

Abstract
The coupling reaction between hindered thioacids and isonitriles is developed and described. The mechanism for the formation of the thiopyruvamide products is explored, and the method is applied to a selection of substrates.
Recently, our laboratory described a novel microwave-mediated coupling reaction between carboxylic acids and isonitriles leading to the formation of N-formyl amides.1 These N-formyl amides are readily converted to biologically important linkages such as amides, N-hydroxymethyl amides and N-methyl amides. 2 Further experimental 3 and computational 4 investigations showed that E and/or Z formimidate carboxylate mixed anhydrides (FCMAs), formed from the merger of 1 and 2, underwent [1,3]-O→N acyl migration to generate 3 via microwave thermolysis at 150 °C (Scheme 1). We have also reported on the coupling of carboxylic thioacids with isonitriles to provide N-thioformyl amides at ambient temperature.5 In this communication, we disclose some unexpected results in the context of attempts to extend the above findings to highly hindered substrates.
Scheme 1.

We first explored the feasibility of coupling t-butyl carboxylic acid 4 and t-butyl isonitrile 5 (Scheme 2A). Unfortunately, microwave irradiation of 4 with 5 in 1,2-dichloroethane (DCE) at 160 °C for 30 minutes did not afford N-formyl imide 8. Adjustments of reaction conditions such as temperature, reaction time, and molar ratios of reactants were not fruitful. The decreased reactivity observed in attempts to merge these hindered reactants may be ascribed to several causes. First, the formation of FCMA intermediate 6 might not occur with neopentylic coupling partners. Second, the bulky nature of the tert-butyl groups could undermine the critical [1, 3]-O→N migration en route to 8. The former of these possibilities was tested by introducing a nucleophile that could intercept the putative FCMA intermediate (6). Indeed, when the coupling reaction was repeated in the presence of p-methoxy benzylamine (9), amide 10 as well as formamide 11 were observed (Scheme 2B). The formation of these products suggested to us that FCMA intermediate 6 does form under these conditions, but that the rearrangement to provide the expected N-formyl imide 8 does not proceed.
Scheme 2.

We then turned our attention to the use of thiocarboxylic acids based on the following rationale: our precedents cited above5, 6 suggested that conversion of the thio-FCMA intermediate (13, Scheme 3A) to product is more rapid than that of the oxy-FCMA (6/7). Moreover, [1,3]-S→N acyl migration of the thio-FCMA might be more facile than is the case for its oxa analog.
Scheme 3.

Investigation of the coupling of hinderd thioacids began by treating t-butyl thioacid (12) with t-butyl isonitrile (5) at ambient temperature (Scheme 3A). Interestingly, when 2 equivalents of thioacid and 1 equivalent of isonitrile were employed, a 1:1 ratio of thioanhydride 15 7 and thioformamide 16 8 were formed, as determined by 1H NMR analysis of the crude reaction mixture. Subsequent purification afforded thioanhydride 15 in 60% yield and thioformamide 16 in 88% yield.9 The formation of these products was suggestive of successful formation of a thio-FCMA intermediate. Thus, we anticipated that microwave heating might facilitate the sluggish acyl shift, enabling access to N-thioformyl imine 14. However, heating two equivalents of thioacid 12 and one equivalent of isonitrile 5 in DCE in the microwave at 155 °C for 40 minutes did not lead to the formation of N-thioformyl imide 14. 10 Interestingly, upon reversing the stoichiometry of the starting materials (i.e. a 2:1 molar ratio of isonitrile 5: thioacid 14 was used) and repeating the reaction, a new coupling product was produced following microwave-induced thermolysis at 155°. Although two carbonyl-type signals were observed in the 13C NMR, the anticipated thioformyl signal in the 1H NMR was substantially shifted upfield and broadened. X-Ray analysis of a single crystal indicated that the product was not the N-thioformyl imide expected on the basis of precedent, but rather the known thiopyruvamide 17, 11 which had been formed in 43% yield.12
Intrigued by this unexpected result, we sought a better understanding of the mechanism for the formation of thiopyruvamide 17. In light of the rapid formation of thioanhydride 15 and thioformamide 16 at ambient temperatures, we investigated the role that these components might play in the transformation. Reaction of 15 and 16 in DCE at 150 °C in the microwave led only to low-yielding formation of a DCE insertion product10 and recovered starting materials (Scheme 4A). However, addition of 0.1 equivalents of isonitrile 5 led to detection of thiopyruvamide 17. Suspicious about the role of the excess isonitrile in both this transformation and the formation of 17 in Scheme 3B, thioanhydride 15 was subjected to the action of one equivalent of isonitrile 5 in DCE in the microwave at 150 °C (Scheme 4B). Indeed, thiopyruvamide 17 was obtained in 21% yield with 48% recovery of anhydride 15. Thus, we propose that in the reaction of thioacid 12 and isonitrile 5, thiopyruvamide 17 is formed by in-situ generation of thioanhydride 15. A mechanism for this transformation, which can be readily envisioned, is one where thioanhydride 15 is attacked by isonitrile 5 to generate intermediate 18 and an equivalent of thiocarboxylate 19. Addition of thiocarboxylate 19 to 18 would then afford adduct 20, which upon attack of a nucleophilic species (i.e., thiocarboxylate, isonitrile, or water), would lead to thiopyruvamide 17.
Scheme 4.

The generality of this interesting finding in the reaction of hindered partners 12 and 5 was probed further. Indeed, adamantyl thioacid 21 13 reacted with t-butyl isonitrile 5 to form thiopyruvamide 22 in 40% yield (Scheme 5). Similarly, t-butyl thioacid 12 and adamantyl isonitrile 23 were coupled to form thiopyruvamide 24 in 43% yield. Finally, the bis-adamantyl thiopyruvamide 25 could be reached from 21 and 23 in 42% yield. Based on the mechanisms proposed in Scheme 3A and Scheme 4, the maximum theoretical yield of the thiopyruvamide products is 50% (i.e., 1 mmol of thioacid will produce 0.5 mmol of thioanydride, the limiting reagent).
Scheme 5.

During the course of our investigations, we sought to cleave thiopyruvamide 17 using hydrolysis and methanolysis conditions. To our surprise, treatment of thiopyruvamide 17 with excess sodium methoxide (5–10 equivalents) in methanol, at either ambient temperature or even at reflux, did not afford any fragmentation products. Similarly, subjecting the thiopyruvamide to the action of lithium hydroxide in THF/H2O at 60 °C resulted in no apparent reaction. However, following treatment with sodium hydride in THF at reflux, 14 thiopyruvamide 17 suffered conversion to 16 in 88% yield (Scheme 6). One formalism for this unexpected transformation involves an initial deprotonation of the thiopyruvamide to give rise to delocalized anion 26. Attack of thiolate upon the carbonyl carbon would give rise to a thioepoxide intermediate 27 15 which could isomerize to thio-FCMA anion 28 en route to the FCMA intermediate (13). Hydrolysis of 13 would then yield thioformamide 16. We emphasize that the progression in Scheme 6 is intended as a mnemonic and is certainly not based on experimental evidence.
Scheme 6.

In summary, we have described the activation of extremely hindered thioacids with hindered isonitriles giving rise to thiopyruvamides (see 17, 22, 24 and 25). Presumably, these reactions proceed via putative thioformamidate carboxylate mixed anhydrides. At ambient temperature, such intermediates progress to thioanhydrides and thioformamides. Following microwave-induced thermolytic intervention, the thiopyruvamides are produced. Mechanistic investigations revealed that the active components in the formation of thiopyruvamides are actually the initially-formed thioanhydride and excess isonitrile. For operational simplicity, the thioacid and isonitrile may be employed directly. Efforts to incorporate these mechanistic insights in our overall isonitrile program are ongoing.
Supplementary Material
Acknowledgments
This work was supported by the NIH (CA103823 to S.J.D.). Special thanks to Dana Ryan for assistance with the preparation of the manuscript. We thank Dr. Pavel Nagorny, Dr. Jianglong Zhu, Dr. Xuechen Li and Dr. Cindy Kan for their helpful discussions. We thank Dr. George Sukenick, Ms. Hui Fang, and Ms. Sylvi Rusli of the Sloan-Kettering Institute’s core facility for mass spectral analysis and assistance with NMR spectroscopy. We are grateful to Dr. Louis Todaro of Hunter College for X-ray crystallographic analysis and Nathaniel Sherden for preparation of the ORTEP image.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Li X, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:5446–5448. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li X, Danishefsky SJ. Nature Protocols. 2008;3:1666–1670. doi: 10.1038/nprot.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee J, Gilon C, Hoffman A, Kessler H. Acc. Chem. Res. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- 3.(a) Li X, Yuan Y, Berkowitz WF, Todaro LJ, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:13222–13224. doi: 10.1021/ja8047078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li X, Yuan Y, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:13225–13227. doi: 10.1021/ja804709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Hou J-L, Ajami D, Rebek J., Jr J. Am. Chem. Soc. 2008;130:7810–7811. doi: 10.1021/ja802288k. [DOI] [PubMed] [Google Scholar]; (b) Restorp P, Rebek J., Jr J. Am. Chem. Soc. 2008;130:11850–11851. doi: 10.1021/ja803854r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jones GO, Li X, Hayden AE, Houk KN, Danishefsky SJ. Org. Lett. 2008;10:4093–4096. doi: 10.1021/ol8016287. [DOI] [PubMed] [Google Scholar]
- 5.Yuan Y, Zhu J, Li X, Wu X, Danishefsky SJ. Tetrahedron Lett. 2009;50:2329–2333. doi: 10.1016/j.tetlet.2009.02.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chupp JP, Leschinsky KL. J. Org. Chem. 1975;40:66–71. [Google Scholar]
- 7.Masumoto H, Tsutumi H, Kanda T, Komada M, Murai T, Kato S. Sulfur Lett. 1989;10:103–115. [Google Scholar]
- 8.(a) Kato S, Takagi T, Katada T, Mizuta M. Angew. Chem. 1977;89:820. [Google Scholar]; (b) Stowell JC, Ham BM, Esslinger MA, Duplantier AJ. J. Org. Chem. 1989;54:1212–1213. [Google Scholar]
- 9.The thioanhydride is not fully recovered owing to decomposition to the component thioacid during the chromatography event.
- 10.Small amounts (≤15% yield) of products formed by reaction with the solvent (chloride displacement) were observed in this case.
- 11.Adiwidjaja G, Günther H, Jürgen V. Liebigs Ann. Chem. 1983:1116–1132. [Google Scholar]
-
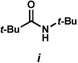
12.Extended microwave mediated thermolysis led to amide i (17:i = 1.6:1 in 29% overall yield). The mechanism of formation i, which formally would require extrusion of C=S from 17 is not established. However, it has not been shown that i in fact arises from 17 as opposed to some other pathway.
- 13.Toru T, Nishigaki M, Seko T, Kanefusa T, Maekawa E. Synthesis. 1985;9:878–879. [Google Scholar]
- 14.During reflux, the solvent was allowed to evaporate over 14 hours by the flow of nitrogen over the reaction mixture. No 4 Tetrahedron Letters reaction occured at the initial concentration (0.02M) after as much as 6 hours.
- 15.A similar intermediate is proposed in the following computational report: Norris KE, Bacskay GB, Gready JE. J. Comput. Chem. 1993;14:699–714.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.