz E., Fitzgerald K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roberts T.L., Idris A., Dunn J.A., Kelly G.M., Burnton C.M., Hodgson S., Hardy L.L., Garceau V., Sweet M.J., Ross I.L., et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 62.Allen I.C., Scull M.A., Moore C.B., Holl E.K., McElvania-TeKippe E., Taxman D.J., Guthrie E.H., Pickles R.J., Ting J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barton G.M., Kagan J.C., Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 64.Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 65.Kawane K., Ohtani M., Miwa K., Kizawa T., Kanbara Y., Yoshioka Y., Yoshikawa H., Nagata S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 66.Zheng L., Dai H., Zhou M., Li M., Singh P., Qiu J., Tsark W., Huang Q., Kernstine K., Zhang X., et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat. Med. 2007;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- 67.Pisitkun P., Deane J.A., Difilippantonio M.J., Tarasenko T., Satterthwaite A.B., Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 68.Tian J., Avalos A.M., Mao S.Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 69.Lande R., Gregorio J., Facchinetti V., Chatterjee B., Wang Y.H., Homey B., Cao W., Su B., Nestle F.O., Zal T., et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 70.Nejentsev S., Walker N., Riches D., Egholm M., Todd J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldbach-Mansky R., Dailey N.J., Canna S.W., Gelabert A., Jones J., Rubin B.I., Kim H.J., Brewer C., Zalewski C., Wiggs E., et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]