

Abstract
A chiral phosphine catalyzes the addition of a carbon nucleophile to the γ position of an electron-poor allene (amide-, ester-, or phosphonate-substituted), in preference to isomerization to a 1,3-diene, in good ee and yield. This strategy provides an attractive method for the catalytic asymmetric γ functionalization of carbonyl (and related) compounds.
During the past several decades, the development of effective chiral catalysts that generate a new carbon–carbon bond and a new stereocenter α or β to a carbonyl group has been the focus of intense investigation.1 In contrast, little progress has been described in corresponding catalytic enantioselective functionalizations of the γ position.2 In 1992, Trost reported that phosphines catalyze the isomerization of electron-poor alkynes and allenes to 1,3-dienes (eq 1).3,4 Soonafter, he established that in the case of substrates that lack a δ hydrogen (and therefore cannot isomerize to a 1,3-diene) phosphines promote the addition of an array of nucleophiles to the γ position (eq 2).5
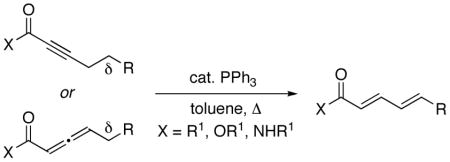 |
(1) |
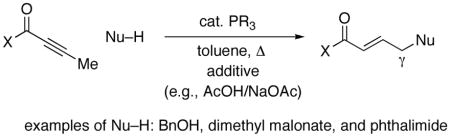 |
(2) |
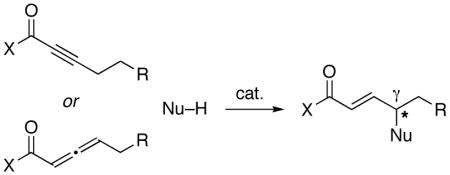
Clearly, the utility of phosphine-catalyzed γ additions would be greatly enhanced if such processes could be achieved with higher homologues (eq 3), in preference to isomerization (eq 1) (Figure 1). This substantial enlargement in scope would be accompanied by a second significant challenge: controlling the absolute configuration of the γ stereocenter, which could be complicated by issues such as the E/Z geometry of critical intermediates and the reversibility of key elementary steps (Figure 1).6
Figure 1.

Possible mechanisms for phosphine-catalyzed reactions of electron-poor alkynes and allenes: Isomerization and γ addition (for the sake of simplicity, only one E/Z isomer is illustrated and all of the elementary steps are drawn as irreversible).
 |
(3) |
To date, progress in addressing these two challenges has been limited. With respect to achieving addition rather than isomerization, phosphine-catalyzed intermolecular γ addition has only been accomplished with nitrogen nucleophiles (albeit in ≤30% yield),7 although intramolecular additions of oxygen nucleophiles have been described.5a,8 With regard to asymmetric catalysis to generate a γ stereocenter, just one success has been reported (intramolecular γ additions of oxygen nucleophiles).8,9
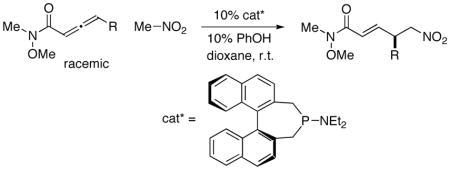
Thus, there are no examples of the use of a carbon nucleophile in a phosphine-catalyzed γ addition of the type illustrated in eq 3,10 as well as no reports of enantioselective intermolecular additions to produce a γ stereocenter for any family of nucleophiles (carbon, nitrogen, or oxygen). We were therefore pleased to determine that, through the appropriate choice of catalyst and reaction conditions, both of these deficiencies can be remedied (Table 1, entry 1).11 Specifically, phosphepine 1 catalyzes the γ addition of nitromethane to a racemic allene that bears a Weinreb amide12 in good ee and yield at room temperature. Phosphepine 1 has been reported to serve as a chiral ligand for rhodium-catalyzed hydrogenations and hydroformylations, but to the best of our knowledge it has not previously been employed as a nucleophilic catalyst.13,14
Table 1.
Catalytic asymmetric γ addition of a carbon nucleophile to an allene: Effect of reaction parameters.
 | |||
|---|---|---|---|
| Entry | change from the “standard conditions” | ee (%)a | yield (%)b |
| 1 | none | 93 | 83 |
| 2 | 2 instead of 1 | – | <2 |
| 3 | 3 instead of 1 | 67 | 51 |
| 4 | 4 instead of 1 | 68 | 51 |
| 5 | 5 instead of 1 | −83 | 47 |
| 6 | (S)-MONOPHOS instead of 1 | – | <2 |
| 7 | (R,R)-Et-DUPHOS instead of 1 | – | <2 |
| 8 | (R)-BINAP instead of 1 | – | <2 |
| 9 | quinidine instead of 1 | – | <2 |
| 10 | no PhOH | 74 | 29 |
| 11 | AcOH instead of PhOH | – | <2 |
| 12 | toluene instead of dioxane | 94 | 46 |
| 13 | CH2Cl2 instead of dioxane | 92 | 35 |
| 14 | 1.5 equiv instead of 5.5 equiv of MeNO2 | 94 | 48 |
All data are the average of two experiments.
A negative value for the ee signifies that the enantiomer of the illustrated product is formed preferentially.
The yield was determined by GC analysis with the aid of a calibrated internal standard.
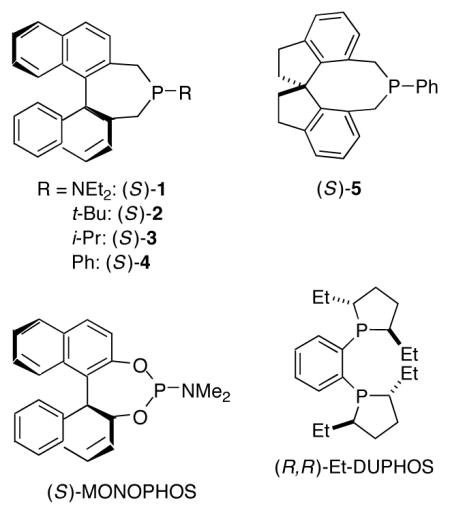
Related phosphepines are less effective as enantioselective catalysts for the γ addition of nitromethane to the allenamide (Table 1, entries 2–4),15 as are a range of other chiral phosphines and amines (e.g., entries 5–9). In the absence of an additive, a lower ee and yield are observed (entry 10), and the other additives that we have examined are less useful than phenol (e.g., entry 11).16 A smaller amount of the γ-addition product is observed in solvents such as toluene and CH2Cl2 (entries 12 and 13). Finally, the use of less nitromethane leads to a diminished yield (entry 14).
Under a standard set of conditions, phosphepine 1 serves as an effective catalyst for the enantioselective addition of nitromethane to an array of allenamides to generate a new carbon–carbon bond and a new γ stereocenter (Table 2). The R substituent can range in size from methyl to sterically demanding isopropyl, and it can bear a variety of functional groups.17,18
Table 2.
Phosphine-catalyzed asymmetric γ additions of nitromethane to allenamides.
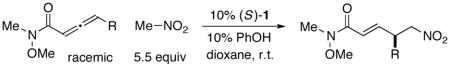 | |||
|---|---|---|---|
| entry | R | ee (%) | yield (%)a |
| 1 | Me | 97 | 94 |
| 2 | n-Pr | 93 | 81 |
| 3 | 87 | 73 | |
| 4b | i-Pr | 81 | 62 |
| 5 | (CH2)4OTBS | 92 | 57 |
| 6 | (CH2)3CO2Me | 93 | 75 |
| 7 | (CH2)5CO2Me | 92 | 82 |
| 8 | 92 | 83 | |
| 9 | 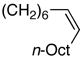 |
93 | 84 |
All data are the average of two experiments.
Yield of purified product.
15% 1 was used.
These new phosphine-catalyzed asymmetric carbon–carbon bond-forming processes are not limited to allenes substituted with a Weinreb amide. In a preliminary study, we have determined that ester- and phosphonate-activated allenes also undergo γ addition of nitromethane with useful efficiency (Table 3). To the best of our knowledge, allenylphosphonates have not previously been employed as substrates in phosphine-catalyzed γ additions.
Table 3.
Phosphine-catalyzed asymmetric γ additions of nitromethane to electron-poor allenes.

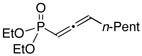
All data are the average of two experiments.
Yield of purified product.
Conditions: 3 equiv PhOH, 60 °C.
During the course of a phosphepine-catalyzed γ addition, the allene starting material remains racemic (i.e., no evidence for kinetic resolution), and the ee of the product is essentially constant (eq 4). Furthermore, 31P NMR studies establish that the resting state of the catalyst is “free” phosphepine 1, not a derivative such as a phosphonium salt (e.g., one of the intermediates illustrated in Figure 1), an observation that can be accommodated by the pathway outlined in Figure 1
 |
(4) |
The development of methods for the catalytic asymmetric functionalization of carbonyl compounds in the γ position has the potential to complement the impressive accomplishments that have been reported for functionalization of the α and the β positions; to date, comparatively few such γ functionalizations have been described. In view of the ready accessibility of allenes,19 the use of chiral phosphines to catalyze γ additions of nucleophiles represents an attractive strategy for addressing this deficiency. However, due to the facility of isomerization to a 1,3-diene (eq 1), there had been only limited success in achieving phosphine-catalyzed additions of nucleophiles to allenes (or alkynes) that create a γ stereocenter; in particular, there had been no reports with carbon-based nucleophiles. In this investigation, we have determined that under the appropriate conditions such processes can be accomplished not only in useful yield, but also with good enantioselectivity. The product of the γ addition is an α,β-unsaturated carbonyl compound that is poised for stereoselective functionalization of the α and β positions. Additional studies of phosphine-catalyzed γ additions are underway.
Supplementary Material
Acknowledgments
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, grant R01-GM57034), Merck Research Laboratories, and Novartis. We thank Dr. Nicolas Marion for assistance, Evonik Degussa for providing samples of phosphepines 1–4, and Prof. Qi-Lin Zhou for providing a precursor to phosphine 5. Funding for the MIT Department of Chemistry Instrumentation Facility has been furnished in part by NIH IS10RR13886 and NSF DBI–9729592.
Footnotes
Supporting Information Available: Experimental procedures and compound characterization data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For example, see reviews of: aldol reactions: Carreira EM, Fettes A, Marti C. Org Reactions. 2006;67:1–216.conjugate additions of Grignard reagents: Harutyunyan S, den Hartog T, Geurts K, Minnaard AJ, Feringa BL. Chem Rev. 2008;108:2824–2852. doi: 10.1021/cr068424k.
- 2.For example, see a review of catalytic asymmetric vinylogous aldol reactions: Denmark SE, Heemstra JR, Jr, Beutner GL. Angew Chem, Int Ed. 2005;44:4682–4698. doi: 10.1002/anie.200462338.
- 3.Trost BM, Kazmaier U. J Am Chem Soc. 1992;114:7933–7935.For a review, see: Kwong CKW, Fu MY, Lam CSL, Toy PH. Synthesis. 2008:2307–2317.
- 4.For reviews and leading references to nucleophilic catalysis by phosphines, see: Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035–1050.Ye LW, Zhou J, Tang Y. Chem Soc Rev. 2008;37:1140–1152. doi: 10.1039/b717758e.Lu X, Zhang C, Xu Z. Acc Chem Res. 2001;34:535–544. doi: 10.1021/ar000253x.
- 5.(a) Trost BM, Li CJ. J Am Chem Soc. 1994;116:10819–10820. [Google Scholar]; (b) Trost BM, Li CJ. J Am Chem Soc. 1994;116:3167–3168. [Google Scholar]; (c) Trost BM, Dake GR. J Org Chem. 1997;62:5670–5671. [Google Scholar]
- 6.For some key early examples of asymmetric nucleophilic catalysis with chiral phosphines, see: kinetic resolutions of alcohols, see: Vedejs E, Daugulis O, Diver ST. J Org Chem. 1996;61:430–431. doi: 10.1021/jo951661v.Vedejs E, Daugulis O. J Am Chem Soc. 1999;121:5813–5814.Morita-Baylis-Hillman reactions: Hayase T, Shibata T, Soai K, Wakatsuki Y. Chem Commun. 1998:1271–1272.couplings of allenes with an unsaturated partner: Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J Am Chem Soc. 1997;119:3836–3837.
- 7.(a) Trost BM, Dake GR. J Am Chem Soc. 1997;119:7595–7596. [Google Scholar]; (b) Liu B, Davis R, Joshi B, Reynolds DW. J Org Chem. 2002;67:4595–4598. doi: 10.1021/jo016154u. [DOI] [PubMed] [Google Scholar]; (c) Virieux D, Guillouzic AF, Cristau HJ. Tetrahedron. 2006;62:3710–3720. [Google Scholar]
- 8.Chung YK, Fu GC. Angew Chem, Int Ed. 2009;48:2225–2227. doi: 10.1002/anie.200805377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enantioselective phosphine-catalyzed intermolecular γ additions of prochiral carbon nucleophiles to 2-butynoates and 2,3-butadienoates (for which there is no possibility of isomerization to a 1,3-diene) have been investigated. Such processes generate a stereocenter in the δ, rather than the γ, position (up to 81% ee). See: Chen Z, Zhu G, Jiang Q, Xiao D, Cao P, Zhang X. J Org Chem. 1998;63:5631–5635.
- 10.For phosphine-catalyzed additions of carbon nucleophiles to 2-butynoates and 2,3-butadienoates, see: (a) Reference 5b. Zhang C, Lu X. Synlett. 1995:645–646.(c) Reference 9. Alvarez-Ibarra C, Csaky AG, de la Oliva CG. J Org Chem. 2000;65:3544–3547. doi: 10.1021/jo9918192.Lu C, Lu X. Org Lett. 2002;4:4677–4679. doi: 10.1021/ol0270733.
- 11.Conditions that have been reported by others for phosphine-catalyzed γ additions to 2-butynoates and 2,3-butadienoates (e.g., Ref. 5b and Ref. 10d) lead to significant undesired isomerization (to form the 1,3-diene), but little or none of the desired γ addition.
- 12.For a review of the synthetic utility of Weinreb amides, see: Balasubramaniam S, Aidhen IS. Synthesis. 2008:3707–3738.
- 13.For applications of phosphepine 1 as a chiral ligand, see: Chi Y, Zhang X. Tetrahedron Lett. 2002;43:4849–4852. (includes a synthesis of 1)Junge K, Oehme G, Monsees A, Riermeier T, Dingerdissen U, Beller M. J Organomet Chem. 2003;675:91–96.Erre G, Enthaler S, Junge K, Gladiali S, Beller M. J Mol Catal A: Chem. 2008;280:148–155.
- 14.For applications of phosphepine 2 as a chiral nucleophilic catalyst, see: initial studies: Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234–12235. doi: 10.1021/ja053277d.Wilson JE, Fu GC. Angew Chem, Int Ed. 2006;45:1426–1429. doi: 10.1002/anie.200503312.leading references to recent investigations: Pinto N, Fleury-Bregeot N, Marinetti A. Eur J Org Chem. 2009:146–151.
- 15.For the use of phosphine 5 as a chiral nucleophilic catalyst, see Ref. 8.
- 16.For some examples of the use of additives in phosphine-catalyzed γ additions, see Ref. 5.
- 17.Notes: (a) For all of the phosphine-catalyzed asymmetric γ additions illustrated in Tables 2 and 3, only the E isomer of the product is observed (>20:1 E:Z selectivity). (b) Under our standard conditions, phosphepine 1 does not serve as an effective enantioselective catalyst for corresponding γ additions of nitroethane and nitrocyclohexane. (c) After exposure of solid phosphepine 1 to air for 40 days at room temperature, no phosphine oxide was detected by 31P NMR spectroscopy. (d) The phosphine oxide derivative of 1 does not catalyze these γ additions. (e) In a gram-scale reaction (1.05 g of product), the γ addition illustrated in entry 2 of Table 2 proceeds in 93% ee and 77% yield. (f) In a preliminary study, γ addition to the sterically demanding t-butyl-substituted allene (Table 2, R = t-Bu; 15% of phosphepine 1) proceeded in 40% ee and ~80% yield (according to 1H NMR spectroscopy). (g) An initial investigation of a phosphepine-catalyzed γ addition of nitromethane to a cyano-substituted allene furnished the desired product in high ee (≥90%) but modest yield (~45%; 5:1 E:Z). (h) Under our standard conditions, when R = Ph (Table 2), the γ addition proceeds very slowly. (i) For the reactions depicted in Table 2, only a small amount of isomerization to the 1,3-diene was typically observed (≤5%). (j) The configurations of two of the γ-addition products were determined by correlation with compounds of known stereochemistry (see the Supporting Information).
- 18.(a) General procedure. In a glovebox, catalyst (S)-1 (29 mg, 0.075 mmol; 0.10 equiv) and phenol (7.0 mg, 0.075 mmol; 0.10 equiv) were added to an oven-dried 20-mL vial. These solids were dissolved in anhydrous dioxane (15 mL), and then nitromethane (225 μL, 4.15 mmol; 5.5 equiv) and the allene (0.75 mmol; 1.0 equiv) were added via syringe. The vial was capped and removed from the glovebox, and the reaction mixture was stirred at room temperature for 15 h. The solvent was then evaporated, and the product was purified by flash chromatography. (b) Glovebox-free procedure. On a benchtop, catalyst (S)-1 (43.5 mg, 0.113 mmol; 0.15 equiv; with 10% (S)-1, a small amount of unreacted allene was observed after 15 h) and phenol (10.5 mg, 0.113 mmol; 0.15 equiv) were added to an oven-dried 20-mL vial. The vial was capped with a septum, and then it was evacuated and refilled with argon (three cycles). Next, anhydrous dioxane (15 mL), nitromethane (225 μL, 4.15 mmol; 5.5 equiv), and the allene (0.75 mmol; 1.0 equiv) were added in order via syringe through the septum. The reaction mixture was stirred at room temperature for 15 h. The solvent was then evaporated, and the product was purified by flash chromatography.
- 19.For example, the allenamide illustrated in Table 1 is synthesized in one step from N-methoxy-N-methyl-2-(triphenylphosphoranylidene)-acetamide and pentanoyl chloride (both reactants are commercially available).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.