

Abstract
Here we propose a simple statistical algorithm for rapidly scoring loci associated with disease or traits due to recessive mutations or deletions using genome-wide single nucleotide polymorphism genotyping case–control data in unrelated individuals. This algorithm identifies loci by defining homozygous segments of the genome present at significantly different frequencies between cases and controls. We found that false positive loci could be effectively removed from the output of this procedure by applying different physical size thresholds for the homozygous segments. This procedure is then conducted iteratively using random sub-datasets until the number of selected loci converges. We demonstrate this method in a publicly available data set for Alzheimer′s disease and identify 26 candidate risk loci in the 22 autosomes. In this data set, these loci can explain 75% of the genetic risk variability of the disease.
Keywords: disease network, homozygous segments, risk loci, statistical algorithm, whole-genome screening
Introduction
Advances in whole-genome single nucleotide polymorphism (SNP) assay technology have provided a powerful array of tools for simultaneously scoring common genetic variation. However, it is often difficult to identify loci associated with disease because of the large number of tests carried out and the associated conservative multiplicity adjustment, such as Bonferroni method. We are interested in identifying such loci associated with a disease likely due to recessive mutation or gene deletions.
High density SNP analysis readily reveals the presence of large homozygous segments in unrelated subjects (Hinds et al, 2005; Simon-Sanches et al, 2007; Wang et al, 2007). The probability of a randomly selected SNP locus being homozygous (‘AA' or ‘BB') based on data from HapMap is about 0.65 (Hinds et al, 2005; Rabbee and Speed, 2006) and this may lend itself to autozygosity mapping in ostensibly outbred populations; however, traditional autozygosity mapping methods (Lander and Botstein, 1987; Mueller and Bishop, 1993; Gschwend et al, 1996) based on consanguineous relationships are not appropriate for unrelated individuals. To identify loci with possible recessive effects of relatively high penetrance in outbred populations, large sample sizes are needed for genotyping. Some recent studies on homozygosity analysis of SNP assays have been attempted using different approaches (Woods et al, 2004; Lencz et al, 2007; Miyazawa et al, 2007). However, they either have some familial relationship requirements (Woods et al, 2004; Miyazawa et al, 2007) or a high false positive rate (Lencz et al, 2007).
In the context of SNP genotyping, it is often not easy to distinguish heterozygous genomic deletion from homozygosity; thus a segment with all loci genotyped being ‘AA' or ‘BB' in a pedigree genotype file could be either a region of genuine homozygosity or effective hemizygosity caused by genomic deletion. We call such a region ‘apparently homozygous region' (AH). By carrying out an appropriate association analysis on AHs, one can detect not only the possible recessively mutated loci from some common ancestor but also deletions (Hunter, 2005; Klein et al, 2005; Van Eyken et al, 2007).
In this paper, we propose a simple statistical algorithm for genome-wide AH analysis (GAHA) of case–control data in unrelated subjects. It can robustly identify loci that are associated with disease by efficiently removing false positive loci. We demonstrate this method in a publicly available data set for Alzheimer's disease (AD) (Coon et al, 2007), consisting 502 627 SNP loci genotyped in unrelated 859 cases and 552 neurologically normal controls. A total of 26 loci from the 22 autosomes are identified and they explain 75% of the genetic risk variability of the disease.
Results and discussion
AH size threshold
In the context of the current data, it is not appropriate to use the number of loci as a measure of AH size as previously reported (Lencz et al, 2007) because of its dependence on SNP density. Here we use the number of nucleotide basepairs between the first and last loci of an AH as a measure of AH size.
Let C be a size threshold of AHs. We are interested in identifying loci proportions of which are significantly different between controls and cases in AHs with sizes ⩾C. As seen in Figure 1, for example, there are n1 cases and with a given C we count the proportion of the locus SNP-1 on AHs p1=(number of AHs containing SNP-1)/n1. Similarly, for n0 controls, we find the proportion p0 of the same locus. Using p1 and p0, we compute z-statistic for proportional test as described in Materials and methods. The locus is selected for further screening if ∣z∣⩾z1−α/2, where α is the level of significance. The test statistic z follows a standard normal distribution asymptotically as n0 and n1 increase with each greater than 30.
Figure 1.

Scheme for computing the proportion of a locus on AHs. For a given chromosome of a subject, the symbols (•, ○) represent SNP loci. The shaded segments denote AHs with size greater than or equal to a pre-selected threshold C. The proportion of a locus on AHs is computed as p= (the number of AHs containing this locus)/(the total number of individuals), for example p1=4/6 for SNP-1.
We investigated the power for selecting loci based on α, AH percentage difference between cases and controls, and AH size threshold C through simulation. The relationships between z value and AH percentage difference with various C are shown in Supplementary Figure 1. At a significance level α=0.001, the powers to detect candidate loci were computed accordingly. We define that a candidate locus is detectable if the power>0.8. Our results showed that at a significance level α=0.001, we could detect a locus on AHs⩾C with a difference of 30% between cases and controls using C=10 kb, or only of 7% using C=1 Mb.
On the basis of above significance level α and a moderate C value, typically thousands of loci could be selected with a large false positive rate from data of unrelated subjects. A key step is to efficiently remove these falsely associated loci from the candidate list. If we knew the minimum size of risk loci, then we would set it as C and consider only AH⩾C, leading to a lower false positive rate. However, such a C value is unknown. One approach is to use multiple values of C as discussed below. In convention, define C=1 for considering AHs with size ⩾1.
Algorithm for screening risk loci
We propose to use multiple C values for screening risk loci. Suppose we choose C1 and C2, with C1<C2, for selecting candidate loci with ∣z∣⩾z1−α/2. It should be noted that the distance between C1 and C2 must be larger than the minimum distance between loci of the platform and may be chosen by referring to some public genotyping parameters (for example, the average distance between loci is ∼9 kb in Affymetrix 500K GeneChip, and a median distance is ∼3 kb in Illumina HumanHap550 BeadChip according to Gunderson et al, 2005; Steemers and Gunderson, 2007). Let S1 be the set containing the loci selected with C1 and S2 with C2, respectively. As the true AHs with size ⩾C2>C1 will remain using either C1 or C2, the loci, not in S1⋂S2, should be more likely false positives and thus be removed. For example, in the AD data using a significance level α=0.001, among the 25 086 loci on chromosome 1, there were 18 loci selected using C=10 kb and 12 loci using C=30 kb, respectively, with only three being common loci in both sets. In general, we set C={Ci, i=1, 2,…, L} with C1<C2<…<CL to cover a wide range of AHs and let S be the set containing all loci common in adjacent sets S={S1⋂S2, S2⋂S3,…, SL−1⋂SL}. This loci-selecting procedure is called ‘procedure of adjacent-C-selection' (PACS).
The PACS can efficiently remove false positive loci, however, for a real data set in unrelated individuals with large genetic variation, the selected loci usually still contain some false positives, many of which could be removed through further ‘purification'. To achieve this, ideally we should repeat the above steps using an independent data set from the same population to get another candidate set. Then identify the common loci from both sets. This new candidate set contains fewer false positive loci, which could be further removed by repeating above steps iteratively until the number of candidate loci converges. Although it is generally not realistic to do so, we could do the ‘purification' using random subsets from the full data set as described below.
Let nk*=[f × nk]>30 be the size of a random subset from the full data set of size nk, where k=1 for cases and k=0 for controls, and f be a constant with 0<f<fmax, fmax= (mink (nk) − 1)/mink (nk. The randomly and independently chosen n1* cases and n0* controls form a random case–control sub-data set for further removing the false positive loci from the candidate set using the same set of C values as applied to the full data set.
Let S be the set containing the selected loci from the full data set and S* be that from the first random sub-data set. Let S1*=S*⋂S containing the common loci in both sets and N1=∣S1*∣ be the number of loci in S1*. Next we generate a new S* from the second random sub-data set and let S2*=S1*⋂S* with N2=∣S2*∣. Repeating these steps to update the candidate loci set until the number of Nt, t=1,2,………, converges to a constant integer Nc with Nc =0 if the null hypothesis of no difference between p1 and p0 is true and Nc>0 if the alternative hypothesis p1≠p0 is true. For a given f, there are
possible ways for selecting case–control subset, which should be much larger than the number required for reaching convergence at an appropriate level of significance. The above GAHA algorithm is summarized in Box 1.
The false positive rate of a locus in the final set should be ⩽α. The false negative rates of loci selection in a random subset were estimated under the same settings for the full data set (Supplementary Table 2).
Application to AD data set
Set C={1, 10 kb, 30 kb, 50 kb, 100 kb, 140 kb, 250 kb, 500 kb, 1 Mb} and α=0.001. We identified 607 loci from 4054 loci whose ∣z∣⩾z1−α/2 (Figure 2A) from the 22 autosomes in the AD data set (Coon et al, 2007).
Figure 2.

The plot of z versus nucleotide basepair of chromosome 19 in the AD data set: (A) before and (B) after the procedure of adjacent-C-selection, (C) the most significant region—the peak locus is rs4420638, (D) the most significant region with two loci on APOE (↓).
The most significant AH region was on 19q13.2 (see Figure 2B) with positive z values suggesting significantly more AHs in controls than in cases. This region, covering the whole apolipoprotein E (APOE) gene, contains four loci including rs4420638 (Figure 2C), which is in linkage disequilibrium with APOE (Coon et al, 2007). However, there were no genotypes within APOE in the AD data. We added available genotyping information (Coon et al, 2007) of two loci on APOE, rs429358 and rs7412, to the AD data. The two APOE loci define the ɛ2/ɛ3/ɛ4 genotypes. Figure 2D shows the APOE loci indeed on the AH region where the majority controls have the ɛ3 genotype, supporting the observation that APOE ɛ3 is protective against the disease when compared with ɛ4 (Farrer et al, 1997).
To further reduce the false positive rate within this list, we chose f=0.9 for generating random subsets, each with 773 cases and 497 controls. The use of f=0.9 may not be the statistically optimal choice; it is, however, the best we tried. The convergence of the loci number is shown in Figure 3. There were 26 loci in the final list (Figure 3B) (Table I). Based on a logistic regression model fit, the percent variation of the genetic risk explained by these 26 loci was 75.3%. Model selection removed 10 confounder loci and retained 16 loci (each with P-value<0.05), including rs4420638, in the reduced model with 74.8% of the genetic risk variation explained (Supplementary Table 3, 4).
Figure 3.

Convergence of the loci number. (A) At a level of significance α=0.001, a total of 607 loci (□) were selected from the 4054 loci for which ∣z∣⩾ z1−α/2 (Δ) by applying the procedure of adjacent-C-selection in the AD data set. Random case–control subsets were generated using f=0.9 and used in screening iteration (○). (B) The enlarged plot showing the convergence of selected loci to the number 26.
Table 1.
List of candidate loci associated with AD from the 22 autosome of the AD SNP genotype data (Coon et al, 2007)
| CHR | SNP ID | Locationa | Function | Gene | Gene ID | Effect |
|---|---|---|---|---|---|---|
| aIn nucleotide basepair. | ||||||
| bLoci remained in the model on logistic regression selection with a P-value<0.05. | ||||||
| cLoci in homozygous regions containing candidate loci of recessive genetic lesion causing AD (Clarimón et al, 2008). | ||||||
| dGenes are on known functional pathways and networks as revealed by the use of Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com). | ||||||
| eA SNP in Affymetrix 500K GeneChip, but without NCBI ID. | ||||||
| 1 | rs17325887b,c | 69998761 | Intron | LRRC7d | 57554 | Risk |
| 1 | rs7520521c | 70020703 | Intron | LRRC7d | 57554 | Risk |
| 1 | rs1913269b,c | 70052194 | Intron | LRRC7d | 57554 | Risk |
| 1 | rs10754339b | 117491795 | mRNA–UTR | VTCN1d | 79679 | Protect |
| 1 | rs16842422b | 196366613 | –66918 | LOC647195 | 647195 | Protect |
| 2 | rs7582851 | 192032391 | –392328 | LOC647167 | 647167 | Protect |
| 3 | rs6784615b | 52481466 | Intron | NISCHd | 11188 | Protect |
| 4 | rs9994615 | 40786592 | Intron | APBB2d | 323 | Risk |
| 4 | rs10015784b | 40793978 | Intron | APBB2d | 323 | Risk |
| 5 | rs1602843b,c | 86324342 | 0 | COL24A1d | 255631 | Risk |
| 5 | rs2913719b | 163947773 | 2403 | LOC440700 | 440700 | Protect |
| 6 | rs13213247b | 81572755 | –91974 | LOC729817 | 729817 | Risk |
| 6 | rs16892285 | 81592721 | –72008 | LOC729817 | 729817 | Risk |
| 6 | rs13193950 | 81593433 | –71296 | LOC729817 | 729817 | Risk |
| 6 | rs156232b | 104979509 | 481535 | LOC642337 | 642337 | Risk |
| 10 | rs10827687b | 36999313 | –39887 | GRIK3d | 2899 | Risk |
| 10 | rs10824310b | 53698470 | Intron | PRKG1d | 5592 | Risk |
| 10 | rs10740548 | 54877234 | –2797 | C1orf175d | 374977 | Risk |
| 11 | rs1038891b,c | 40895642 | 0 | RIMS3d | 9783 | Risk |
| 12 | rs1354470b | 59088188 | –32939 | LOC645757 | 645757 | Risk |
| 12 | rs7967572 | 73396068 | 51514 | KRT8P21 | 126811 | Risk |
| 18 | rs1785928b | 31979929 | Coding non-synonymous | ELP2d | 55250 | Risk |
| 19 | rs11879589 | 50065116 | Intron | PVRL2d | 5819 | Protect |
| 19 | rs4420638b | 50114786 | Locus region | APOC1d | 341 | Protect |
| 19 | e | 50150075 | ||||
| 19 | rs204907 | 50153836 | Intron | CLPTM1d | 1209 | Protect |
The APOE ɛ4 was carried by ∼40% of the later-onset AD cases (Poirier et al, 1993; Laws et al, 2003). Recall that rs4420638 is in linkage disequilibrium with APOE, we found that the percent genetic risk variation explained by this locus alone was 34.2%. However, when rs4420638 was excluded from the reduced model, the percentage genetic risk variation explained by the remaining 15 loci was decreased only by 2.9% (from 74.8% to 71.9%). This suggests these loci explain the genetic risk variation of AD as a group. Several of the 26 loci identified in this screening were also found in homozygous regions identified in an early onset AD study of a consanguineous family (Clarimón et al, 2008), suggesting that one of these regions harbors a recessive genetic lesion causing AD.
The 26 loci are on 20 genes of which 13 are in known functional pathways or networks as revealed from an Ingenuity Pathway Analysis (Ingenuity Systems, www.ingenuity.com) (Supplementary Pathway/Network analysis). On the basis of the correlations among the 20 genes and AD status of subjects, we construct an AD genetic network (Supplementary Figure 2).
Summary
We propose a statistical method for GAHA of SNP case–control data in unrelated subjects to identify risk loci that are most likely associated with a disease or abnormality due to recessive mutation or deletion. The main novelty of this method over other approaches is to minimize the false positive rate of the risk candidates. We remove the false positive loci by selecting the common loci with different size thresholds of homozygous segments and repeating these steps iteratively using random sub-data sets until the number of selected loci converges. Furthermore, this method allows selects risk loci from a wider AH size range. By demonstrating of the method using a publicly available AD SNP assay data set, we identified 26 candidate risk loci from the 22 autosomes.
Materials and methods
Notes
Suppose there are n SNP loci genotyped on a given chromosome (an autosome). We view the sequences of SNP loci on a chromosome as linked regions either being heterozygous or AHs. Let H be a set such that H={h1, h2,…, hm} where hi denotes the number of AHs containing i consecutive SNP loci genotyped, and m is the maximum number of consecutive SNP loci. The probability of a randomly selected SNP locus on AHs with SNP number being equal to or larger than a predetermined integer k is 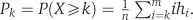 .
.
Data
A SNP genotype data set of late-onset AD(500K Affymetrix) was downloaded from a publicly available website, http://www.neuron.org, to demonstrate our method. This data set consists of 502 627 SNP loci genotyped in unrelated 859 cases and 552 neurologically normal controls.
Proportion test
We are interested in identifying loci at which the proportion of a SNP locus, on AHs with size equal to or larger than a given threshold C, is significantly different between controls and cases. Our null hypothesis is that the SNP at a given locus has the same probability of being on AHs with size ⩾C in the control and case groups. The test statistic in a standard proportion test is
 |
and follows a Gaussian distribution under the null hypothesis, where the p0 is the proportion of the locus on AHs for the n0 control subjects and the p1 is that for the n1 cases. We define z=0 when both p0=0 and p1=0. For a given level of significance α, a locus is selected if ∣z∣⩾z1−α/2. This test requires large sample size (n0, n1>30).
Logistic regression
In logistic regression using the selected loci as predictor variables, let xij=1 if the ith locus of the jth subject is on an AH with size being equal to or larger than C=10 kb and xij=0 otherwise. Logistic regression is carried out using SAS 9.0.
Declaration
The views expressed in this article do not represent those of the US Food and Drug Administration.
Outline of the GAHA algorithm.
(1) For case–control SNP data with n1 cases and n0 controls, choose a level of significance α, set AH thresholds C={Ci, i=1, 2,…, L} with C1<C2<…<CL, and then find AHs with size Ci, i=1, 2,…, L, for each subject
(2) Compute z at each locus and select it if ∣z∣⩾z1-α/2. Perform the PACS and let Sold be the set of selected loci and Nold=∣Sold∣. Chose 0 < f < mink{nk}−1/mink{nk}, and ℓ=0
(3) Randomly select a case–control sub-dataset from (1) with n1* = [f × n1] >30 cases and n0* = [f × n0] >30 controls. Find AHs for each subject at given C, then compute z at each locus and select it if ∣z∣⩾ z1-α/2
(4) Carry out the PACS and let S* be the set containing all the loci selected from the sub-dataset. Find Snew=Sold⋂S* Nnew=∣Snew∣
(5) 
Supplementary Material
Supplementary figures S1–2, Supplementary tables S1–4, Pathway/Network Analysis
Acknowledgments
This study was supported by the Intramural Program of the National Institute on Aging, National Institutes of Health and Department of Health and Human Services, project number AG000950-07. This study used high-performance computational capabilities of the Biowulf Systems at the National Institutes of Health, Bethesda, MD (http://helix.nih.gov).
Footnotes
The authors declare that they have no conflict of interest.
References
- Clarimón J, Djaldetti R, Lleó A, Guerreiro RJ, Molinuevo JL, Paisán-Ruiz C, Gómez-Isla T, Blesa R, Singleton A, Hardy J (2008) Whole genome analysis in a consanguineous family with early onset Alzheimer's disease. Neurobiol Aging, doi:10.1016/j.neurobiolaging.2008.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Hu Lince D, Zismann VL, Beach TG, Leung D, Bryden L, Halperin RF, Marlowe L, Kaleem M, Walker DG, Ravid R, Heward CB, Rogers J, Papassotiropoulos A, Reiman EM, Hardy J et al. (2007) A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer′s disease. J Clin Psychiatry 68: 613–618 [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. J Am Med Assoc 278: 1349–1356 [PubMed] [Google Scholar]
- Gschwend M, Levran O, Kruglyak L, Ranade K, Verlander PC, Shen S, Faure S, Weissenbach J, Altay C, Lander ES, Auerbach AD, Botstein D (1996) A locus for Fanconi anemia on 16q determined by homozygosity mapping. Am J Hum Genet 59: 377–384 [PMC free article] [PubMed] [Google Scholar]
- Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS (2005) A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet 37: 549–554 [DOI] [PubMed] [Google Scholar]
- Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR (2005) Whole-genome patterns of common DNA variation in three human populations. Science 307: 1072–1079 [DOI] [PubMed] [Google Scholar]
- Hunter DJ (2005) Gene–environment interactions in human diseases. Nat Rev Genet 6: 287–298 [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308: 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Botstein D (1987) Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236: 1567–1570 [DOI] [PubMed] [Google Scholar]
- Laws SM, Hone E, Gand S, Martins RN (2003) Expanding the association between the APOE gene and the risk of Alzheimer′s disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J Neurochem 84: 1215–1236 [DOI] [PubMed] [Google Scholar]
- Lencz T, Lamberta C, DeRosse P, Burdick K, Morgan TV, Kane JM, Kucherlapati R, Malhotra AK (2007) Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci USA 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa H, Kato H, Awata T, Kohda M, Iwasa H, Koyama N, Tanaka T, Huqun, Kyo S, Okazaki Y, Hagiwara K (2007) Homozygosity haplotype allows a genome-wide search for the autosomal segments shared among patients. Am J Hum Genet 80: 1090–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller RF, Bishop DT (1993) Autozygosity mapping, complex consanguinity, and autosomal recessive disorders. J Med Genet 30: 798–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S (1993) Apolipoprotein E polymorphism and Alzheimer′s disease. Lancet 342: 697–699 [DOI] [PubMed] [Google Scholar]
- Rabbee N, Speed TP (2006) A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics 22: 7–12 [DOI] [PubMed] [Google Scholar]
- Simon-Sanches J, Scholz S, Fung HC, Matarin M, Hernandez D, Gibbs JR, Britton A, Wavrant de Brieze F, Peckham E, Gwinn-Hardy K, Crawley A, Keen JC, Nash J, Borgaonkar D, Hardy J, Singleton A (2007) Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet 16: 1–14 [DOI] [PubMed] [Google Scholar]
- Steemers FJ, Gunderson KL (2007) Whole genome genotyping technologies on the BeadArray platform. Biotechnol J 2: 41–49 [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant S, Hakonarson H, Bucan M (2007) PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17: 1665–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG, Valente EM, Bond J, Roberts E (2004) A new method for autozygosity mapping using single nucleotide polymorphisms (SNPs) and EXCLUDEAR. J Med Genet 41: e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyken E, Van Camp G, Van Laer L (2007) The complexity of age-related hearing impairment: contributing environmental and genetic factors. Audiol Neurootol 12: 345–358 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures S1–2, Supplementary tables S1–4, Pathway/Network Analysis