

Abstract
Considerable data support a role for glycinergic ventromedial medulla neurons in the mediation of the postsynaptic inhibition of spinal motoneurons necessary for the motor atonia of rapid-eye movement (REM) sleep in cats. These data are, however, difficult to reconcile with the fact that large lesions of the rostral ventral medulla do not result in loss of REM atonia in rats. In the present study, we sought to clarify which medullary networks in rodents are responsible for REM motor atonia by retrogradely tracing inputs to the spinal ventral horn from the medulla, ablating these medullary sources to determine their effects on REM atonia and using transgenic mice to identify the neurotransmitter(s) involved. Our results reveal a restricted region within the ventromedial medulla, termed here the “supraolivary medulla” (SOM), which contains glutamatergic neurons that project to the spinal ventral horn. Cell-body specific lesions of the SOM resulted in an intermittent loss of muscle atonia, taking the form of exaggerated phasic muscle twitches, during REM sleep. A concomitant reduction in REM sleep time was observed in the SOM-lesioned animals. We next used mice with lox-P modified alleles of either the glutamate or GABA/glycine vesicular transporters to selectively eliminate glutamate or GABA/glycine neurotransmission from SOM neurons. Loss of SOM glutamate release, but not SOM GABA/glycine release, resulted in exaggerated muscle twitches during REM sleep that were similar to those observed after SOM lesions in rats. These findings, together, demonstrate that SOM glutamatergic neurons comprise key elements of the medullary circuitry mediating REM atonia.
Introduction
Muscle atonia is a defining characteristic of rapid eye movement (REM) sleep. Chase and colleagues have previously shown that muscle atonia during REM sleep is caused by glycine-mediated postsynaptic inhibition of spinal motoneurons (Chase et al., 1989; Chase and Morales, 1990). Other studies investigating the nature of supraspinal control of REM atonia have identified the dorsolateral pons and the medial medulla as two brainstem regions containing neural circuitry critical for producing REM atonia (for review, see Lai and Siegel, 1998, Chase and Morales, 2005). For example, in 1965 Jouvet and Delorme first reported that electrolytic lesions of the subceruleus region, located in the dorsolateral pons, resulted in REM sleep without atonia in cats (Jouvet and Delorme, 1965). REM sleep without atonia was characterized as a variety of simple and complex motor behaviors, which Jouvet and Delorme termed “oneiric behavior.” Recent studies from our laboratory have revealed the detailed nature of the pontine circuitry responsible for REM atonia (Lu et al., 2006). Specifically, we showed that spinally projecting, glutamatergic neurons located in the rat sublaterodorsal nucleus (SLD) are critical for producing REM atonia. On the other hand, many studies have suggested that the ventromedial medulla (VMM) could be the relay for the inhibitory actions of the pontine inhibitory area on spinal motor neurons (Sakai et al., 1981; Chase and Morales, 1990). The VMM has been hypothesized to contain premotor neurons, presumptively GABA/glycinergic, responsible for inhibition of spinal motor neurons (Holstege and Bongers, 1991; Fort et al., 1993; Rampon et al., 1996; Stornetta and Guyenet, 1999). In general support of this hypothesis, electrical and pharmacological stimulation of the medial medulla in decerebrate rats and cats produces muscle atonia (Lai and Siegel, 1988, 1992, 1997; Hajnik et al., 2000). A subset of medial medulla neurons in dogs is active only during periods of complete muscle atonia (Siegel et al., 1991) and chemical lesions of this region produced REM without atonia in cats (Schenkel and Siegel, 1989; Holmes and Jones, 1994). Moreover, glycine-immunoreactive neurons in the VMM express c-Fos during carbachol-induced REM sleep atonia (Morales et al., 2006). Collectively, these studies suggest that glycinergic neurons in the VMM may mediate motor atonia during REM sleep. Importantly, however, recent studies in rats have challenged this notion, instead suggesting that VMM neurons and GABA/glycine may not be critical in producing muscle atonia during REM sleep (Lu et al., 2006; Brooks and Peever, 2008). For example, normal REM atonia has been reported after large lesions of the medial medulla in rats (Lu et al., 2006), suggesting that REM atonia circuitry might be different in felines and rodents. Moreover, microdialysis application of the glycine antagonist, strychnine, and/or the GABAA antagonist, bicuculline, did not reverse the masseter muscle atonia during REM sleep in rats (Brooks and Peever, 2008). This latter study has proven particularly provocative and has challenged and stimulated debate over the long-standing hypothesized role of GABA/glycine in REM atonia (Chase, 2008; Funk, 2008; Kubin, 2008; Soja, 2008). In the present study, we sought to reexamine the supraspinal medullary networks responsible for motor atonia in rodents by retrogradely tracing inputs to the spinal ventral horn from the medulla, ablating these medullary sources to determine their effects on REM atonia and using transgenic mice to reveal the nature of the neurotransmitter(s) involved.
Materials and Methods
Animals
Pathogen-free adult male Sprague Dawley rats (300–350 g, Harlan) were used for experiments 1 and 2. For experiment 3, we used two different transgenic mouse models (25–35 g), which had lox P sites flanking exon 2 of either the vesicular glutamate transporter 2 (Vglut2) gene or the vesicular GABA transporter (Vgat) gene (Tong et al., 2007, 2008). The mice and rats were housed in individual cages. The cages were housed inside isolation chambers, which provided ventilation, computer-controlled lighting (12/12 light-dark cycle, lights on at 7:00 A.M.; 200 lux), an ambient temperature of 22 ± 1°C, and visual isolation. Care of the rats and mice in the experiment met National Institutes of Health standards, as set forth in the Guide for the Care and Use of Laboratory Animals, and all protocols were approved by the Beth Israel Deaconess Medical Center and Harvard Medical School Institutional Animal Care and Use Committees.
Experiment 1: Tracing studies
Surgery.
Under chloral hydrate anesthesia (7% in saline, 350 mg/kg), five rats received stereotaxic injections of Fluorogold [FG; 20 nl of a 5% (w/v) solution, Invitrogen) into the ventral horn of spinal cord (C8–T1 level). The injections were performed using a fine glass pipette (1 mm glass stock, tapering slowly to a 10–20 μm tip) and compressed air delivery system as described previously (Elmquist and Saper, 1996; Scammell et al., 1998).
Tissue preparation.
Seven days after the FG injections, the rats were killed by deep anesthesia with 500 mg/kg chloral hydrate and perfused through the heart with saline (100 ml), followed by 500 ml of neutral phosphate buffered formalin (Fischer Scientific). Brains were removed and incubated overnight in 20% sucrose and 0.01% sodium azide at 4°C until they sank. The brains were then sectioned in the coronal plane on a freezing microtome into four series of 40 μm sections and were stored at 4°C in PBS with 0.01% sodium azide.
In situ hybridization and immunohistochemistry.
Sections were washed in PBS, and incubated in primary antiserum (Goat anti-FG, 1:1000 dilution, Vector Labs) diluted in PBS containing 0.3% Triton X-100 and 0.2% sodium azide for 1 d at room temperature. Sections were then washed in PBS and incubated in biotinylated secondary antiserum (Donkey anti-goat IgG, 1:1000, Vector) in PBS for 1 h, washed in PBS, and incubated in ABC reagents for 1 h. Sections were then washed again and incubated in a solution of 0.06% 3,3-diaminobenzidine tetrahydrochloride (DAB, Sigma) and 0.02% H2O2.
For double staining of VGLUT2 mRNA and FG, we used a method we have previously described (Marcus et al., 2001; Chou et al., 2002; Lu et al., 2002, 2006) to label FG with an immunoperoxidase brown color and then VGLUT2 mRNA by autoradiography. Briefly, sections (40 μm thickness) were first stained immunohistochemically for FG (brown DAB reaction), as above, except that all solutions were DEPC treated to destroy RNAase. The sections were then acetylated and hybridized overnight (55°C) with a 35S-labeled cRNA probe synthesized from a plasmid containing the complete coding sequence of VGLUT2 mRNA. After a succession of 1 h washes [2× SSC/1 mm dithiothreitol (DTT), 50°C; 0.2× SSC/1 mm DTT, 55°C; 0.2× SSC/1 mm DTT, 60°C], the tissue was treated with RNAase-A (Boehringer Mannheim) and washed under conditions of increasing stringency, including a 30 min wash at 60°C in 0.1× SSC. The tissue was then dehydrated in alcohol and air-dried. The sections were exposed to x-ray film (Eastman-Kodak) for 2–3 d, and the slides were dipped in Kodak NTB-2 emulsion and exposed for 1 month. Slides were developed in Kodak D-19, fixed, and then dehydrated and coverslipped using Permaslip mounting medium (Alban Scientific).
Experiment 2: Orexin-saporin lesions of medial medullary regions
Surgery.
Under anesthesia, the rats (n = 35) were placed in the stereotaxic apparatus. A fine glass pipette containing orexin-saporin (0.1% solution, Advanced Targeting Systems) was lowered to one of six predetermined targets, including: supraolivary medulla [SOM, anteroposterior (AP) −12.8 dorsoventral (DV) −8.6 lateral (L) 0.0, 120 nl], rostral dorsal paragigantocellularis (rDPGi; AP −11.6 DV −7.5 L 0.0, 300 nl), caudal dorsal paragigantocellularis (cDPGi; AP −12.8, DV −7.8, L 0.0, 120 nl) medullary parvicellular reticular nucleus (PcRt; AP −12.75, DV −6.3 L ± 1.4, 120 nl), rostral ventral medulla (RVM; AP −11.9, DV −8.8, L 0, 120 nl) or lateral paragigantocellularis (LPGi; AP −12.8, DV −8, L ± 1.0, 200 nl) as per the rat atlas of Paxinos and Watson (2005). The orexin-saporin solution was injected using the compressed air delivery system described previously (Elmquist and Saper, 1996; Scammell et al., 1998). For the collection of sleep–wake data (i.e., EEG/EMG recordings), four EEG screw electrodes were implanted into the skull, in the frontal (two) and parietal bones (two) of each side, and two flexible EMG wire electrodes were placed into the neck muscles. The free ends of the leads were soldered into a socket that was attached to the skull with dental cement, and the incision was then closed by wound clips (Lu et al., 2000, 2002).
Sleep–wake recording and analysis.
After 2 weeks of postoperative recovery, the rats were connected via flexible recording cables to a commutator, which in turn was connected to a Grass polygraph and computer. The rats were habituated to the cables for 1 d. Continuous recording of the EEG/EMG and time-lock video began after the 24 h habituation period and continued uninterrupted for 48 h. The digitized EEG/EMG data of each rat was divided into 12 s epochs and visually scored as wake, non-REM (NREM) sleep or REM sleep. Scoring was done before histological examination, and so the scorers were unaware of the extent of the lesions. For staging, NREM sleep was identified by a preponderance of high-amplitude, low-frequency (<4 Hz) EEG activity and relatively low and unchanging EMG activity, whereas wakefulness was characterized by a preponderance of low-amplitude, fast EEG activity and highly variable muscle tone on EMG. REM sleep was identified by very low EMG activity and a low-amplitude monotonous EEG containing a predominance of theta range (4–7 Hz) EEG activity. When two states (for example, NREM sleep and wake) occurred within a 12 s epoch, the epoch was scored for the state that predominated (Lu et al., 2000). The percentage of time spent in wake, NREM sleep and REM sleep, and frequency and the episode durations of each stage were calculated. These individual data for each group was compared with those of control animals using Student's t test.
EMG quantification.
Integrated EMG for every 12 s epochs of the entire 24 h data was calculated for control and SOM-lesioned animals using SleepSign software. The average integral EMG values during different sleep–wake stages (i.e., wake, NREM and REM) were then calculated. The integrated EMG values during different sleep–wake states in SOM-lesioned animals were compared with the corresponding integrated EMG values of control animals using Student's t test. The integrated EMG values during REM sleep in both groups of animals were compared with those values during NREM sleep of the same group of animals. Finally, the integrated EMG values were calculated for all REM epochs without any phasic muscle activity (twitches), i.e., nonphasic REM sleep in SOM-lesioned animals and were compared with those from control animals using Student's t test.
Histology.
On completion of the recordings, the rats were killed by deep anesthesia with 500 mg/kg chloral hydrate and perfused through the heart with saline (100 ml), followed by 500 ml of neutral phosphate buffered formalin (Fischer Scientific). Brains were removed, incubated overnight in 20% sucrose and 0.01% sodium azide at 4°C until they sank. The brains were then sectioned in the coronal plane on a freezing microtome into four series of 40 μm sections and were stored at 4°C in PBS with 0.01% sodium azide.
One series was mounted on gelatin-coated slides, washed in H2O, and washed in PBS. Sections were then incubated in 0.25% thionin in 0.1 m acetate buffer solution for 2 min, differentiated in graded ethanols, and delipidated in xylene before being coverslipped using Permaslip mounting medium (Alban Scientific).
Experiment 3: Conditional knock-out of Vglut2 gene or Vgat from SOM in mice
Microinjection and recordings.
Experiment 3 was performed using mice with loxP sites flanking exon-2 of either the Vglut2 or Vgat gene (n = 4 each). To selectively eliminate glutamate and GABA/glycine neurotransmission by SOM neurons, we injected an adeno-associated viral (AAV) vector containing Cre recombinase (Cre) into the SOM (AP −7.0, DV 5.8, L 0.0) (Paxinos and Franklin, 2001) of the conditional Vglut2 and Vgat mice, respectively. When exposed to Cre, the second exon is deleted, and the gene is no longer capable of expressing functional protein and hence ultimately prevents neurotransmitter (glutamate or GABA/glycine) release from SOM neurons (Tong et al., 2007, 2008). As vector-injection controls, we also injected AAV containing green fluorescent protein (GFP) into the SOM of Vglut2 mice (n = 3). Injections of AAV-Cre or AAV-GFP into the SOM of these mice were done using a compressed air delivery system (Elmquist and Saper, 1996; Scammell et al., 1998). After injections of either AAV-Cre or AAV-GFP, the mice were implanted with biotelemetry units (TL11M-F20EET, Data Sciences International) for the chronic recording of sleep-wake. Briefly, the biotelemetry units were placed intraperitoneally, and the leads from the transmitter were routed subcutaneously to the skull. Two tiny holes (one in the frontal bone and another one on the parietal bone) were drilled and two EEG leads from the transmitters were secured in such a way that these leads were touching the dura mater. Two EMG leads were secured on to the neck muscles and skin was sutured. After 14 d of surgery and a day of habituation to the recording setup, the telemetric recording of EEG/EMG form these animals were performed for 48 h. Analysis of sleep–wakefulness and quantification of EMG were similar to the one performed for rat recordings.
In situ hybridization and immunohistochemistry. On completion of the recordings, the mice were deeply anesthetized with chloral hydrate and perfused transcardially with saline (50 ml) followed by 10% formalin (200 ml). The brains were cut in the coronal plane on a freezing microtome into three evenly spaced series of 30 μm sections. One series from each brain was processed either for VGLUT2 or for VGAT in situ hybridization, another one for Cre-immunohistochemistry (Lazarus et al., 2007), and the third for Nissl staining (Lu et al., 2000). One series of sections from the animals that received AAV-GFP were processed for GFP-immunohistochemistry as described previously (Lu et al., 2006), instead of Cre-immunohistochemistry.
Results
Tracing studies
After FG injections into the spinal cord ventral horn, we observed intense FG labeling in a confined region of the medulla, immediately above the inferior olive and extending through the level of the beginning of area postrema (Fig. 1A–D). This region included the ventral gigantocellularis (GiV) and medullary gigantocellular reticular field immediately dorsal to GiV, and therefore contained the gigantocellular reticular nucleus, the nucleus paramedianus, and the nucleus raphe obscurus along the midline. Double labeling of these sections with VGLUT2 riboprobe showed that ∼61.3% of all the FG-immunoreactive neurons within the SOM (378 of 616 neurons counted from 3 sections each in 2 animals where injections were restricted to the spinal cord ventral horn) contained VGLUT2 mRNA (Fig. 1E, F). We have termed this confined region of the medulla the SOM (Fig. 1A–C) based on the following criteria: (1) the presence of VGLUT2-containing neurons; (2) direct projections to the spinal cord ventral horn; and (3) function (see Lesion studies below).
Figure 1.

A–C, Coronal sections (A, B) and sagittal section (C) from the rat atlas of Paxinos and Watson (2005) showing the extent of the SOM (shaded area); the SOM region included the GiV and medullary gigantocellular reticular nucleus (Gi), nucleus paramedianus, and nucleus raphe obscurus that lies immediately dorsal to GiV. Inset in C shows the FG injection site in the ventral horn of the spinal cord at C8–T1 level. D, Retrogradely labeled (FG-immunoreactive) neurons (brown) within the SOM after spinal ventral horn injections of FG; E, double-labeling of FG-ir neurons (brown) for the presence of VGLUT2 mRNA showed that an average of 61% of FG-immunoreactive neurons also contained VGLUT2 mRNA (black puncta) thus marking them as glutamatergic. F, High power photomicrograph of the boxed region in E. Arrows indicate double-labeled (FG and VGLUT2) neurons in the SOM and arrowheads indicate single-labeled (FG) neurons. Inset in F shows enlarged view of two neurons underneath the asterisk—one single-labeled (FG) and one double-labeled (FG and VGLUT2). GiA, Gigantocellular reticular nucleus, α part; IO, inferior olive; py, pyramidal tract; IRt, intermediate reticular nucleus. Scale bars: D–F, 100 μm.
Lesion studies
Cell-body specific lesions of the SOM and surrounding medullary areas were made using orexin-saporin. Nissl-stained sections of these brains revealed complete loss of neurons in the site of injection (see Fig. 3B). The extent of lesions in 8 animals, which had lesions restricted to the SOM, is depicted schematically in Figure 2. Analysis of sleep–wake and the video recordings showed that the animals with SOM lesions had high amplitude phasic muscle activity during REM sleep. These high-amplitude phasic muscle activities were rapid, jerky, and visible (in the video) twitches that ranged in frequency from 0.5 to 16 per min of REM sleep (on average ∼5 myoclonic jerks/min of REM sleep) (Fig. 3G). Several animals also demonstrated abrupt, violent phasic activity including accelerating themselves into the side of the cage while in REM sleep. SOM lesions, however, did not result in any coordinated behavior such as walking while in REM sleep, such as that observed after SLD lesions (Lu et al., 2006). Thus, whereas most of the movements seen in the SOM-lesioned rats were rapid and jerky, slow head rising or whole body movements were also observed occasionally after SOM lesions. Quantitative analysis of the EMG showed that the integrated EMG during NREM and REM sleep in these animals were higher than that of control animals whereas the waking EMG tone was not significantly different (Fig. 3C–E). The integrated EMG during REM sleep in SOM-lesioned animals was higher than that observed during NREM sleep, but this did not reach statistical significance (Fig. 3C). This increase in EMG tone during REM was mainly caused by the phasic twitches and the various body movements observed during REM sleep (Fig. 3G). However, the tonic or nonphasic muscle atonia during REM sleep was still present as evident from the EMG records (Fig 3G). We next compared the integral EMG of all nonphasic REM epochs (which comprised 38% of total REM sleep time in SOM-lesioned animals) in the SOM-lesioned animals with that of control animals. This analysis revealed that muscle tone during nonphasic REM sleep was in fact higher in SOM-lesioned animals (12.52 ± 2.1 mV·s) compared with control animals (4.41 ± 0.67 mV·s). The magnitude of the change in muscle tone from NREM to nonphasic REM sleep, calculated as the ratio of EMG tone of these two distinct stages, was not however significantly different between SOM-lesioned (54.51 ± 7.84%) and control animals (51.82 ± 4.51%).
Figure 3.

A, B, Nissl staining of rat brain section at the level of SOM in control animal showing healthy cells after sham injections (A) and loss of these cells from the SOM region after orexin-saporin injections (B). Scale bars, 250 μm. C, Integrated EMG values (mV·s) during different stages of sleep–wakefulness (i.e., wake, NREM and REM sleep) in control and SOM-lesioned animals. The integrated EMG values during NREM and REM in SOM-lesioned animals were significantly higher than that of control animals. Values are mean ± SEM, **p < 0.01. However, the magnitude of depression of EMG tone from NREM to nonphasic REM was not significantly altered (see Results, Lesion studies), suggesting the SOM-lesioned animals could decrease muscle tone during REM as effectively as control animals, although they were unable to sustain this muscle atonia throughout the complete REM episode. D, E, EEG and EMG traces during wakefulness in control and SOM-lesioned animals, respectively, showing the waking EMG in SOM-lesioned animals were not different from control animals. F, EEG and EMG traces from a control animal during NREM and REM sleep displaying complete muscle atonia in neck muscles during REM sleep. G, EEG and EMG traces from a SOM-lesioned animal during NREM and REM sleep. These animals displayed intermittent loss of atonia, taking the form of exaggerated phasic muscle twitches (as indicated by arrows) while in REM sleep.
Figure 2.

Coronal sections from Paxinos and Watson (2005) atlas showing the extent of the orexin-saporin lesions in eight animals that had complete SOM lesions. Often the lesions extended slightly to adjoining regions. However, lesions of the surrounding regions (data not shown in the figure) had no effect on muscle atonia or REM sleep.
Although SOM-lesioned animals displayed alterations in NREM and REM muscle activity, the EMG tone during wakefulness was comparable to that of control animals (Fig. 3C–E). With the exception of a rotarod test, in which SOM-lesioned animals performed comparably with control animals (R. Vetrivelan and J. Lu, unpublished observations), the SOM-lesioned animals were not tested for changes in motor behaviors. Importantly, however, time-lock video capture of the SOM-lesioned animals did not reveal any motoric deficits or abnormal motor behaviors compared with the control animals.
In addition to the exaggerated phasic activity during REM sleep, SOM lesions also produced large reductions (46%) in REM sleep (Table 1). The total sleep (REM + NREM) time, however, remained unaffected. This decrease in REM sleep was primarily attributable to a reduction in the frequency of the REM episodes (45.38 ± 6.47 in SOM-lesioned animals vs 74.6 ± 7.22 in the control animals). The mean duration of REM sleep was unaffected in the SOM-lesioned animals (74.25 ± 5.98 s in SOM-lesioned animals vs 85 ± 6.69 s in the control animals).
Table 1.
Changes in percentages of sleep–wake stages after the lesions of SOM and surrounding regions
| Lesion area | Wake (%) | NREM sleep (%) | REM % |
|---|---|---|---|
| Control (no lesions) (n = 6) | 48.83 ± 1.72 | 43.93 ± 1.46 | 7.23 ± 0.43 |
| SOM (n = 8) | 46.79 ± 1.98 | 49.21 ± 2.12 | 4.00 ± 0.31*** |
| RVM (n = 4) | 49.5 ± 0.42 | 44.3 ± 0.91 | 6.21 ± 0.49 |
| PcRt (n = 4) | 49.19 ± 3.49 | 43.64 ± 2.92 | 7.17 ± 1.05 |
| rDPGi (n = 6) | 49.59 ± 1.28 | 44.17 ± 0.86 | 6.25 ± 0.43 |
| cDPGi (n = 3) | 47.93 ± 0.6 | 45.88 ± 0.93 | 6.18 ± 0.61 |
| LPGi (n = 4) | 50.77 ± 2.40 | 41.29 ± 3.08 | 7.93 ± 0.92 |
Values are mean ± SEM. ***p < 0.001 compared with the controls.
In contrast to SOM-lesioned animals, lesions in the surrounding regions that included the RVM, PcRt, rDPGi, cDPGi or LPGi altered neither the amounts of REM sleep (Table 1) nor the muscle atonia during REM sleep (data not shown).
Conditional knock-out of neurotransmitters from SOM
Injection of AAV-Cre into the SOM of floxed Vglut2 and Vgat mice resulted in complete loss of VGLUT2 signals and VGAT signals (assessed by in situ hybridization) (Fig. 4B, D) in the SOM. In addition, the spread of injections in all the mice brains was verified by Cre immunolabeling (Fig. 4A,C).
Figure 4.
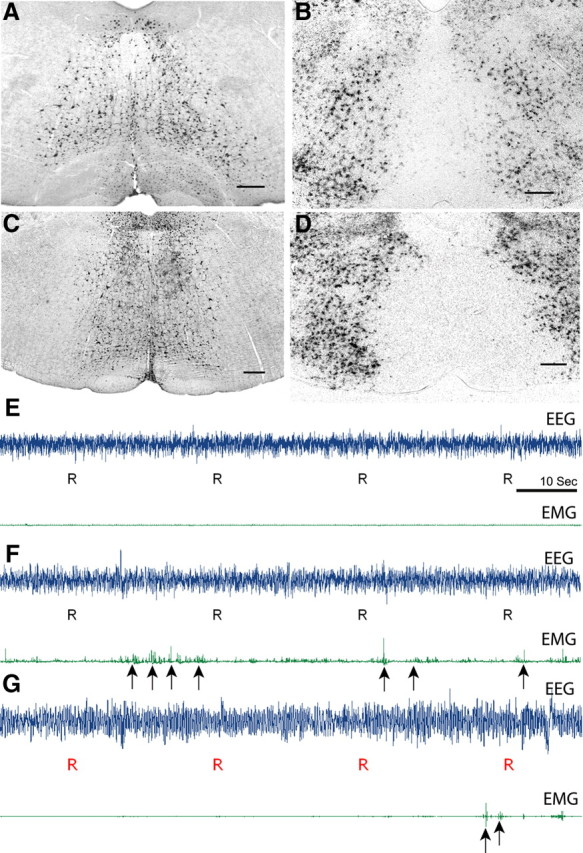
Focal and selective elimination of glutamate and GABA/glycine release from the SOM was achieved using mice with lox-P modified alleles of either the glutamate or GABA/glycine vesicular transporters. Injection of AAV-Cre into the SOM in these mice produces a nonfunctioning transporter protein and ultimately prevents neurotransmitter (glutamate or GABA) release from SOM neurons. A, C, Injection sites into the SOM in a floxed Vglut2 mouse (A) and Vgat mouse (C) as evidenced by Cre-immunostaining. B, D, Loss of VGLUT2 mRNA signals (B) and VGAT mRNA signals (D) in the corresponding region as evidenced by loss of in situ hybridization signals (black puncta) in the Vglut2 and Vgat mice, respectively. E–G, EEG, EMG traces during REM sleep in a control mouse injected with AAV-GFP (E) and from a floxed Vglut2 mouse (F) and a floxed Vgat mouse (G) after the injection of AAV-Cre. Specific elimination of Vglut2 from the SOM neurons resulted in exaggerated phasic muscle twitches (indicated by arrows) during REM sleep. In contrast, elimination of Vgat neurotransmission had minor effects on REM atonia. Scale bar, 300 μm.
Analysis of EEG/EMG recordings showed that selective elimination of glutamate release from SOM neurons after AAV-Cre injections into floxed Vglut2 mice produced exaggerated phasic muscle activity during REM sleep similar to that observed after SOM lesions in rats (Fig. 4F). The integral EMG values during REM sleep were higher in floxed-Vglut2 mice after the injection of AAV-Cre (8.25 ± 1.94 mV · s), when compared with the mice that received AAV-GFP injections (4.45 ± 1.42 mV·s). The drop in muscle tone during the transition from wake to NREM was still present in cre-injected Vglut2 mice and was comparable to control animals. However, the drop in muscle tone during the transition from NREM to REM was less pronounced in these mice, mainly because of exaggerated muscle twitches observed during REM sleep (Fig. 4). The magnitude of decrease in the integral EMG values between NREM and REM was 0.2 ± 0.41 mV·s in the floxed Vglut2 mice that received AAV-Cre injection compared with 3.00 ± 0.58 mV·s in the mice that received AAV-GFP injections into the SOM. In addition, the integral EMG during nonphasic REM sleep in the cre-injected Vglut2 mice (5.45 ± 2.5 mV·s) was not significantly different from that of control mice that received AAV-GFP into the SOM (4.41 ± 1.45 mV·s).
The floxed Vgat mice that received AAV-Cre injections into the SOM also displayed brief phasic muscle twitches (Fig. 4G) although the integral EMG values during REM sleep in these mice (3.37 ± 0.57 mV·s) were comparable to those of control mice (4.45 ± 1.42 mV·s). The drop in muscle tone during the transition from wake to NREM and from NREM to REM was also not significantly different from control mice. Finally, the integral EMG tone during wakefulness in floxed Vglut2 and Vgat mice that received cre injections into the SOM (24.1 ± 5.7 mV·s and 33.06 ± 5.47 mV·s respectively) did not differ from that of control mice that received AAV-GFP into the SOM (33.9 ± 4.8 mV·s). In addition, we did not observe any abnormal motor behavior during wakefulness in these mice.
There was no change in any of the sleep–wake parameters between the mice that received AAV-GFP and AAV-Cre (both floxed Vglut2 and Vgat). The percentage of REM sleep in these mice was comparable to those of control mice (7.32 ± 1.48% in floxed Vglut2 mice and 6.74 ± 0.15% in floxed Vgat mice and 6.92 ± 0.40% in control mice).
Discussion
Using combined retrograde labeling and in situ hybridization, we have shown that the medullary neurons projecting to the ventral horn of the spinal cord are mostly glutamatergic. Cell-body specific lesions of the medullary region containing these glutamatergic neurons, termed here the SOM, resulted in increased phasic muscle activity during REM sleep and a concomitant reduction in the amount of REM sleep. Interestingly, muscle tone during NREM sleep was also higher in the SOM-lesioned animals. Selective and focal elimination of glutamatergic, but not GABA/ glycinergic, neurotransmission by SOM neurons produced exaggerated phasic muscle activity during REM sleep in mice, similar to that observed in rats with cell-body specific SOM lesions. Loss of neither SOM glutamatergic neurotransmission nor SOM GABA/glycinergic neurotransmission, however, altered total REM sleep time.
SOM REM atonia control
The myoclonic twitches and increased phasic activity in the neck EMG observed after SOM lesions suggests that the integrity of the SOM is critical for the maintenance of muscle atonia and for suppressing phasic twitching during REM sleep in rats. This region which represents an anatomically restricted rostral–caudal segment of the gigantocellular reticular field and corresponds to the “medullary inhibitory area” proposed by Magoun and Rhines (1946) has previously been implicated in the regulation of REM sleep atonia in cats (Lai and Siegel, 1988). In addition, neurotoxic lesions of this medullary region in cats resulted in REM without atonia (Schenkel and Siegel, 1989; Holmes and Jones, 1994). This is the first study to show a similar phenomenon in rats. Interestingly, this is different from what we had previously reported when we placed lesions in the RVM (Lu et al., 2006). This apparent incongruity is however readily explained by the fact that the RVM is located ∼1 mm rostral to the SOM. This explanation is also consistent with the fact that the RVM projects primarily to the dorsal horn (Skagerberg and Björklund, 1985). In contrast, the SOM region projects heavily to the ventral horn and a major percentage of the projection neurons are glutamatergic, as shown in our present tracing studies. Although we injected FG at the C8-T1 level for our tracing experiments, the reticulospinal neurons in this medullary region has been shown to project to the entire length of the spinal cord (Holstege, 1996). Moreover, only the lesions restricted to the SOM, but not RVM or surrounding regions, resulted in increased phasic muscle activity during REM in rats showing that medullary neurons involved in REM atonia control are confined to the SOM.
Although myoclonic twitches and occasional whole body movements were observed during REM sleep, the nonphasic or tonic component of muscle atonia was still observed in the SOM-lesioned animals. The EMG tone during nonphasic REM sleep was higher in SOM-lesioned animals; however, the EMG tone was also higher during NREM sleep. Hence, the SOM or medial medulla in general may not be the ‘final common pathway’ for the REM atonia process, at least in rats, as has been hypothesized in cats. Nevertheless a critical role for the SOM in the REM atonia process in rats is still supported by the dramatic increase in the number of phasic twitches in the SOM-lesioned animals. We hypothesize that the increased number of phasic twitches in the SOM-lesioned animals occurs because the excitatory drives responsible for these twitches can more readily “break through” the nonphasic REM atonia in the absence of the SOM. Conversely, it can also be argued that the increase in both REM twitches and EMG tone during nonphasic REM may be related to a general enhancement of EMG tone across states. The lack of an increase in EMG tone during wakefulness and the absence of any abnormal motor behavior in SOM-lesioned animals do not, however, support this hypothesis.
Arguably the most important finding of this study is that the SOM neurons responsible for suppressing REM related phasic muscle activity are primarily glutamatergic. The VMM, of which the SOM is a part, contains a mixture of excitatory and inhibitory descending projections. It has been previously proposed that descending projections from the medial medulla inhibit spinal motor neurons during REM sleep using the inhibitory neurotransmitters GABA and glycine. Although both glutamatergic and GABA/glycinergic neurons in the SOM project to the spinal cord (Holstege and Bongers, 1991; Stornetta and Guyenet, 1999) (the present study), selective and focal elimination of glutamate, but not GABA/glycine, release from SOM neurons in mice resulted in exaggerated phasic muscle activity similar to that observed after SOM lesions. Hence, it appears that glutamatergic neurons in this region are important for REM atonia control. Interestingly, very few neurons in the ventromedial medullary SOM region project directly to motoneurons in the spinal ventral horn. Rather, a considerable percentage of these neurons project to interneurons (Drew and Rossignol, 1990; Takakusaki et al., 2001). It has been further demonstrated that reticulospinal neurons in the medulla evoked disynaptic IPSPs in extensor and flexor motoneurons (Takakusaki et al., 1989, 1994). The interneurons that mediate this motor inhibition are located in the ventromedial portion of lamina VII and a large percentage of these interneurons are monosynaptically excited from the medial medulla (Takakusaki et al., 2001, 2003). Consistent with the electrophysiological findings, c-Fos expression is increased in these interneurons during carbachol-induced motor inhibition (Yamuy et al., 1991). Our model therefore predicts that glutamatergic SOM neurons excite the GABA/glycinergic interneurons within the spinal ventral horn to control REM atonia.
Another important issue that needs to be considered is that the loss of neither glutamate nor GABA/glycine from SOM neurons completely eliminated REM atonia. To this end, we offer the following two possible explanations: (1) Direct glutamatergic projections from the SLD to spinal cord may be sufficient to generate nonphasic REM atonia (Lu et al., 2006). While the SLD projects to the SOM, these projections appear to arise from different SLD neurons than those projecting to spinal cord interneurons. For example, when two different retrograde tracers were simultaneously injected into the spinal ventral horn and SOM, we found very few doubly labeled neurons in the SLD (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that separate neuronal population in the SLD project to the SOM and spinal cord and therefore comprise two independent pathways involved in REM motor atonia control (Fig. 5). (2) Generation and maintenance of REM atonia are under the influence of many cell groups (in addition to SLD and SOM) and neurotransmitters (in addition to glutamate and GABA/glycine) (Siegel, 2005a). For example, in addition to active inhibition by GABA/glycine, dysfacilitation of serotonergic and noradrenergic neurotransmission has been demonstrated in the depression of hypoglossal motoneuronal activity during REM sleep (Fenik et al., 2004). Hence, simultaneous withdrawal of one or all of the neurotransmitters, e.g., norepinephrine and serotonin, along with the activation of GABA/glycinergic spinal cord interneurons by the SLD and SOM glutamatergic neurons might collectively orchestrate the REM atonia process.
Figure 5.

Two independent pathways emanating from the SLD participate in REM atonia control. The first pathway comprises the direct projections to spinal cord ventral horn and the second pathway is via a relay in the SOM. Both pathways use the neurotransmitter glutamate and excite GABA/glycinergic interneurons in the spinal cord to produce motor inhibition. Many other brainstem structures and neurotransmitters, such as serotoninergic and noradrenergic neurotransmission and dysfacilitation thereof, as suggested by Siegel (2005a) (data not shown in the figure), might be involved in generation and maintenance of the REM atonia along with these two pathways.
SOM REM sleep control
The dramatic reduction in total REM sleep time observed after SOM lesions indicates an important role for this region in REM control. As discussed in the previous section, animals with SOM lesions also displayed myoclonic jerks and exaggerated phasic twitches while in REM sleep, which could possibly interrupt this stage of sleep and produce an apparent reduction in total REM sleep time. We feel this explanation is unsatisfactory however, because of the following observations: (1) it was the frequency of REM sleep episodes and not episode duration that was affected by the lesions; and (2) glutamate knock-out from the SOM in mice (see Results and below), despite producing exaggerated muscle twitches, did not produce a significant decrease in REM sleep.
The large reduction in REM sleep percentage in SOM-lesioned animals raises concerns over the concept of a unitary REM sleep center in the dorsolateral pons, as suggested by previous studies (Jones, 1991; Siegel, 2005b; Lu et al., 2006; Luppi et al., 2006). Many previous studies have also shown the importance of medullary structures in the executive mechanisms of REM sleep. For example, REM sleep is totally abolished after transections at the level of the pontomedullary junction both in cats and rats (Webster et al., 1986, Gottesmann et al., 1995). Moreover, pontine carbachol administration does not elicit REM sleep after these transections (Vanni-Mercier et al., 1991). Large quisqualic acid lesions in the medial medullary reticular formation in cats also produced a reduction in REM sleep (Holmes and Jones, 1994).
Unlike SOM lesions, conditional knock-out of glutamate or GABA/glycine from SOM neurons did not result in a reduction in REM sleep time. On the other hand, Goutagny et al., (2008) and Sapin et al. (2009) have recently demonstrated increased Fos expression in GABAergic neurons in several medullary regions including the SOM region, DPGi, and LPGi during REM sleep rebound. Nevertheless, as we show, lesions of the DPGi or LPGi did not alter the amount of REM sleep. Although SOM lesions reduced REM sleep, specific elimination of GABA/glycine from the SOM did not result in a reduction in total REM sleep time, suggesting that GABA/glycinergic neurons in this region do not participate in the executive control of REM sleep. In addition to glutamate and GABAergic neurons, the SOM contains several other neurotransmitter phenotypes such as nociceptinergic, cholinergic, serotonergic, and nitric oxide synthase-containing cell groups (Holmes et al., 1994; Mason, 2001). Future studies are therefore needed to characterize the neuronal cell group in the SOM responsible for REM sleep regulation.
Footnotes
This work was supported by National Institutes of Health Grants NS 051609, HL60292, NS 33987, AG 09975, and NS 062727. We thank Quan Hue Ha and Minh Ha for their excellent technical support.
References
- Brooks PL, Peever JH. Glycinergic and GABA(A)-mediated inhibition of somatic motoneurons does not mediate rapid eye movement sleep motor atonia. J Neurosci. 2008;28:3535–3545. doi: 10.1523/JNEUROSCI.5023-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH. Confirmation of the consensus that glycinergic postsynaptic inhibition is responsible for the atonia of REM sleep. Sleep. 2008;31:1487–1491. doi: 10.1093/sleep/31.11.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH, Morales FR. The atonia and myoclonia of active (REM) sleep. Annu Rev Psychol. 1990;41:557–584. doi: 10.1146/annurev.ps.41.020190.003013. [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR. Control of motoneurons during sleep. In: Kryger MK, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Ed 4. Philadelphia: Saunders; 2005. pp. 154–168. [Google Scholar]
- Chase MH, Soja PJ, Morales FR. Evidence that glycine mediates the postsynaptic potentials that inhibit lumbar motoneurons during the atonia of active sleep. J Neurosci. 1989;9:743–751. doi: 10.1523/JNEUROSCI.09-03-00743.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–990. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew T, Rossignol S. Functional organization within the medullary reticular formation of intact unanesthetized cat. I. Movements evoked by microstimulation. J Neurophysiol. 1990;64:767–781. doi: 10.1152/jn.1990.64.3.767. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Saper CB. Activation of neurons projecting to the paraventricular hypothalamic nucleus by intravenous lipopolysaccharide. J Comp Neurol. 1996;374:315–331. doi: 10.1002/(SICI)1096-9861(19961021)374:3<315::AID-CNE1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Fenik V, Davies RO, Kubin L. Combined antagonism of aminergic excitatory and amino acid inhibitory receptors in the XII nucleus abolishes REM sleep-like depression of hypoglossal motoneuronal activity. Arch Ital Biol. 2004;142:237–249. [PubMed] [Google Scholar]
- Fort P, Luppi PH, Jouvet M. Glycine-immunoreactive neurones in the cat brain stem reticular formation. Neuroreport. 1993;4:1123–1126. [PubMed] [Google Scholar]
- Funk GD. Are all motoneurons created equal in the eyes of REM sleep and the mechanisms of muscle atonia? Sleep. 2008;31:1479–1482. doi: 10.1093/sleep/31.11.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesmann C, Gandolfo G, Zernicki B. Sleep-waking cycle in chronic rat preparations with brain stem transected at the caudopontine level. Brain Res Bull. 1995;36:573–580. doi: 10.1016/0361-9230(94)00247-x. [DOI] [PubMed] [Google Scholar]
- Goutagny R, Luppi PH, Salvert D, Lapray D, Gervasoni D, Fort P. Role of the dorsal paragigantocellular reticular nucleus in paradoxical (rapid eye movement) sleep generation: a combined electrophysiological and anatomical study in the rat. Neuroscience. 2008;152:849–857. doi: 10.1016/j.neuroscience.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Hajnik T, Lai YY, Siegel JM. Atonia-related regions in the rodent pons and medulla. J Neurophysiol. 2000;84:1942–1948. doi: 10.1152/jn.2000.84.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes CJ, Jones BE. Importance of cholinergic, GABAergic, serotonergic and other neurons in the medial medullary reticular formation for sleep-wake states studied by cytotoxic lesions in the cat. Neuroscience. 1994;62:1179–1200. doi: 10.1016/0306-4522(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Holmes CJ, Mainville LS, Jones BE. Distribution of cholinergic, GABAergic and serotonergic neurons in the medial medullary reticular formation and their projections studied by cytotoxic lesions in the cat. Neuroscience. 1994;62:1155–1178. doi: 10.1016/0306-4522(94)90351-4. [DOI] [PubMed] [Google Scholar]
- Holstege JC. The ventro-medial medullary projections to spinal motoneurons: ultrastructure, transmitters and functional aspects. Prog Brain Res. 1996;107:159–181. doi: 10.1016/s0079-6123(08)61864-6. [DOI] [PubMed] [Google Scholar]
- Holstege JC, Bongers CM. A glycinergic projection from the ventromedial lower brainstem to spinal motoneurons. An ultrastructural double labeling study in rat. Brain Res. 1991;566:308–315. doi: 10.1016/0006-8993(91)91715-d. [DOI] [PubMed] [Google Scholar]
- Jones BE. Paradoxical sleep and its chemical/structural substrates in the brain. Neuroscience. 1991;40:637–656. doi: 10.1016/0306-4522(91)90002-6. [DOI] [PubMed] [Google Scholar]
- Jouvet M, Delorme F. Locus coeruleus et sommeil paradoxal. C R Soc Biol. 1965;159:895–899. [Google Scholar]
- Kubin L. Adventures and tribulations in the search for the mechanisms of the atonia of REM sleep. Sleep. 2008;31:1473–1476. doi: 10.1093/sleep/31.11.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Medullary regions mediating atonia. J Neurosci. 1988;8:4790–4796. doi: 10.1523/JNEUROSCI.08-12-04790.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Corticotropin-releasing factor mediated muscle atonia in pons and medulla. Brain Res. 1992;575:63–68. doi: 10.1016/0006-8993(92)90423-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Brainstem-mediated locomotion and myoclonic jerks. II Pharmacological effects. Brain Res. 1997;745:265–270. doi: 10.1016/s0006-8993(96)01180-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Muscle atonia and REM sleep. In: Mallick BN, Inoue S, editors. Rapid eye movement sleep. New Delhi: Narosa; 1998. pp. 69–90. [Google Scholar]
- Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, Saper CB. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci. 2007;10:1131–1133. doi: 10.1038/nn1949. [DOI] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–3842. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bjorkum AA, Xu M, Gaus SE, Shiromani PJ, Saper CB. Selective activation of the extended ventrolateral preoptic nucleus during rapid eye movement sleep. J Neurosci. 2002;22:4568–4576. doi: 10.1523/JNEUROSCI.22-11-04568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Gervasoni D, Verret L, Goutagny R, Peyron C, Salvert D, Leger L, Fort P. Paradoxical (REM) sleep genesis: the switch from an aminergic-cholinergic to a GABAergic-glutamatergic hypothesis. J Physiol Paris. 2006;100:271–283. doi: 10.1016/j.jphysparis.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Magoun HW, Rhines R. An introductory mechanism in the bulbar reticular formation. J Neurophysiol. 1946;9:165–171. doi: 10.1152/jn.1946.9.3.165. [DOI] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- Mason P. Contributions of the medullary raphe and ventromedial reticular region to pain modulation and other homeostatic functions. Annu Rev Neurosci. 2001;24:737–777. doi: 10.1146/annurev.neuro.24.1.737. [DOI] [PubMed] [Google Scholar]
- Morales FR, Sampogna S, Rampon C, Luppi PH, Chase MH. Brainstem glycinergic neurons and their activation during active (rapid eye movement) sleep in the cat. Neuroscience. 2006;142:37–47. doi: 10.1016/j.neuroscience.2006.05.066. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. New York: Academic; 2005. [DOI] [PubMed] [Google Scholar]
- Rampon C, Luppi PH, Fort P, Peyron C, Jouvet M. Distribution of glycine-immunoreactive cell bodies and fibers in the rat brain. Neuroscience. 1996;75:737–755. doi: 10.1016/0306-4522(96)00278-3. [DOI] [PubMed] [Google Scholar]
- Sakai K, Sastre JP, Kanamori N, Jouvet M. State-specific neurons in the ponto-medullary reticular formation with special reference to the postural atonia during paradoxical sleep in the cat. In: Pompeiano O, Ajmone Marsan C, editors. Brain mechanisms and perceptual awareness. New York: Raven; 1981. pp. 405–429. [Google Scholar]
- Sapin E, Lapray D, Bérod A, Goutagny R, Léger L, Ravassard P, Clément O, Hanriot L, Fort P, Luppi PH. Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS ONE. 2009;4:e4272. doi: 10.1371/journal.pone.0004272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE, Griffin JD, Elmquist JK, Saper CB. Microinjection of a cyclooxygenase inhibitor into the anteroventral preoptic region attenuates LPS fever. Am J Physiol. 1998;274:R783–R789. doi: 10.1152/ajpregu.1998.274.3.R783. [DOI] [PubMed] [Google Scholar]
- Schenkel E, Siegel JM. REM sleep without atonia after lesions of the medial medulla. Neurosci Lett. 1989;98:159–165. doi: 10.1016/0304-3940(89)90503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM. Control of muscle tone across the sleep-wake cycle. In: Parmeggiani PL, Velluti RA, editors. The physiologic nature of sleep. London: Imperial College; 2005a. pp. 281–302. [Google Scholar]
- Siegel JM. REM sleep. In: Kryger MK, Roth T, Dement WC, editors. Principles and pactice of seep mdicine. Ed 4. Philadelphia: Saunders; 2005b. pp. 120–135. [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer HM, Paul R, Shiromani P, Dement WC, Mignot E, Chiu C. Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science. 1991;252:1315–1318. doi: 10.1126/science.1925546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skagerberg G, Björklund A. Topographic principles in the spinal projections of serotonergic and non-serotonergic brainstem neurons in the rat. Neuroscience. 1985;15:445–480. doi: 10.1016/0306-4522(85)90225-8. [DOI] [PubMed] [Google Scholar]
- Soja PJ. Glycine-mediated postsynaptic inhibition is responsible for REM sleep atonia. Sleep. 2008;31:1483–1486. doi: 10.1093/sleep/31.11.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Guyenet PG. Distribution of glutamic acid decarboxylase mRNA-containing neurons in rat medulla projecting to thoracic spinal cord in relation to monoaminergic brainstem neurons. J Comp Neurol. 1999;407:367–380. [PubMed] [Google Scholar]
- Takakusaki K, Ohta Y, Mori S. Single medullary reticulospinal neurons exert postsynaptic inhibitory effects via inhibitory interneurons upon alpha-motoneurons innervating cat hindlimb muscles. Exp Brain Res. 1989;74:11–23. doi: 10.1007/BF00248276. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Shimoda N, Matsuyama K, Mori S. Discharge properties of medullary reticulospinal neurons during postural changes induced by intrapontine injections of carbachol, atropine and serotonin, and their functional linkages to hindlimb motoneurons in cats. Exp Brain Res. 1994;99:361–374. doi: 10.1007/BF00228973. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Kohyama J, Matsuyama K, Mori S. Medullary reticulospinal tract mediating the generalized motor inhibition in cats: parallel inhibitory mechanisms acting on motoneurons and on interneuronal transmission in reflex pathways. Neuroscience. 2001;103:511–527. doi: 10.1016/s0306-4522(00)00586-8. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Kohyama J, Matsuyama K. Medullary reticulospinal tract mediating a generalized motor inhibition in cats: III. Functional organization of spinal interneurons in the lower lumbar segments. Neuroscience. 2003;121:731–746. doi: 10.1016/s0306-4522(03)00542-6. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, Choi CS, Zigman JM, Shulman GI, Sherwin RS, Elmquist JK, Lowell BB. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5:383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanni-Mercier G, Sakai K, Lin JS, Jouvet M. Carbachol microinjections in the mediodorsal pontine tegmentum are unable to induce paradoxical sleep after caudal pontine and prebulbar transections in the cat. Neurosci Lett. 1991;130:41–45. doi: 10.1016/0304-3940(91)90222-f. [DOI] [PubMed] [Google Scholar]
- Webster HH, Friedman L, Jones BE. Modification of paradoxical sleep following transections of the reticular formation at the pontomedullary junction. Sleep. 1986;9:1–23. doi: 10.1093/sleep/9.1.1. [DOI] [PubMed] [Google Scholar]
- Yamuy J, Mancillas JR, Tran RT, Morales FR, Chase MH. C-fos-like expression in the lumbar spinal cord of the cat during carbachol-induced atonia. Sleep Res. 1991;20A:76. [Google Scholar]