

Abstract
Configurational entropy is thought to influence biomolecular processes, but there are still many open questions about this quantity, including its magnitude, its relationship to molecular structure, and the importance of correlation. The mutual information expansion (MIE) provides a novel and systematic approach to computing configurational entropy changes due to correlated motions from molecular simulations. Here, we present the first application of the MIE method to protein-ligand binding, using multiple molecular dynamics simulations (MMDSs) to study association of the UEV domain of the protein Tsg101 and an HIV-derived nonapeptide. The current investigation utilizes the second-order MIE approximation, which treats correlations between all pairs of degrees of freedom. The computed change in configurational entropy is large and is found to have a major contribution from changes in pairwise correlation. The results also reveal intricate structure-entropy relationships. Thus, the present analysis suggests that, in order for a model of binding to be accurate, it must include a careful accounting of configurational entropy changes.
Keywords: thermodynamics, correlation, mutual information expansion (MIE), multiple molecular dynamics simulation (MMDS), translational/rotational entropy
1. Introduction
When a receptor and ligand bind each other, changes in their fluctuations bring about a change in configurational entropy [1]. This entropy contribution is thought to play a major role in binding, with multifaceted implications. For example, configurational entropy may determine whether protein-protein association is specific or promiscuous [2], unusually small entropy losses appear to account for the ultra-high affinity achieved by a novel host-guest complex [3], and it has been suggested that high affinities might be achieved by ligands that induce increased receptor entropy [4]. As a consequence, there is growing interest in measuring [5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18], calculating [2, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31], and ultimately controlling changes in configurational entropy on binding. These efforts promise to advance our ability to design molecules with desired functions, such as drugs, receptors, and biopharmaceuticals.
Several experimental methods provide information about entropy changes on binding. Isothermal titration calorimetry gives the change in total entropy, though not the configurational part. Neutron scattering can yield at least the vibrational part [32, 1] of the configurational entropy through the vibrational density of states, and has opened an intriguing view of changes in vibrational entropy when a protein binds a small molecule [33, 34]. Finally, generalized order parameters from NMR spectroscopy [35, 36] can be used to estimate changes in configurational entropy [5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18], and the ability to relate entropy to structure in this approach makes it particularly informative. For example, a pioneering application attributed cooperative calcium binding by Calbindin to a reduction in fluctuations at the second binding site due to binding at the first [5]. Still, entropies obtained from order parameters come with several caveats.
One issue is that order parameters may not be available for all bond vectors, so a comprehensive picture of the configurational entropy may be unavailable [11]. Another is that the equations relating order parameters to entropy make somewhat arbitrary assumptions regarding the probability distribution of the bond vectors [6, 11, 13, 16], such as the “diffusion-in-a-cone” model. Finally, although correlations among bond-vectors could affect the entropy, information on these correlations is not typically available [11, 13, 16]. A quasiharmonic analysis [37] of ubiquitin indicated only modest correlation effects on the entropy [38], but the quasiharmonic approximation can be unreliable for the multimodal conformational distributions characteristic of proteins [39, 40]. Another early study of this issue reported minimal correlations among bond-vectors in a molecular dynamics (MD) simulation of a calmodulin-peptide complex [13], but omitted pairs of bond-vectors expected to correlate strongly, such as the amide N-H bonds in two neighboring residues. It is also worth noting that the Pearson correlation coefficient, which was used to measure correlation, can detect only linear correlations [41], while nonlinear correlations also can lower entropy.
Computer simulations also can provide information on configurational entropy. The quasiharmonic approximation [37] extracts configurational entropy from a molecular dynamics simulation. However it was originally introduced to account for the anharmonicity of a single energy well and becomes unreliable for molecules that sample multiple energy wells [40]. The Nearest-Neighbor approach [42, 43, 44] also estimates entropy from simulation data, and it can provide numerically reliable results even for rather high-dimensional data, but not for a system as complex as a protein. Histogram approaches [45, 46, 47, 48, 49], methods based upon thermodynamic integration [25] and the hypothetical scanning method [50, 51] can provide changes in configurational entropy when combined with an implicit solvent model, although they may not be suitable for analyzing the entropic contributions of specific motions and correlations. Finally, the Mining Minima free energy method has provided insights into entropy changes for host-guest systems [22], but it requires further adapation or approximation to be applied to a protein, because of the potential for a combinatorial explosion of local energy wells for so large a molecule.
The mutual information expansion (MIE) offers another approach to extracting the configurational entropy from molecular simulations [29]. It accounts for the multimodal nature of molecular distributions and, like NMR order parameter measurements, connects individual entropy contributions to individual conformational variables. Its mutual information terms furthermore account for both linear and nonlinear correlations. For example, the second-order MIE includes pairwise mutual information terms,
| (1) |
where N is the number of conformational variables (e.g., torsion angles), Si is the entropy associated with variable i, and Iij is the mutual information between coordinates i and j. The mutual information, in turn, is defined as [52]
| (2) |
where Sij is the joint entropy associated with variables i and j. This second-order MIE has yielded good accuracy for small molecules and a bimolecular complex, except in the case of a highly correlated ring system in which more complicated correlations are to be expected [29, 44]. Here, we describe the first application of the MIE method to a protein, the human tumor susceptibility gene 101 protein (Tsg101).
Tsg101 plays an important role in the cellular vacuolar sorting pathway [53, 54], which involves binding of its ubiquitin E2 variant (UEV) domain to the Pro-Ser-Ala-Pro (PSAP) motif of the hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) [55]. The 145-residue UEV domain of TSG101 comprises 4 alpha helices and 4 beta strands[54, 56], and has a binding groove formed by the S2/S3 hairpin, the N-terminal third of the vestigial active site loop, and the C-terminus [56]. This groove binds not only PSAP, but also its threonine variant, PTAP, [54, 55, 57, 58, 59, 60], which occurs in the Ebola Vp40 protein [54, 55, 57, 59, 60] and in the late domain of the p6 region of the HIV Gag protein. Binding to the Tsg101 protein allows these viruses to hijack the normal cellular machinary for vesicle formation and use it instead for viral budding [54, 55, 58, 59, 60]. The failure of viral budding in cells lacking Tsg101 [58] has led to identification of Tsg101 as a candidate target for novel HIV therapies.
The present work uses the MIE to study changes in configurational entropy when the UEV domain of Tsg101 (henceforth termed Tsg101, for brevity) binds the HIV-derived PEPTAPPEE peptide (henceforth termed PTAP). The MIE calculations are based upon multiple molecular dynamics simulations (MMDSs), on the microsecond scale, of the free and bound species. A central result is that correlations within and between the two molecules induced by binding strongly contribute to the overall loss of configurational entropy.
2. Results
We start by reporting entropy changes in the absence of correlations by analyzing first-order MIE terms (Section 2.1) associated with the bond-length, bond-angle, and bond-torsion degrees of freedom (henceforth denoted bonds, angles, and torsions, respectively). Next, we examine the entropic consequences of changes in pairwise correlation on binding by analyzing the pairwise mutual information terms of the second-order MIE (Section 2.2). We then examine the shifts in the probability distribution functions (PDFs) that cause these entropy changes (Section 2.4) and interpret the numerical data in terms of the structure of the complex (Section 2.5). All contributions to the configurational entropy are reported in kcal mol−1 (1 kcal mol−1 = 4.184 kJ mol−1).
2.1. Changes in entropy at first order: neglect of correlations
The first-order MIE yields the entropy change under the approximation that correlations are unchanged by binding. The absence of correlation means that these results are additive, and Table 1 breaks them down by molecule (PTAP and Tsg101) and degrees of freedom (torsions, angles, and bonds). The total first-order entropy loss gives a free energy penalty of 14 kcal mol−1, which is substantial on the scale of protein-ligand binding free energies. Most of this change, 12 kcal mol−1, comes from reduced motion of Tsg101 while only the remaining 2 kcal mol−1 comes from reduced motion in the PTAP ligand. It is not surprising that these entropy changes are dominated by the torsions (12 kcal mol−1), because these soft degrees of freedom are more easily perturbed than the hard angle and bond degrees of freedom. Similarly, the angles, which are softer than bonds, contribute more to the entropy change. The small change in translational and rotational (trans/rot) entropy means that the coordinates which define the relative position and orientation of the two bound molecules (Figure 1) fluctuate significantly in the bound state. Note that the reported change in trans/rot entropy includes the term in Equation 5, which equals about 7 kcal mol−1 at 300K.
Table 1.
First-order approximations to entropy changes upon binding, reported as free-energy contributions −T ΔS (1) in kcal mol−1. The trans/rot value includes the contribution of in the unbound state.
| Bond-Stretches | Bond-Angles | Torsions | Total | |
|---|---|---|---|---|
| Ligand | 0.2 | 0.1 | 1.6 | 1.9 |
| Receptor | 0.5 | 1.3 | 10.4 | 12.1 |
| Trans/Rot | 0.3 | |||
| Total | 0.7 | 1.4 | 12.0 | 14.3 |
Figure 1.

Schematic representation of the 6 translational/rotational (trans/rot) degrees of freedom used to define the relative position and orientation of the receptor and ligand within the Tsg101•PTAP molecular complex. Defining the pseudo bond B (dashed line) creates two pseudoangles and three pseudodihedrals.
The first-order entropies are reasonably well converged, as shown in Figure 2, whose panels a and b correspond, respectively, to the bottom row and right-most column of Table 1. Thus, the variation in −T S (1) over the final quarter of the convergence plot is less than about 2 kcal mol−1. The angle and bond entropies are particularly well converged, and most of the uncertainty in the total entropy comes from the soft torsional degrees of freedom, which are significantly influenced by complex, time-varying noncovalent forces and can adopt multimodal forms (Section 2.4).
Figure 2.

Convergence plots of −T ΔS (1) (in kcal mol−1) as a function of the number of MD snapshots. The data shown indicate the entropy contributions from (a) sets of coordinates and (b) molecular components. These correspond, respectively, to the bottom row and right-hand column of Table 1.
2.2. Entropic consequences of changes in pairwise correlation
The second-order MIE uses pairwise mutual information terms to account for correlations between pairs of conformational variables. Such correlations can only lower the entropy, but binding could, in principle, lead to either increases or decreases in pairwise correlation. Therefore, it is not clear a priori whether these pairwise terms will favor or oppose binding. Table 2 presents the computed changes in pairwise mutual information for binding of Tsg101 and PTAP in the form of a symmetric matrix connecting different groupings of conformational variables: the torsions, angles, and bonds of PTAP, the trans/rot variables, and the torsions, angles and bonds of Tsg101, respectively. Each table entry is the summed change of all pairwise mutual information terms between two groups of variables, or, on the diagonal, within one group of variables. A positive entry in the table opposes binding. Figures 3, 4, and 6 provide convergence plots corresponding to each entry in Table 2.
Table 2.
Total pairwise mutual information contributions to the change in configurational entropy on binding, reported as free-energy contributions (T Δ I2) in kcal mol−1.
| Peptide |
Tsg101 |
|||||||
|---|---|---|---|---|---|---|---|---|
| T | A | B | T/R | T | A | B | ||
| Peptide | T | 3.9 | 1.9 | 0.1 | 2.2 | 16.6 | 3.4 | 1.2 |
| A | — | 0.3 | 0.0 | 0.4 | 3.9 | 1.4 | 0.5 | |
| B | — | — | 0.0 | 0.0 | 1.3 | 0.5 | 0.2 | |
| T/R | — | — | — | 0.9 | 3.1 | 0.3 | 0.0 | |
| Tsg101 | T | — | — | — | — | 2.4 | 0.3 | 0.0 |
| A | — | — | — | — | — | 0.1 | 0.1 | |
| B | — | — | — | — | — | — | 0.0 | |
Figure 3.

Convergence plots of ΣT ΔI2 (kcal mol−1) as a function of the number of MD snapshots, or the peptide-peptide (Frames a – f), peptide-trans/rot (Frames g – i), and trans/rot-trans/rot (Frame j) correlations.
Figure 4.

Convergence plots of ΣT ΔI2(kcal mol−1) as a function of the number of MD snapshots, for the peptide-Tsg101 (Frames a – i) and trans/rot-Tsg101 (Frames j – l) correlations. The dashed lines correspond to contributions from all Tsg101 residues and the solid lines correspond to the top 15 contributing residues of Tsg101 (see text).
Figure 6.

Convergence plots of ΣT ΔI2 (kcal mol−1) as a function of the number of MD snapshots, the Tsg101-Tsg101 correlations. The dashed lines correspond to contributions from all Tsg101 residues, and the solid lines correspond to the top 15 contributing residues (see text).
We first report the relatively well-converged entropy changes that result from changes in correlation involving only the PTAP ligand and the trans/rot variables (Section 2.2.1). We then analyze the less well-converged changes in entropy from changes in correlation involving Tsg101 (Sections 2.2.2 and 2.2.3).
2.2.1. Correlations involving only PTAP and the trans/rot variables
Correlations within PTAP rise on binding, as evident from the positive numbers in the three upper left rows and columns of Table 2. The net effect is an entropy penalty of 6 kcal mol−1, which adds to the first-order contribution of 2 kcal mol−1 from these degrees of freedom. The correlation contribution corresponds to about 0.7 kcal mol−1 per residue of the peptide and results almost entirely from torsions, with a small contribution from angles. Figure 3 shows that these internal peptide contributions to the entropy are reasonably well converged.
Binding also increases correlations involving the trans/rot variables. Increased correlation among trans/rot coordinates generates an entropy penalty of about 1 kcal mol−1, and increased correlation between trans/rot coordinates and PTAP internal coordinates adds another 2.6 kcal mol−1 (Table 2). Both of these contributions are reasonably converged, as shown in Figure 3g – j.
2.2.2. Correlations of Tsg101 with PTAP and the trans/rot variables
The total changes in mutual information due to correlations of Tsg101 with PTAP and the trans/rot variables are large and not well converged (dashed lines in Figure 4). This numerical issue stems chiefly from the fact that these mutual information terms are rigorously zero before binding, while, after binding, they are sums of many pairwise mutual information terms that cannot be negative. Thus, although most of the individual pairwise mutual information terms appear to be converging to values very near zero (data not shown), any residual lack of convergence on their part sums to a large uncancelled mutual information term. We therefore identify the Tsg101 residues that correlate most strongly with PTAP and limit attention to the comparatively robust mutual information terms associated with them.
Most intermolecular PTAP-Tsg101 residue pairs have small mutual informations, as shown in Figure 5. Thus, only 4 of the 1296 pairs have a mutual information greater than about 0.3 kcal mol−1, and these involve residues 68 and 69 in the binding cleft of Tsg101 and residues 4 and 5 at the center of PTAP; binding pocket residues 55, 65 and 140–144 also have relatively large mutual informations with individual PTAP residues. We identified the most important Tsg101 residues in this regard by summing the mutual informations of each Tsg101 residue with all residues of PTAP. The distribution of these sums is displayed in the inset of Figure 5, which shows that only about 10%, or 15 residues, have a net mutual information with PTAP of 0.5 kcal mol−1 or more; the locations of these residues are presented in Section 2.5. The Tsg101-PTAP mutual informations for only these Tsg101 residues converge reasonably well (solid lines in Frames a – i of Figure 4) and are presented in Table 2. The torsion-torsion contribution is large, at about 17 kcal mol−1, and adding contributions from angles and bonds yields a net entropy contribution of about 30 kcal mol−1 from new receptor-ligand correlations on binding.
Figure 5.

The main figure shows the top and side views of a contour plot of the peptide-Tsg101 pairwise mutual information contributions, summed by residue and reported as T I2 (kcal mol −1). The inset provides the cumulative distribution of the fraction of Tsg101 residues contributing a mutual information, summed over peptide residues, greater than a given value of T ΔI2.
Similar considerations apply to the mutual information terms between the trans/rot degrees of freedom and the internal TSG101 degrees of freedom: Figure 4 (panels j – l) shows inadequate convergence when all Tsg101 residues are considered (dashed line) and reasonable convergence for the 15 Tsg101 residues identified above (solid line). The correlations between trans/rot variables and the key 15 residues of Tsg101 generate a net entropy penalty of about 3.5 kcal mol−1 on binding (Table 2). It is important to note that the key 15 residues include Thr 57, which is used to define the geometry of the trans/rot coordinates (Figure 1).
2.2.3. Receptor-receptor correlations
The binding-induced changes in mutual information within Tsg101 itself have better convergence properties (Figure 6a–f, dashed lines) than those between Tsg101 and PTAP (Section 2.2.2). This results from wholesale cancellation, between the bound and unbound states, of the small, residual positive mutual information terms that were problematic for Tsg101-PTAP mutual information terms. Although the Tsg101-Tsg101 results still are not solidly converged, they point to a substantial entropic penalty from increased receptor-receptor correlation on binding. Increased torsion-torsion correlations generate a penalty of ~11 kcal mol−1, with an additional penalty of nearly 6 kcal mol−1 from correlations involving the other conformational variables. However, the loss of configurational entropy per residue, 0.1 kcal mol−1, is considerably smaller than that found for peptide-peptide correlations, 0.7 kcal mol−1 per residue. This makes sense because virtually all of the PTAP ligand interacts directly with TSG101, but only part of Tsg101 comes into contact with PTAP.
As these results are not reliably converged, we also made a more conservative estimate of the contribution of Tsg101-Tsg101 mutual information to the entropy change on binding by focusing on the 15 residues that correlate most with the PTAP ligand (Section 2.2.2). Much better convergence is observed (Figure 6, solid lines), and the numerical results, provided in the lower right-hand entries of Table 2, sum to about 3 kcal mol−1 worth of entropy loss.
2.3. Second-order estimate of the change in configurational entropy on binding
The second-order MIE estimate of the change in configurational entropy on binding is given by Equation 1, where the first term corresponds to the first-order entropy change estimated in Section 2.1, and the second term corresponds to the sum of the correlation contributions estimated in Sections 2.2.1, 2.2.2 and 2.2.3. The first-order data listed in Table 1 and the conservative mutual information terms based upon the top 15 receptor residues (Table 2) sum to about 59 kcal mol−1, a large entropy penalty for binding of PTAP to Tsg101.
2.4. Probability distribution functions before and after binding
The numerical changes in configurational entropy analyzed above were calculated from the PDFs of the molecules’ torsions, angles, and bonds. We now examine sample PDFs before and after binding and connect these with their numerical contributions to the entropy change. We focus on torsional distributions, as they make the most significant entropic contributions.
2.4.1. One-dimensional PDFs
Figure 7 graphs the one-dimensional probability distributions before (dashed) and after (solid) binding for the four torsions in the ligand and the receptor whose entropies decrease most upon binding. The corresponding entropy decreases (−T ΔS (1) in kcal mol−1) are noted in the upper right of each frame, and the torsions are also identified. The entropy losses in these cases appear to result not only from a reduction in the number of probability peaks (modes) upon binding, but also narrowing of the remaining peaks. For example, if the entropy change in Figure 7e resulted purely from a reduction in the number of peaks from two to one, the entropic penalty would amount to . The actual entropy penalty of 0.8 kcal mol−1 is about double that, because the single peak which remains after binding is narrower than either of the two peaks before binding. Similarly, the entropy penalty of about 0.4 kcal mol−1 in Figure 7h is entirely attributable to narrowing of a single mode in the PDF.
Figure 7.

Changes in PDFs of the torsion angles of PTAP (a–d) and Tsg101 (e–h) that lose the most uncorrelated entropy on binding. Dashed lines: free state. Solid lines: bound state. The heavy bonds indicate the central bond of the dihedral. The entropic free energy changes, −T ΔS, (kcal mol−1) are given in the upper-right corner of each frame.
2.4.2. Two-dimensional PDFs
We now examine the joint PDFs of several pairs of torsion angles with large changes in mutual information on binding. Each panel in Figure 8 shows the PDF before (red) and after (blue) binding for a pair of torsions between Tsg101 and the PTAP ligand (left), within the ligand (middle) and within TSG101 (right). The corresponding one-dimensional marginal PDFs are also shown, along with diagrams identifying the torsional variables. It is helpful to recognize that two variables are uncorrelated and have a mutual information of zero when their pairwise PDF equals the product of their one-dimensional marginal PDFs. (Note that the mutual information can be nonzero even if their Pearson correlation coefficient is 0 [41].) In such cases, the number of peaks in their two-dimensional distribution is the product of the number of peaks in their one-dimensional marginals. Because the uncomplexed molecules are completely uncorrelated, we constructed the red, two-dimensional PDFs in Figure 8a,b as products of the corresponding one-dimensional marginals, so these are examples of fully uncorrelated two-dimensional PDFs with zero mutual information. Substantial correlation is evident in many of the blue, two-dimensional PDFs based on the absence of product peaks. (Some small peaks in the uncorrelated PDFs also are not visible because of the contouring levels used.)
Figure 8.

Changes in the joint (contours) and marginal(line graphs) PDFs of torsional pairs with largest increases in mutual information on binding for Tsg101-PTAP pairs (a, b), PTAP-PTAP pairs (c, d) and Tsg101-Tsg101 pairs (e, f). Red: before binding; blue: after binding. The central bonds of the dihedrals are identified with heavy lines and the mutual information changes are reported as free energy contributions (kcal mol−1) in the upper-right corner of each frame.
Because the quasiharmonic approximation (QHA) is sometimes used to estimate entropy, it is of interest to examine it in the context of some of the PDFs from the Tsg101-PTAP system. The QHA approximates multidimensional PDFs with Gaussian distributions whose means and covariances match those of the actual distribution [37]. Figure 9a, b and c compares these Gaussians (orange and green) with the actual PDFs (blue and red) for three increasingly complex PDFs before and after binding. The QHA provides an excellent match to the original PDFs if they are unimodal and approximately Gaussian in shape (as in Frame a), but yields diffuse and comparatively featureless distributions when the original PDFs are multimodal (as in Frames b and c). Table 3 provides the numerical values of the entropy contributions from the actual PDFs and the QHA, which is known to provide an upper limit of the entropy [39], though not necessarily of entropy changes. As might be expected, the QHA entropy changes match the MIE results for Frame a. In Frames b and c, the QHA entropy changes compare much more poorly to the MIE values, particularly for Frame b.
Figure 9.

Comparison of the actual histogrammed PDFs (red, blue) with PDFs estimated by the QHA (orange, green), for three sample torsional pairs. Red and orange: before binding. Blue and green: after binding.
Table 3.
Comparison of pairwise entropy components (kcal mol−1) computed with the MIE and QHA methods. The results correspond to the distributions in Figure 6.
| MIE |
QHA |
||||||
|---|---|---|---|---|---|---|---|
| Frame | Unbound | Bound | Delta | Unbound | Bound | Delta | |
| a | −T Sφ1 | −0.1 | −0.1 | 0.0 | −0.1 | −0.1 | 0.0 |
| −T Sφ2 | 0.0 | −0.2 | −0.2 | −0.1 | −0.3 | −0.2 | |
| −T Sφ1φ2 | 0.0 | 0.0 | 0.0 | −0.1 | −0.1 | 0.0 | |
| b | −T Sφ1 | −0.5 | −0.5 | 0.0 | −0.8 | −1.1 | −0.3 |
| −T Sφ2 | −0.4 | −0.4 | 0.0 | −0.7 | −1.2 | −0.5 | |
| −T Sφ1φ2 | −0.9 | −0.5 | 0.4 | −1.5 | −1.7 | −0.2 | |
| c | −T Sφ1 | −0.5 | −0.6 | −0.1 | −1.2 | −1.1 | 0.1 |
| −T Sφ2 | −0.9 | −1.0 | −0.1 | −0.9 | −1.0 | −0.1 | |
| −T Sφ1, φ2 | −1.3 | −1.4 | −0.1 | −2.2 | −2.1 | 0.1 | |
2.5. Structural analysis of entropy changes on binding
The numerical entropy changes are now analyzed in the context of molecular structure. Residue numbers of Tgs101 used in this article are decremented by 1 relative to those reported in Reference 56, because the reported NMR structure excludes the N-terminal Met residue.
2.5.1. First-order entropy changes on binding
One might expect that the residues in the receptor that interact most closely with the ligand would demonstrate large changes in both entropy and correlation. This relationship can indeed be seen in Figure 10. The Tsg101 residues with the largest first-order entropy changes, blue in Figure 10b, significantly overlap those considered to be in direct contact with the peptide [56], blue in Figure 10a. However, the residues in the topmost loop of the receptor undergo substantial drops in entropy although they do not contact the bound PTAP. This observation may be rationalized by the fact that the loop is flanked by Tyr 62 and Tyr 67, which intimately contact the bound peptide and effectively pin the loop, thus reducing its mobility. Indeed, the largest first-order entropy changes in Tsg101 correspond to χ1 and χ2 of Tyr 67, which control the rotation of this side-chain’s phenolic ring and lose a total of ~1.5 kcal mol−1 worth of uncorrelated entropy on binding. These entropy losses are consistent with the changes in the first-order PDFs of these torsions (Figure 7e and f), and with the observation of Pornillos and co-workers [56] that Pro 6 of the ligand becomes interposed between Tyr 62 and Tyr 67, thus restraining their phenolic rings. This analysis provides a physical justification for the large entropy losses associated with these torsions and allows for a more detailed understanding of the interactions between the receptor and the ligand.
Figure 10.
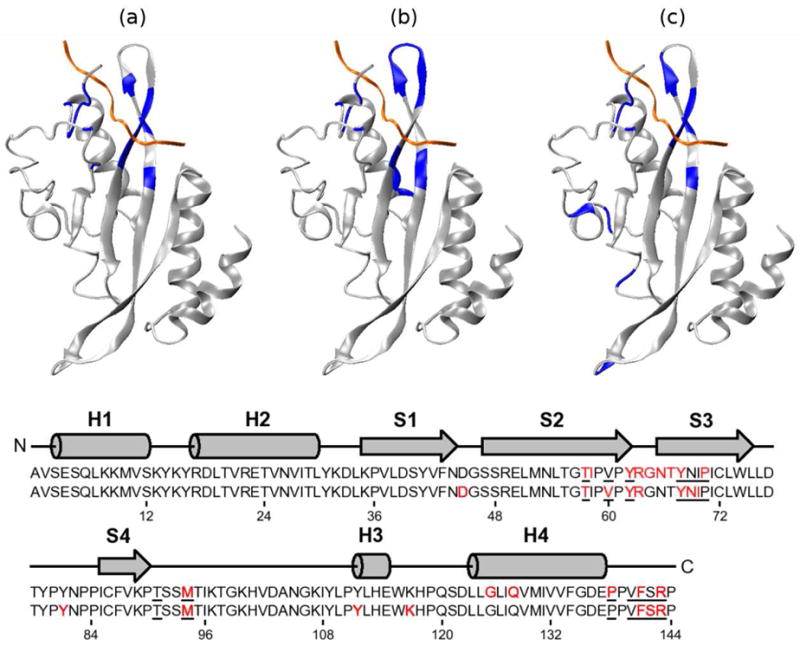
Structure-entropy relationships of Tsg101. The top row of figures diagrams (a) the backbones considered to contact the PTAP ligand [56], (b) the 15 residues with the largest uncorrelated (first-order) entropy changes, and (c) the 15 residues with the largest pairwise mutual informations with the PTAP ligand as a whole. The mutual information value assigned to a given TSG101 residue is the sum of the mutual information values of all of its atoms with all ligand atoms. The bottom shows the secondary structure and sequence of Tsg101. The red residues in the top and bottom sequences correspond, respectively, to those highlighted in (b) and (c) The underlined residues correspond to the contact points shown in (a).
Figure 11 provides a broader view of first-order entropy changes of residues at the surface of Tsg101 from the “front” (a), “back” (b), and “top” (c) of the complex, where each residue is colored according to its first-order torsional entropy change upon binding. The red end of the scale corresponds to increased entropy (increased motion) and the blue end of the scale to decreased entropy (decreased motion) on binding. (Note that the color scale is not symmetric, and that the the magnitude of entropy increases is roughly five times smaller than that of the decreases.) As expected residues located in or near the binding pocket show large decreases in entropy. More interestingly, a large region on the “back” of the receptor shows an unexpected increase in entropy on binding. These increases in entropy appear to be reasonably well-converged, with less than 0.4 kcal mol−1 variation for over the final half of the data set for the 13 residues with the greatest increase in entropy, as demonstrated in the inset of Figure 11.
Figure 11.
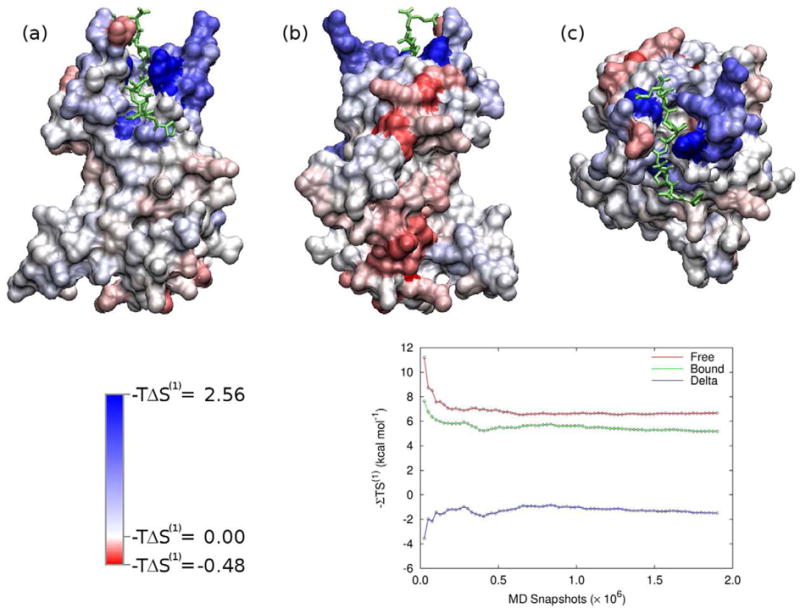
Surface plot of Tsg101 colored by −T ΔS (1) per residue. Red indicates a increase in entropy (increased motion) and blue indicates a decrease in entropy (decreased motion) on binding. The views are (a) “front”, (b) “back”, and (c) “top”. The ligand is colored green. The inset shows the convergence of the entropy associated with the red stripe along the “back” of the receptor.
2.5.2. Pairwise mutual information
The pairwise mutual information terms provide a great deal of information about correlations between specific structural elements. This information is now analyzed in several different ways.
We first examine the 15 Tsg101 residues with the greatest summed mutual informations with PTAP (Section 2.2.2), as shown in Figure 10c. As might be expected, this analysis does a better job of picking out the Tsg101 residues in the binding site that contact PTAP (Figure 10a) than the first-order entropy changes do (Figure 10b). In particular, the residues constituting the loop at the “top” of the complex are no longer shaded as in Frame b, consistent with their absence from the binding site. Thus, although these residues show large reductions in their first-order entropy on binding, they do not show substantial increases in correlation with PTAP. Interestingly, Tsg101 residue 45 also is found to correlate significantly with PTAP, although it is located diametrically opposite the binding site (Figure 10c). This result is well converged, with a variation of less than 0.04 kcal mol−1 over the last two-thirds of the data (data not shown), but the entropy contribution in question is only 0.6 kcal/mol. (For comparison, the correlations of one of the binding site residues with PTAP contributes over 4 kcal/mol.) The mechanism that couples residue 45 with PTAP is not clear, and we cannot definitely exclude the possibility of an artifact, but it is worth pointing out that the type IIβ-turn containing residue 45 is linked to the binding site by one of the protein’s long central β strands.
The complete network of pairwise correlations in the complex is examined in Figure 12, which provides heat plots of residue-residue distance (a) and of absolute residue-residue mutual information values in the complex (b). (The changes on binding are presented below.) Most of the mutual information values are small, less than 0.1 kcal mol−1 per residue, with a relatively small number of residue pairs contributing most substantially to the second-order correlation term. Many of the larger mutual information contributions lie along the diagonal, indicating that the correlations are strongest within residues and between successive residues along the peptide chain. Strong correlations are also evident between the receptor and ligand, as seen previously in Figure 5, where the correlations between residues 68 and 69 with ligand residues 4 and 5 are particularly prominent. The mutual information matrix in Figure 12b is similar in form to the distance plot in Figure 12a, indicating, not surprisingly, that the motions of residues that are close in space tend to correlate more than those of distant residues.
Figure 12.
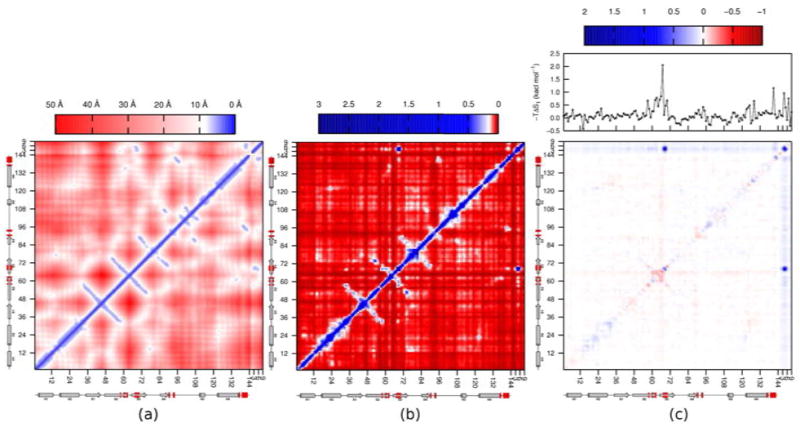
Heat plots of (a) Cα-Cα distances in the Tsg101•PTAP complex; (b) pairwise mutual information contributions summed by residue; and (c) change in pairwise mutual information summed by residue, with the change in first-order entropy per residue shown above. The secondary structure of Tsg101, with the contact residues [56] highlighted in red, is shown below each frame. All values are reported as T I2 in kcal mol−1.
The network of changes in mutual information on binding is presented in Figure 12c, along with the first-order entropy changes for each residue, above the heat plot. Most residue pairs undergo very small changes in mutual information upon binding; nonetheless, a vague outline of the underlying structure of the distance plot (Figure 12a) is evident. The regions corresponding to receptor/ligand correlation appear as strips of blue at the top and right sides of the plot (which are essentially equivalent to Figure 5), indicating an increase of mutual information upon binding. Several regions of increased mutual information occur within Tsg101, predominantly along the diagonal. Intriguingly, these indicate increased intraresidue and neighbor-neighbor correlation in the complex relative to the free state. Perhaps surprisingly, there are also regions where mutual information, and hence correlation, decrease upon binding. These correspond chiefly to the receptor residues located in the binding region and the loop between residues 62 and 67. These decreases in correlation might result in part from the overall decrease in motion of these residues on binding (see one-dimensional plot at top of Figure 12c). However, this does not seem to be a full explanation, because other residue pairs show increases in correlation even though their first-order entropies decrease upon binding. It thus appears that binding causes some Tsg101 residues to abandon their correlations with each other in exchange for new correlations with PTAP.
3. Discussion
This first application of the MIE to a protein provides a novel accounting of the changes in configurational entropy when Tsg101 binds the HIV-derived PTAP peptide. In addition to numerical results, we are able to probe structure-entropy relationships, and gain insight into the contribution of correlations to the change in entropy on binding. We now discuss the reliability and significance of the results, followed by lessons learned regarding strengths, weaknesses, and prospects for further development and use of the MIE to study proteins.
3.1. Configurational entropy change in context
The present calculations indicate that losses in configurational entropy strongly oppose binding in this system. This is broadly consistent with the view, derived from NMR studies, that changes in protein dynamics can substantially influence protein thermodynamics [61]. The best estimate of the entropic penalty at 300K is ~60 kcal mol−1. This quantity accounts for changes in the fluctuations and pairwise correlations of torsions, angles, and bonds, as well as the trans/rot degrees of freedom. Due to convergence issues, discussed below, we regard this as a semiquantitative estimate, but it does not seem unreasonable. It is about twice the value computed with the M2 method for the contribution of ligand motions alone on binding of amprenavir to HIV protease [1], and it is plausible that incorporating the protein contribution, as done here but not in the prior study, would roughly double the entropic cost. The present results can also be compared with prior analyses of small receptors that bind a tripeptide guest molecule [62], whose computed entropic penalties range from 19 to 33 kcal mol−1. These are smaller than those found for Tsg101, but the host-guest systems are much smaller as well: they pertain to a tripeptide rather than a nonapeptide, and a small host molecule rather than a 144 residue protein.
One may furthermore combine the computed entropy change for Tsg101 and PTAP with the measured binding free energy of about −7 kcal mol−1 [54, 58] to address the energetics of binding. The change in free energy on binding can be written as
| (3) |
where ΔU and ΔW are, respectively, the change in mean potential and solvation energy on binding [1]. Thus, combining the present results with the experimental binding free energy indicates a change in the mean potential plus solvation free energy of −67 kcal mol−1. Again, this value may be compared with the host-guest systems mentioned above, whose computed values of ΔU + ΔW range from −26 to −41 kcal mol−1. It is reasonable that a larger energetic contribution is obtained for the present system, given that its interfacial surface area on binding, 1250 Å2 [63], is 2- to 4-fold greater than those of the host-guest systems.
Note that the entropy changes considered here are purely configurational, and so exclude the change in solvation entropy on binding. The total entropy change on binding equals the sum of the configurational and solvation entropy changes [1], and it has been argued that the change in solvation entropy usually favors binding [19]. We would therefore anticipate that a calorimetric study of Tsg101 binding to PTAP would yield a total entropy change considerably less unfavorable than our estimated configurational entropy penalty of 60 kcal mol−1.
3.2. Entropic consequences of correlation
Entropy is a measure of uncertainty, and correlations lower the configurational entropy, because, if two degrees of freedom are correlated, then knowing the value of one reduces the uncertainty regarding the other. Some change in correlation on binding is expected; indeed, it is the essence of binding that the motions of two molecules become correlated. However, it has not been clear whether changes in correlation on binding significantly influence binding thermodynamics.
The present study provides strong evidence that changes in correlation on binding can lead to large changes in entropy. This result is best shown by the well-converged data restricted to the peptide ligand and its trans/rot degrees of freedom. When correlations are neglected, the reduced fluctuations of the peptide degrees of freedom on binding lead to an entropy penalty of only ~2 kcal mol−1. However, the increased correlation among these degrees of freedom generates an additional ~10 kcal mol−1 opposing binding. The largest contribution comes from new correlations among peptide torsions, which may stem from “crankshaft” motions of the bound ligand [11] that the free ligand is not constrained to execute. This would be consistent with the observation that some of the largest mutual information contributions come from adjacent backbone torsions (e.g., Figure 8). New correlations involving the trans/rot coordinates also contribute strongly. These result from correlations of the trans/rot coordinates with ligand torsions, and also among the trans/rot coordinates themselves. The appearance of strong correlations involving the trans/rot coordinates implies that changes in translational and rotational entropy on binding cannot be cleanly separated from other entropic contributions. Not surprisingly, the calculations point to even greater binding-induced correlations between the peptide and the protein. Even when attention is limited to the 15 most significant protein residues, the resulting entropic penalty is over 20 kcal mol−1. This results chiefly from correlations between Tsg101 and PTAP torsions, but angles and trans/rot motions also contribute substantially.
These conclusions differ from a prior suggestion that correlations do not contribute strongly to configurational entropy [13]. However, the two studies may be reconcilable. For one thing, the prior study did not check for correlations between pairs of torsions that are neighbors in the bonding sequence, but we find that some of the strongest changes in correlation involve such pairs. Also, the previous study used the Pearson correlation coefficient to identify correlations, but this may miss some correlations because it is sensitive only to linear relationships. Indeed, one can construct PDFs for which the Pearson correlation coefficient is zero but the mutual information is not [41].
3.3. Structure and entropy
The MIE connects the change in configurational entropy on binding to specific structural elements and interactions. Not surprisingly, the interfacial residues of Tsg101, the residues of PTAP, and the trans/rot degrees of freedom are found to make the greatest contributions to the entropy change. At a finer level of detail, those residues which form intimate new interactions on binding, such as Pro 7 of the ligand and Tyr 62 and Tyr 67 of the protein, play a particularly important role. The intricate structure-entropy relationships observed here indicate that a computer model of binding must treat configurational entropy in some detail in order to reach the level of accuracy needed for reliable molecular design.
Although the pattern is primarily one of decreased configurational entropy due to interactions between the two molecules, there are also interesting exceptions. For example, we find that binding reduces correlations within the binding-site loop of Tsg101, thereby reducing the entropy penalty. In addition, we observe an increase in fluctuations, and hence in the first-order entropy, along a surface of the protein far from the binding site. Such observations are contrary to the perhaps intuitive expectation that adding a ligand will simply tighten down the protein and produce a global reduction in configurational entropy. However, they are consistent with NMR studies of proteins showing that ligand-binding can tighten the binding site while making other regions of the protein more mobile [10, 11, 14, 64, 65, 66, 67, 68, 69]. In fact, it has been proposed that there is a general “conformational relay” mechanism that balances increased rigidity of some residues against increased flexibility of their neighbors [14]. We suggest that this scenario occurs when a protein can optimize either its internal interactions away from the binding site or its interactions with a bound ligand, but not both. When no ligand is present, the binding site remains flexible and the rest of the protein is kept rigid by its internal interactions. When the ligand is present, the conformation of the binding site adjusts and becomes less flexible, but this change disrupts internal interactions in the rest of the protein and makes it more mobile. However, a conformational relay could also work in the opposite sense, so that increased rigidity at one site leads to increased rigidity elsewhere [5]. This will occur if the formation of stabilizing interactions with the ligand is structurally compatible with formation of energetically stabilizing interactions elsewhere. These two senses of the conformational relay could lead to entropic allostery [5, 21, 70, 71] between multiple binding sites on a protein that is either cooperative or anticooperative.
3.4. The mutual information expansion
The MIE provides a theoretical framework for studying entropy which accounts, at least in principle, for the network of correlations that characterizes molecular motions and provides informative links between structure and thermodynamics. It also has the merit, unlike the quasiharmonic approximation, of accounting for the multimodal nature of molecular distribution functions. On the other hand, it is computationally nontrivial and some second-order mutual information terms computed here did not converge adequately. We circumvented this issue by focusing on those residues that make the most robust contribution to the peptide-protein mutual information. The mutual information signal for these residues rises above the noise, is reasonably well-converged, and provides informative results. However, this focused approach means we are not yet able to assess the entropic contributions of longer-ranged peptide-protein pairs with weaker correlations. In addition, the present study makes an approximation by truncating the MIE at second order. This is enough to provide a series of interesting observations regarding the role of correlation as a determinant of changes in configurational entropy, but there may be more to learn by going to higher order in the MIE. We now discuss the numerical challenges posed by the MIE, sources of error, and the prospects for improving convergence and extending the calculations to higher order.
Computing the MIE at second order requires converging a large number of pairwise mutual information terms. The first requirement for converging each pairwise term is that the molecular simulation must adequately sample the pairwise marginal PDF for the two corresponding conformational variables. This can be time-consuming even for a pair of uncorrelated torsions. Consider, for example, the two unbound torsion angles illustrated in Figure 8d. Converging the joint histogram requires sampling from the four or five predominant peaks of the two-dimensional joint PDF, which means that each torsion must undergo multiple transitions between its modes. To the extent that this sampling is incomplete, the joint histogram will appear overly correlated and the mutual information will be too large. The second requirement for converging each pairwise term is that the corresponding two-dimensional histogram must be populated well enough to generate an accurate picture of the actual PDF. If the histogram is too rarified, because of too few conformational samples per bin, the distribution effectively becomes rough and therefore underestimates the actual entropy.
The present study uses multiple MD simulations (MMDSs) carried out in parallel to generate a large number of conformational samples. Nonetheless, the full pairwise results are not well converged. This is largely because the pairwise mutual information can only be positive or zero, so that even if two torsions, say, are uncorrelated, any imprecision in the calculation will result in a falsely positive mutual information, and the errors of many such pairs will be additive. For protein-protein pairs, significant cancellation still occurs between the free and bound states. However, no cancellation occurs for the many peptide-protein pairs. Thus, even though the mutual informations of most of these pairs converge to near zero, their sum is large. As noted above, we have worked around this problem by focusing on those peptide-protein mutual information terms which clearly rise above the noise. In the future, we believe it will be possible to reduce the noise level by applying more powerful entropy estimators to this data set or other simulations of similar size. Options include kernel [72], spline [73] and Nearest-Neighbor (NN) [42, 43, 44, 74, 75] approaches. Recent numerical studies indicate that a combined MIE-NN approach offers markedly superior convergence properties than the histogram-based MIE approached used here, albeit at a higher computational cost [44].
It is also worth considering the reliability of the entropy contributions that do appear to be well converged. Thus, the force field and simulation protocol can affect the reliability of these results, and there is no guarantee that the runs were long enough to fully populate all combinations of energy wells in the pairwise PDFs and overcome all conformationally important energy barriers. Encouragingly, the present simulations appear to satisfactorily reproduce distance restraints provided by nuclear Overhauser effect data, as will be reported separately. Nonetheless, extended simulations of these and other systems with increasingly reliable energy models will be of great value in assessing the reliability and generality of the present results.
Finally, it is important to consider the level of the MIE required to completely characterize the configurational entropy change in a binding process. In this investigation, the MIE was truncated at second order, thus accounting for correlations between pairs of degrees of freedom. The second-order truncation has yielded good agreement with a reference computational method for small, acyclic molecules [29], but little is known about whether or when higher-order correlations are important in proteins. This will clearly be an intriguing area for future research. One obstacle to moving to third order will be the difficulty of achieving adequate convergence for third-order mutual information terms; the MIE-NN method mentioned above should be valuable in this regard. Another obstacle will be the very large number of third-order contributions in a molecule as large as a protein. One way to address this would be to limit the set of triplets that are evaluated based, for example, upon the distances or the pairwise correlations among the variables composing the triplet.
4. Conclusions
The central conclusions of this study are as follows.
The observation of large changes in configurational entropy on binding supports the idea that changes in configurational entropy can influence affinity just as strongly as energetic interactions, such as electrostatics, hydrogen bonding and the hydrophobic effect.
We see a detailed picture of structure-entropy relationships, such as the large contributions of the binding-site tyrosines and their trading of protein-protein correlations for protein-peptide correlations. Accounting for such intricacies in computer models of binding should significantly increase accuracy and thus lead to enhanced tools for molecular design.
We also find that increases in correlation on binding reduce the entropy at least as much as changes in the fluctuations of individual coordinates. This suggests that analysis of NMR order parameters may tend to underestimate entropy losses. The importance of correlation also provides a caution against studying the entropic contributions of a few conformational variables, such as the trans/rot coordinates, in isolation.
Important questions remain, including the effects of weak, long-range correlations and the importance of higher-order correlations in the expansion.
Continued development of computational approaches like the MIE, as well as enhanced sampling methods, more robust density estimation, and applications to other systems, should provide further insight into the role of configurational entropy in binding and the rules that govern it.
5. Theory and Methods
The standard change in free energy on receptor-ligand binding can be written as [1, 76]
| (4) |
where U and W, respectively, represent the potential energy and the solvation free energy of the receptor and ligand; the angle brackets indicate Boltzmann averages; T is the absolute temperature; and is the configuration entropy, the focus of this study. Note that the change in mean solvation free energy on binding, Δ〈W〉, includes an entropic component, which is not addressed in the present study. Instead, we focus on the change in configurational entropy, which is a measure of the fluctuations of the translational, rotational and internal degrees of freedom of the receptor and ligand.
5.1. The Mutual Information Expansion (MIE)
The configuration of a molecule composed of M atoms can be defined by a set of 3M − 6 internal coordinates, x = (x1, …, x3M−6), and its configurational entropy at standard concentration can be expressed as [1, 29]
| (5) |
The first term in the right hand side of Equation 5 accounts for the molecule’s freedom of rotation and translation at standard concentration C° (generally 1 mol L−1 = 6.022×10−4 molec. Å−3), and is trivial to compute [77]. We will set it aside and focus on the second term in the expression. The function ρ(x) is the (3M −6)-dimensional probability density (or distribution) function (PDF) of the molecule’s internal coordinates and J(x) is the Jacobian determinant, required to correct the volume element of the integral for the transformation from Cartesian to internal coordinates (Section 5.2). The negative of the integral, which extends over all accessible conformations, is the Shannon entropy [78] associated with the PDF and is closely related to the Gibbs H-function [79, 80]. When multipled by the gas constant, R, this integral becomes a thermodynamic entropy and, therefore, a measure of conformational fluctuations. This entropy will henceforth be termed S:
| (6) |
The MIE allows Equation 6 to be separated into first-order contributions, Sa, associated with fluctuations along a single degree of freedom xa; second-order contributions in the form of mutual information terms, Iab, resulting from correlations between pairs of degrees of freedom xa and xb; and higher-order correlation corrections which will not be used in this work. For full derivations of the expansion terms and discussion of the relation between the mutual information expansion and the PDF, please see the Theory section of Reference 28.
The first-order approximation to S is based on the assumption that all degrees of freedom are uncorrelated; i.e., the motion along one degree of freedom is completely decoupled from the motions along all other degrees of freedom. As a result, in the first-order approximation to the total entropy, here denoted S (1), each degree of freedom xa makes an independent contribution Sa to the total entropy:
| (7) |
Each marginal entropy Sa is computed by applying the Gibbs/Shannon formula to ρ(xa), the marginal probability density in xa,
| (8) |
where J(xa) is the part of the Jacobian associated with xa. The first-order approximation given in Equation 7 is an upper with bound to the configurational entropy. It becomes an equality in the limit of completely uncorrelated motions [81, 82].
The pairwise joint mutual information of coordinates xa and xb, defined as [52]
| (9) |
is a non-negative quantity that serves as a measure of the amount of correlation between the two degrees of freedom. It is worth noting that the pairwise mutual information is zero only if the two degrees of freedom are completely uncorrelated. The pairwise joint entropy, Sab, is computed by applying the Gibbs/Shannon formula to ρ(xa, xb), the two-dimensional joint probability density in xa and xb,
| (10) |
The second-order approximation to the total entropy, S(2), accounts for pairwise correlations between the degrees of freedom by subtracting all unique pairwise mutual information terms from the first-order approximation,
| (11) |
Higher-order correlations can be accounted for in an analogous fashion with higher-order mutual information terms [29, 81], but are not addressed here.
The second-order approximation to S given in Equation 11 can be substituted into Equation 5 to yield the second-order approximation of the standard configurational entropy,
| (12) |
5.2. Bond-angle-torsion coordinate system
MD simulations typically use a Cartesian coordinate system, but Cartesian coordinates are not suitable for use in the MIE method, so a set of consistent internal coordinates must be obtained. The present study uses a bond-angle-torsion (BAT) coordinate system [29, 83, 84, 85, 86, 87, 88] in which the internal coordinates of a molecule with M atoms comprise M − 1 bond-lengths b, M − 2 bond-angles θ, and M − 3 bond-torsions φ. The Jacobian determinant for the transformation from Cartesian to BAT coordinates is [29]
| (13) |
The Jacobian can be factored into separate bond and angle components, respectively
| (14) |
and
| (15) |
where and J(θi) = sin θi. Note that i indexes atoms, rather than degrees of freedom.
The marginal entropies associated with the bond-stretch, angle-bend, and bond-torsion of atom i are:
| (16) |
| (17) |
| (18) |
Analogous expressions apply to the joint entropies; for example, the joint entropy associated with the bond-stretch of atom i and the angle-bend of atom j is given by
| (19) |
Special care must be taken when defining the BAT coordinates of a complex of two molecules. If the first molecule has M1 atoms and the second has M2 atoms, then 3(M1 + M2) − 6 degrees of freedom are required to completely define the system. The first molecule contributes M1 −1 bonds, M1 −2 angles, and M1 −3 dihedrals and the second contributes M2 − 1 bonds, M2 − 2 angles, and M2 − 3 dihedrals. Clearly, six additional degrees of freedom are needed to completely describe the complex. These are added by constructing a pseudobond, B, connecting neighboring atoms in the two molecules (see Figure 1), which then allows creation of two pseudoangles and three pseudotorsions, denoted as Θ1, Θ2, Φ1, Φ2, and Φ3, respectively. These six special degrees of freedom describe the relative position (translation) and orientation (rotation) of the two molecules, and will be collectively referred to as the trans/rot degrees of freedom.
The BAT coordinate system has a tree-like structure, because the BAT coordinates of each atom must be defined in terms of 3 other atoms whose coordinates are already given. The coordinates of the first three atoms of the tree, termed root atoms, cannot be fully specified in terms of other atoms. Instead, they include 6 external coordinates, which define the position and orientation of the molecule as a whole in the lab frame. It is the integration over these coordinates that yields the term in the entropy. Here, the root atoms of Tsg101 and its complex with PTAP are the amine H and N and the Cα of the N-terminal alanine. The root atoms of the free ligand are its amine H and N, and the Cδ of the N-terminal proline. For the complex, the trans/rot degrees of freedom are defined by a pseudobond connecting Oγ of Thr 57 to an amine H in Pro 1, as demonstrated in Figure 1. The complete set of atoms selected to form the trans/rot degrees of freedom are Cα, Cβ, and Oγ from Thr 57 and an amide H, N, and Cδ from Pro 1. Note that none of these pseudo-degrees of freedom has any influence on the energy; they are used only to convert the MD trajectory into BAT coordinates.
Some analyses in this study apportion entropy contributions according to residue. There is some ambiguity in how to assign BAT coordinates to residues, especially for coordinates involving the backbone amide bond. We assigned a bond to a given residue if both atoms were in that residue; if one atom was in residue j and the other in residue j + 1, the bond was arbitrarily assigned to residue j. An angle was assigned to that residue which contained the majority of atoms defining the angle. A torsion was assigned to that residue which contained both central atoms (atoms i + 1 and i + 2 of a torsion involving atoms i, i + 1, i + 2, i + 3). For the special case of the backbone amide, where atoms i + 1 and i + 2 belong to different residues, the torsion was arbitrarily assigned to the residue containing atom i + 1. This assignment method was applied consistently for the receptor, the ligand, their complex.
5.3. Molecular dynamics simulations
Initial coordinates of PTAP (residue IDs 205-213), Tsg101 (residue IDs 2-144), and their complex, were taken from the first of 20 solution NMR-refined conformers of the complex available at the Protein Data Bank [89, 90] (PDB code: 1M4P) [56]. (The terminal Met was absent from the NMR structure, and was therefore not included in the simulations). Topology and coordinate files for the two molecules and their complex were generated with the LINK, EDIT and PARM modules of the AMBER 5.0 program [91]. The Tsg101•PTAP complex was solvated with a preequilibrated box of TIP3P water molecules [92], where any water molecule was removed if it had an oxygen atom closer than 2.2 Å to any solute atom or a hydrogen atom closer than 2.0 Å to any solute atom, or if it lay further than 8.2 Å along the x-, y- or z-axis from any solute atom. This left 5646 TIP3P waters, for a system of 19408 atoms in all. The free receptor was similarly solvated with 5723 waters and neutralized by 3 chloride ions, for a system of 19514 atoms. The free peptide was solvated with 784 waters and neutralized by 3 sodium ions for a system of 2483 atoms. The solvated systems were first energy-minimized for 200 steps to remove any close van der Waals contacts within the system and were then slowly heated from 0 to 300 K at a rate of 10 K ps−1 under constant temperature and volume, and were finally simulated under constant temperature and pressure.
MMDSs were performed according to a published protocol [93] with the SANDER module of AMBER 7.0 [94] and the AMBER force field (frcmod.ff99) [95]. All simulations used the Berendsen coupling algorithm [96], periodic boundary conditions at a constant temperature of 300 K and a constant pressure of 1 atm with isotropic molecule-based scaling, the Particle-Mesh Ewald [97] treatment of long ranged electrostatic interactions, a 1.0 fs time-step, SHAKE bond-length constraints applied to all bonds involving hydrogen atoms, and default values of all other inputs of the SANDER module. Two hundred different 10 ns simulations, each starting with a unique random number seed for the initial velocities of the system, were carried out for each of the free and bound species. Snapshots were saved at 1 ps intervals, for a total of 2×106 snapshots. The snapshots from the first 500 ps of each simulation were discarded to allow for equilibration.
5.4. Data processing
Entropies were calculated from the trajectories with the Algorithm for Computing Configurational ENTropy from Molecular Mechanics (ACCENT-MM) [29], which is freely available for download at http://gilsonlab.umbi.umd.edu/software1a.html. The MMDS trajectory output files were concatenated as though they were part of a single long simulation, to facilite data processing, and the Cartesian coordinates were converted to BAT coordinates and stored. Groups of 2 or 3 atoms in a rigid unit controlled by one torsion angle (e.g., the 3 hydrogens of a methyl group) were treated as one regular torsion plus 1 or 2 phase angles defined relative to the regular torsion [29, 87]. The phase angles have narrow, unimodal PDFs, with minimal correlation to other variables. This approach allows a larger fraction of the torsional correlation to be captured in the first- and second-order MIE approximations [29]. The trans/rot degrees of freedom defined for the complex were processed in conjunction with the regular degrees of freedom using the same methods.
The second-order MIE requires one-dimensional marginal and two-dimensional joint PDFs for all degrees of freedom. Here, the ACCENT-MM code was used to construct these PDFs in the form of one- or two-dimensional histograms with 25 bins of uniform width along each axis. (Tests with 50 bins gave very similar results.) The range of each histogram axis was fit to the observed range of the corresponding variables. For the torsional variables, which are circular, the histogram was fit to the smallest range. Identical ranges and bin widths were used for corresponding one- and two-dimensional histograms to ensure that the marginal densities are consistent between the two levels. The Supplemental Material derives the equations used to compute the one- and two-dimensional entropies from these histograms.
We applied the quasiharmonic approximation [37] (QHA), which approximates the true PDF by a multivariate Gaussian having the same mean and covariances, to several pairs of torsional variables. The marginal and joint entropies, respectively, are given by
| (20) |
and
| (21) |
where σi and Σij, respectively, are the variance and the two-dimensional covariance matrix of variables i and j, and e is the base of the natural logarithm. The circularity of the torsional variables was addressed by using the shortest deviations from the means to compute the variances.
Acknowledgments
This publication was made possible by Grant No. GM61300 (MKG) from the National Institute of General Medical Sciences of the National Institutes of Health and Grant No. W81XWH-08-1-0154 (YPP) from the U. S. Army Medical Research Acquisition Activity. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of General Medical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chang CEA, Chen W, Gilson MK. Ligand configurational entropy and protein binding. Proc Natl Acad Sci USA. 2007;104:1534–1539. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang CEA, McLaughlin WA, Baron R, Wang W, McCammon JA. Entropic contributions and the influence of the hydrophobic environment in promiscuous protein-protein association. Proc Natl Acad Sci USA. 2008;105:7456–7461. doi: 10.1073/pnas.0800452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rekharsky MV, Mori T, Yang C, Ko YH, Selvapalam N, Kim H, Sobransingh D, Kaifer AE, Liu S, Isaacs L, Chen W, Gilson MK, Kim K, Inoue Y. A synthetic host-guest system achieves avidin-biotin affinity by overcoming enthalpy-entropy compensation. Proc Natl Acad Sci USA. 2007;104:20737–20742. doi: 10.1073/pnas.0706407105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crespo A, Fernandez A. Induced disorder in protein-ligand complexes as a drug-design strategy. Mol Pharmaceutics. 2008;5(3):430–437. doi: 10.1021/mp700148h. [DOI] [PubMed] [Google Scholar]
- 5.Akke M, Brüschweiler R, Palmer AG., III NMR Order Parameters and Free Energy: An Analytical Approach and Its Application to Cooporative Ca2+ Binding by Calbindin D9k. J Am Chem Soc. 1993;115:9832 – 9833. [Google Scholar]
- 6.Yang D, Kay LE. Contributions to conformational entropy arising from bond vector fluctuations measured from NMR-derived order parameters: Application to protein folding. J Mol Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Raychaudhuri S, Wand AJ. Insights into the local residual entropy of proteins provided by NMR relaxation. Prot Sci. 1996;5:2647–2650. doi: 10.1002/pro.5560051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bremi T, Brüschweiler R. Locally anisotropic internal peptide backbone dynamics by NMR relaxation. J Am Chem Soc. 1997;119:6672 – 6673. [Google Scholar]
- 9.Zidek L, Novotny MV, Stone MJ. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat Struct Biol. 1999;6:1118–1121. doi: 10.1038/70057. [DOI] [PubMed] [Google Scholar]
- 10.Lee AL, Kinnear SA, Wand AJ. Redistribution and loss of side chain entropy upon formation of a calmodulin-peptide complex. Nat Struct Biol. 2000;7:72 – 77. doi: 10.1038/71280. [DOI] [PubMed] [Google Scholar]
- 11.Stone MJ. NMR relaxation studies of the role of conformational entropy in protein stability and ligand binding. Acc Chem Res. 2001;34:379–388. doi: 10.1021/ar000079c. [DOI] [PubMed] [Google Scholar]
- 12.Arumugam S, Gao G, Patton BL, Semenchenko V, Brew K, Doren SRV. Increased backbone mobility in β-barrel enhances entropy gain driving binding of N-TIMP-1 to MMP-3. J Mol Biol. 2003;327:719–734. doi: 10.1016/s0022-2836(03)00180-3. [DOI] [PubMed] [Google Scholar]
- 13.Prabhu NV, Lee AL, Wand AJ, Sharp KA. Dynamics and entropy of a calmodulin-peptide complex studied by NMR and molecular dynamics. Biochemistry. 2003;42:562–570. doi: 10.1021/bi026544q. [DOI] [PubMed] [Google Scholar]
- 14.Bingham RJ, Findlay JBC, Hsieh S, Kalverda AP, Kjellberg A, Perazzolo C, Phillips SEV, Seshadri K, Trinh CH, Turnbull WB, Bodenhausen G, Homans SW. Thermodynamics of binding of 2-methoxy-3-isopropylpyrazine and 2-methoxy-3-isobutylpyrazine to the major urinary protein. J Am Chem Soc. 2004;126:1675–1681. doi: 10.1021/ja038461i. [DOI] [PubMed] [Google Scholar]
- 15.Homans SW. Probing the binding entropy of ligand-protein interactions by NMR. ChemBioChem. 2005;6:1585–1591. doi: 10.1002/cbic.200500010. [DOI] [PubMed] [Google Scholar]
- 16.Spyracopoulos L. Thermodynamic interpretation of protein dynamics from NMR relaxation measurements. Protein Pept Lett. 2005;12:235–240. doi: 10.2174/0929866053587075. [DOI] [PubMed] [Google Scholar]
- 17.Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325 – 330. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stöckmann H, Bronowska A, Syme NR, Thompson GS, Kalverda AP, Warriner SL, Homans SW. Residual ligand entropy in the binding of p-substituted benzenesulfonamide ligands to bovine carbonic anhydrase II. J Am Chem Soc. 2008;130:12420 – 12426. doi: 10.1021/ja803755m. [DOI] [PubMed] [Google Scholar]
- 19.Brady GP, Sharp KA. Entropy in protein folding and in protein-protein interactions. Curr Opin Struct Biol. 1997;7:215 – 221. doi: 10.1016/s0959-440x(97)80028-0. [DOI] [PubMed] [Google Scholar]
- 20.Fischer S, Smith JC, Verma CS. Dissecting the vibrational entropy change on protein/ligand binding: Burial of a water molecule in bovine pancreatic trypsin inhibitor. J Phys Chem B. 2001;105:8050–8055. [Google Scholar]
- 21.Jusuf S, Loll PJ, Axelsen PH. The role of configurational entropy in biochemical cooperativity. J Am Chem Soc. 2002;124:3490–3491. doi: 10.1021/ja017259h. [DOI] [PubMed] [Google Scholar]
- 22.Chang CE, Gilson MK. Free energy, entropy, and induced fit in host-guest recognition: Calculations with the Second-Generation Mining Minima Alogorithm. J Am Chem Soc. 2004;126:13156–13164. doi: 10.1021/ja047115d. [DOI] [PubMed] [Google Scholar]
- 23.Chen W, Chang CE, Gilson MK. Calculation of cyclodextrin binding affinities: Energy, entropy, and implications for drug design. Biophys J. 2004;87:3035 – 3049. doi: 10.1529/biophysj.104.049494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swanson JMJ, Henchman RH, McCammon JA. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys J. 86;2004:67–74. doi: 10.1016/S0006-3495(04)74084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peter C, Oostenbrink C, van Dorp A, van Gunsteren WF. Estimating entropies from molecular dynamics simulations. J Chem Phys. 2004;120:2652–2661. doi: 10.1063/1.1636153. [DOI] [PubMed] [Google Scholar]
- 26.Hsu STD, Peter C, van Gunsteren WF, Bonfin AMJJ. Entropy calculation of HIV-1 env gp120, its receptor CD4, and their complex: An analysis of configurational entropy changes upon complexation. Biophys J. 2005;88:15–24. doi: 10.1529/biophysj.104.044933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruvinsky AM, Kozintsev AV. New and fast statistical-thermodynamic method for computation of protein-ligand binding entropy substantially improves docking accuracy. J Comput Chem. 2005;26:1089 – 1095. doi: 10.1002/jcc.20246. [DOI] [PubMed] [Google Scholar]
- 28.Lu B, Wong CF. Direct estimation of entropy loss due to reduced translational and rotational motions upon molecular binding. Biopolymers. 2005;79:277–285. doi: 10.1002/bip.20344. [DOI] [PubMed] [Google Scholar]
- 29.Killian BJ, Kravitz JY, Gilson MK. Extraction of configurational entropy from molecular simulations via an expansion approximation. J Chem Phys. 2007;127:024107. doi: 10.1063/1.2746329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuentes G, Ballesteros A, Verma CS. Enthalpic and entropic contributions in the transesterification of sucrose: Computational study of lipases and subtilisin. J Biomolec Struct Dynam. 2007;25:145–155. doi: 10.1080/07391102.2007.10507163. [DOI] [PubMed] [Google Scholar]
- 31.Chang MW, Belew RK, Carroll KS, Olson AJ, Goodsell DS. Empirical entropic contributions in computational docking: Evaluation in APS reductase complexes. J Comput Chem. 2008;29(11):1753–1761. doi: 10.1002/jcc.20936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karplus M, Ichiye T, Pettitt BM. Configurational entropy of native proteins. Biophys J. 1987;52:1083–1085. doi: 10.1016/S0006-3495(87)83303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balog E, Becker T, Oettl M, Lechner R, Daniel R, Finney J, Smith JC. Direct determination of vibrational density of states change on ligand binding to a protein. Phys Rev Lett. 2004;93:028103. doi: 10.1103/PhysRevLett.93.028103. [DOI] [PubMed] [Google Scholar]
- 34.Balog E, Smith JC, Perahia D. Conformational heterogeneity and low-frequency vibrational modes of proteins. Phys Chem Chem Phys. 2006;8:5543 – 5548. doi: 10.1039/b610075a. [DOI] [PubMed] [Google Scholar]
- 35.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546 – 4559. [Google Scholar]
- 36.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559 – 4570. [Google Scholar]
- 37.Karplus M, Kushick JN. Method for estimating the configurational entropy of macromolecules. Macromolecules. 1981;14:325–332. [Google Scholar]
- 38.Prompers JJ, Brüschweiler R. Thermodynamic interpretation of NMR relaxation parameters in proteins in the presence of motional correlations. J Phys Chem B. 2000;104:11416 – 11424. [Google Scholar]
- 39.Schlitter J. Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chem Phys Lett. 1993;215:617 – 621. [Google Scholar]
- 40.Chang CE, Chen W, Gilson MK. Evaluating the accuracy of the quasiharmonic approximation. J Chem Theory Comput. 2005;1:1017. doi: 10.1021/ct0500904. [DOI] [PubMed] [Google Scholar]
- 41.Warner RM. Applied Statistics: From Bivariate through Multivariate Techniques. SAGE Publications; Los Angeles: 2008. [Google Scholar]
- 42.Singh H, Misra N, Hnizdo V, Fedorowicz A, Demchuk E. Nearest neighbor estimates of entropy. Am J Math Manag Sci. 2003;23:301–321. [Google Scholar]
- 43.Hnizdo V, Darian E, Fedorowicz A, Demchuk E, Li S, Singh H. Nearest-neighbor nonparametric method for estimating the configurational entropy of complex molecules. J Comput Chem. 2007;28:655–668. doi: 10.1002/jcc.20589. [DOI] [PubMed] [Google Scholar]
- 44.Hnizdo V, Tan J, Killian BJ, Gilson MK. Efficient calculation of configurational entropy from molecular simulations by combining the mutual-information expansion nearest-neighbor methods. J Comput Chem. 2008;29:1605–1614. doi: 10.1002/jcc.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrenberg AM, Swendsen RH. Optimized monte carlo data analysis. Phys Rev Lett. 1989;63:1195 – 1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 46.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. I. the method. J Comput Chem. 1992;13:1011 – 1021. [Google Scholar]
- 47.Rick SW. Increasing the efficiency of free energy calculations using parallel tempering and histogram reweighting. J Chem Theory Comput. 2006;2:939 – 946. doi: 10.1021/ct050207o. [DOI] [PubMed] [Google Scholar]
- 48.Ohkubo YZ, Brooks CL., III Exploring flory’s isolated-pair hypothesis: Statistical mechanics of helix-coil transitions in polyanaline and the c-peptide from rnase a. Proc Natl Acad Sci USA. 2003;100:13916–13921. doi: 10.1073/pnas.2334257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohkubo YZ, Thorpe IF. Evaluating the conformational entropy of macromolecules using an energy decomposition approach. J Chem Phys. 2006;124:024910. doi: 10.1063/1.2138696. [DOI] [PubMed] [Google Scholar]
- 50.Cheluvaraja S, Meirovitch H. Simulation method for calculating the entropy and free energy of peptides and proteins. Proc Natl Acad Sci USA. 2004;101:9241–9246. doi: 10.1073/pnas.0308201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheluvaraja S, Meirovitch H. Calculation of the entropy and free energy of peptides by molecular dynamics simulations using the hypothetical scanning molecular dynamics method. J Chem Phys. 2006;125:024905. doi: 10.1063/1.2208608. [DOI] [PubMed] [Google Scholar]
- 52.Reza FM. An Introduction to Information Theory. Dover Publications Inc; New York: 1994. [Google Scholar]
- 53.Dupré S, Volland C, Haguenauer-Tsapis R. Membrane transport: Ubiquitylation in endosomal sorting. Curr Biol. 2001;11:R932 – R934. doi: 10.1016/s0960-9822(01)00558-9. [DOI] [PubMed] [Google Scholar]
- 54.Pornillos O, Alam SL, Rich RL, Myszka DG, Davis DR, Sundquist WI. Structure and functional interactions of the TSG101 UEV domain. EMBO J. 2002;21(10):2397–2406. doi: 10.1093/emboj/21.10.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pornillos O, Higginson DS, Stray KM, Fisher RD, Garrus JE, Payne M, He GP, Wang HE, Morham SG, Sundquist WI. HIV gag mimics the TSG101-recruiting activity of the human hrs protein. J Cell Biol. 2003;162(3):425–434. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pornillos O, Alam SL, Davis DR, Sundquist WI. Structure of the TSG101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol. 2002;9(11):812–817. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- 57.VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA. Tsg101, a homologue of ubiquitin-conjugating (e2) enzymes, binds the L domain in HIV type 1 Pr55gag. Proc Natl Acad Sci USA. 2001;98:7724 – 7729. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Côté M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55 – 65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 59.Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nature Med. 2001;7:1313 – 1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 60.Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci USA. 2002;99(2):955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmer AG, III, Massi F. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev. 2006;106:1700 – 1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- 62.Chen W, Chang CE, Gilson MK. Concepts in receptor optimization: Targeting the RGD peptide. J Am Chem Soc. 2006;128:4675 – 4684. doi: 10.1021/ja056600l. [DOI] [PubMed] [Google Scholar]
- 63.Sundquist WI, Schubert HL, Kelly BN, Hill GC, Holton JM, Hill CP. Ubiquitin recognition by the human TSG101 protein. Mol Cell. 2004;13:783–789. doi: 10.1016/s1097-2765(04)00129-7. [DOI] [PubMed] [Google Scholar]
- 64.MacRaild CA, Daranas AH, Bronowska A, Homans SW. Global changes in local protein dynamics reduce the entropic cost of carbohydrate binding in the arabinose-binding protein. J Molec Biol. 2007;368:822–832. doi: 10.1016/j.jmb.2007.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yun SG, Jang DS, Kim DH, Choi KY, Lee HC. NMR relaxation studies of backbone dynamics in free and steroid-bound Delta(5)-3-ketosteroid isomerase from Pseudomonas testosteroni. Biochem. 2001;40:3967–3973. doi: 10.1021/bi0023192. [DOI] [PubMed] [Google Scholar]
- 66.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15NN MR relaxation. Biochem. 1994;33:5984 – 6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 67.Stivers JT, Abeygunawardana C, Mildvan AS, Whitman CP. 15NN MR relaxation studies of free and inhibitor-bound 4-oxalocrotonate tautomerase: Backbone Dynamics and Entropy Changes of an Enzyme upon Inhibitor Binding. Biochem. 1996;35:16036 – 16047. doi: 10.1021/bi961834q. [DOI] [PubMed] [Google Scholar]
- 68.Yu L, Zhu CX, Tse-Dinh YC, Fesik SW. Backbone dynamics of the C-terminal domain of escherichia coli Topoisomerase I in the absence and presence of single-stranded DNA. Biochem. 1996;35:9661 – 9666. doi: 10.1021/bi960507f. [DOI] [PubMed] [Google Scholar]
- 69.Yuan P, Marshall VP, Petzold GL, Poorman RA, Stockman BJ. Dynamics of stromelysin/inhibitor interactions studied by 15N NMR relaxations measurements: Comparison of ligand binding to the S1–S3 and subsites. J Biomol NMR. 1999;15:55 – 64. doi: 10.1023/a:1008398807718. [DOI] [PubMed] [Google Scholar]
- 70.Hawkins RJ, McLeish TC. Coarse-grained model of entropy allostery. Phys Rev Lett. 2004;93:098104. doi: 10.1103/PhysRevLett.93.098104. [DOI] [PubMed] [Google Scholar]
- 71.Tsai CJ, del Sol A, Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J Molec Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moon YI, Rajagopalan B, Lall U. Estimation of mutual infomation using kernal density estimators. Phys Rev E. 1995;52:2318 – 2321. doi: 10.1103/physreve.52.2318. [DOI] [PubMed] [Google Scholar]
- 73.Daub CO, Steuer R, Selbig J, Kloska S. Estimating mutual information using B-spline functions — an improved similarity measure for analyzing gene expression data. BMC Bioinformatics. 2004;5:118. doi: 10.1186/1471-2105-5-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kraskov A, Stögbauer H, Grassberger P. Estimating mutual information. Phys Rev E. 2004;69:066138. doi: 10.1103/PhysRevE.69.066138. [DOI] [PubMed] [Google Scholar]
- 75.Kraskov A, Stögbauer H, Andrezejak RG, Grassberger P. Hierarchical clustering using mutual information. Europhysics Lett. 2005;70:278 – 284. [Google Scholar]
- 76.Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007;36:21–72. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 77.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shannon CE. A mathematical theory of communication. Bell System Tech J. 1948;27:379–423. [Google Scholar]
- 79.Tolman RC. The Principles of Statistical Mechanics. Oxford University Press; London: 1938. [Google Scholar]
- 80.Jaynes ET. Gibbs vs boltzmann entropies. Am J Phys. 1965;33(5):391–398. [Google Scholar]
- 81.Matsuda H. Physical nature of higher-order mutual information: Intrinsic correlations and frustration. Phys Rev E. 2000;62:3096 – 3102. doi: 10.1103/physreve.62.3096. [DOI] [PubMed] [Google Scholar]
- 82.Edholm O, Berendsen HJC. Entropy estimation from simulations of non-diffusive systems. Mol Phys. 1984;51:1011 – 1028. [Google Scholar]
- 83.Potter MJ, Gilson MK. Coordinate systems and the calculation of molecular properties. J Phys Chem A. 2002;106:563–566. [Google Scholar]
- 84.Herschbach DR, Johnston HS, Rapp D. Molecular partition functions in terms of local properties. J Chem Phys. 1959;31(6):1652–1661. [Google Scholar]
- 85.Pitzer KS. Energy levels and thermodynamic functions for molecules with internal rotation.II.unsymmetrical tops attached to a rigid frame. J Chem Phys. 1946;14(4):239–243. [Google Scholar]
- 86.Gô N, Scheraga HA. On the use of classical statisitcal mechanics in the treatment of polymer chain conformation. Macromolecules. 1976;9:535–542. [Google Scholar]
- 87.Abagyan R, Totrov M, Kuznetsov D. ICM — a new method for protein modeling and design: Applications to docking and structure prediction from distrorted native conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]
- 88.Chang CE, Potter MJ, Gilson MK. Calculation of molecular configuration integrals. J Phys Chem B. 2003;107:1048–1055. [Google Scholar]
- 89.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berman HM, Henrick K, Nakamura H. Announcing the worldwide protein data bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. URL http://www.pdb.org. [DOI] [PubMed]
- 91.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Amber, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 92.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 93.Pang YP. Three-dimensional model of a substrate-bound SARS chymotrypsin-like cysteine proteinase predicted by multiple molecular dynamics simulations: Catalytic efficiency regulated by substrate binding. Proteins. 2004;57:747–757. doi: 10.1002/prot.20249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 96.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 97.Darden T, York D, Pedersen L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]