

Abstract
Asthma is a common disease with an increasing prevalence worldwide. Up to 10% of these patients have asthma that is refractory to current therapy. This group have a disproportionate use of health care resources attributed to asthma, have significant morbidity and mortality and therefore represent an unmet clinical need. Asthma is a complex heterogeneous condition that is characterized by typical symptoms and disordered airway physiology set against a background of airway inflammation and remodelling. The inflammatory process underlying asthma is co-ordinated by a cytokine network. Modulating this network with biological therapy presents a new paradigm for asthma treatment. Clinical trials undertaken to date have underscored the complexity of the inflammatory profile and its relationship to the clinical features of the disease and have raised the importance of safety considerations related to these novel therapies. T helper type 2 cytokine blockade remains the most promising strategy, with anti-interleukin-5 reducing asthma exacerbations. Although anti-cytokine therapy is not yet ready for the clinic, the long-awaited possibility of new treatments for severe asthma is moving ever closer.
Keywords: airway hyperresponsiveness, anti-IL-5, anti-TNF-α, asthma, cytokines, exacerbations
Introduction
Asthma affects 300 million people worldwide [1]. Its prevalence is 15–20% in children and 5–10% in adults, and continues to rise. In the majority of cases the disease can be well controlled with inhaled corticosteroid therapy either alone or in combination with long-acting beta-agonists and or leukotrine receptor inhibitors as per international management guidelines [2,3]. However, up to 10% of the asthma sufferers remain poorly controlled in spite of optimal standard therapy. Although these patients represent the minority of people with asthma, they have the greatest morbidity, are at risk of asthma-related death and are responsible for more than 50% of the health care utilization attributed to asthma. Therefore, there is a significant unmet need in this group [4].
Severe asthma can be subdivided further into ‘difficult-to-treat’ or ‘treatment-resistant’ (refractory) asthma. ‘Difficult-to-treat’ asthma is usually a consequence of poor adherence with therapy, co-factors such as co-morbidities including psychosocial factors or persistent exposure to triggers such as smoking. Severe ‘treatment-resistant’ asthma includes patients that remain poorly controlled at Global Initiative for Asthma (GINA) treatment steps IV and V, i.e. treatment with high-dose inhaled corticosteroid therapy and other add-on therapies after aspects of the disease making it ‘difficult-to-treat’ have been managed as completely as possible. This review will not address ‘difficult-to-treat’ asthma further; suffice to say that these issues, where possible, need to be addressed in the management of severe asthma. The American Thoracic Society (ATS) workshop definition of refractory asthma [5] is summarized in Table 1.
Table 1.
Typical clinical features of refractory asthma (adapted from [5]).
| Major criteria |
| Treatment with at least one of the following: |
| •Oral corticosteroids > 50% of the time |
| •High-dose inhaled corticosteroids (> 1200 µg beclomethasone equivalent) |
| Minor criteria |
| •Requirement for daily treatment with long-acting beta-agonists, theophylline or leukotrine antagonists |
| •Daily asthma symptoms requiring rescue medication |
| •Persistent airway obstruction (FEV1 < 80% predicted); diurnal PEF variability > 20% |
| •One or more urgent care visits for asthma per year |
| •Three or more oral steroid bursts per year |
| •Prompt deterioration with > 25% reduction in oral or inhaled corticosteroid dose |
| •Near fatal asthma event in the past |
FEV1, forced expiratory volume in 1 s; PEF, peak expiratory flow.
There is increasing recognition that asthma, and in particular treatment-resistant severe asthma, is a heterogeneous condition [6]. The clinical, physiological and immunopathological domains of the disease often co-exist, but are not necessarily related (summarized in Fig. 1). Novel statistical approaches applied to clinical data sets using data reduction tools such as factor and cluster analysis may assist in the identification of important phenotypes of severe asthma. This approach has found that eosinophilic airway inflammation and symptoms can be closely associated ‘concordant’ disease or dissociated ‘concordant’ disease. There is now a need to dissect the mechanisms that are important in the interplay between the domains of asthma and to unravel the underlying pathobiology of novel phenotypes. This, in turn, will enable us to develop biomarkers and novel therapies. Indeed, biomarkers have been used to direct current therapy [7,8]. This is exemplified by the application of the sputum eosinophil count to target corticosteroid therapy. This strategy leads to a reduction in severe exacerbations in severe disease without an overall increase in corticosteroid therapy across patients, as treatment is appropriately up- and down-titrated. Recent post-hoc analysis of this study has revealed that the success of this approach was due to targeted therapy in the ‘discordant’ phenotypes [6]. This novel management strategy has now become adopted by the British Thoracic Society/Scottish Intercollegiate Guidelines Network (BTS/SIGN) as the ‘gold’ standard in the management of severe asthma. This allows for optimization of current therapy, but fails to address fully the unmet need of severe asthma and new therapies are required urgently.
Fig. 1.
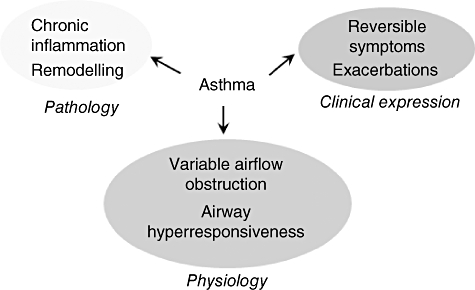
Asthma, a multi-dimensional disease.
Biological therapy has enjoyed great success in some disease areas, most notably anti-tumour necrosis factor (TNF)-α therapy in rheumatoid arthritis [9]. In severe asthma anti-immunoglobulin (Ig)E (Xolair) has provided a new class of treatment and has been demonstrated to improve symptoms, reduce the burden of inhaled corticosteroids and reduce exacerbation frequency [10,11]. For these reasons, anti-IgE is licensed for severe asthmatics with evidence of atopy to a perennial aeroallergen, a serum total IgE within range for therapy and poor asthma control, in spite of optimal standard therapy. In England and Wales, additional criteria related to frequent hospital admissions are included. Anti-IgE has therefore set a new paradigm for the management of severe asthma and has positioned biological therapy at the forefront of drug discovery for this disease area. Not all severe asthmatics have atopy, and in those where this is a major component of their disease, not all respond. Therefore, in the wake of the success of anti-IgE there is considerable enthusiasm to identify new biological therapy for severe asthma. With much of the research into asthma focused upon the role of inflammation in asthma, and in particular the importance of the T helper type 1 (Th1) versus Th2 balance, cytokines as a therapeutic target has become central in the development of biologics.
In this review we shall summarize briefly the role of cytokines in the biology of severe asthma and describe the current successes and failures of anti-cytokine therapies in the clinic.
Cytokines and their role in asthma
Asthma is characterized by the presence of typical day-to-day symptoms of breathlessness, cough and wheeze, punctuated with acute exacerbations together with evidence of variable airflow obstruction and airway hyperresponsiveness. These typical symptoms and disordered airway function occur against a background of airway inflammation and remodelling.
Airway inflammation in asthma is a multi-cellular process involving eosinophils, CD4+ T cells, mast cells and neutrophils [12–17]. This inflammation is restricted largely to the large conducting airways in mild–moderate disease, but in severe asthma the smaller airways are often involved [18]. Asthma is associated commonly with atopy, although asthma does occur in the absence of allergic disease. A key feature of allergic asthma is the recognition of allergens and the subsequent sensitization that leads to a Th2 cytokine response. Dendritic cells in the airway epithelium and submucosa take up and process allergens and present them to T cells in association with important co-stimulatory molecules (reviewed in [19]). Subsequent T cell polarization towards a Th1 or Th2 phenotype is, in part, under the influence of dendritic cell-derived interleukin (IL)-12. Increased IL-12 drives the inflammatory response towards a Th1 bias, whereas in allergic asthma the Th2 phenotype predominates. Once sensitized, T cells are able to home back to sites of allergic inflammation under the control of chemokines via activation of the receptors CCR3, 4, 7 and 8 (reviewed in [20]). The Th2 cells produce Th2 cytokines, the majority of which are produced on the long arm of chromosome 5, namely IL-3, 4, 5, 9 and 13, and granulocyte–macrophage colony-stimulating factor (GM–CSF) (reviewed in [21]). In asthma expression of these cytokines are increased, particularly in severe disease [22–29]. Animal models have positioned these cytokines as critical in allergic sensitization and the development of disease [30–32]. Importantly, in severe disease the inflammatory response is complex and also involves Th1 T cells. These cells secrete TNF-α and interferon (IFN)-γ, among other important mediators. TNF-α expression is also increased in the airway in asthma [33–35] and the TNF-α axis is up-regulated with increased membrane-bound TNF-α on peripheral monocytes [36].
The role of the dendritic cell–T cell axis in allergic sensitization is clear, but there is increasing recognition that other cells are likely to be as, if not more, important in severe asthma. Mast cell numbers are increased in the airway epithelium and in the airway smooth muscle bundle [37–43]. This microlocalization with the airway smooth muscle is a consistent finding and is related closely to the degree of airway hyperresponsiveness [37,41]. In the asthmatic airway mast cells are in an activated state and are an important source of cytokines, chemokines, autocoid mediators, proteases and histamine [15,25,42,43]. Importantly, these cells can be activated via both IgE and non-IgE mechanisms and have been shown to affect airway smooth contractility directly [44–46] and indirectly by up-regulation of airway smooth muscle transforming growth factor (TGF)-β, which in turn drives the airway smooth muscle into a more contractile phenotype via an autocrine activation [47].
In severe disease neutrophils are also increased [16,48,49], and have been implicated in disease [49,50], but whether they play a key role in disease progression or are a consequence of corticosteroid therapy is unclear. Structural cells within the airway, including epithelial cells, fibroblasts, myofibrobalsts, fibrocytes and airway smooth muscle, are also important sources of chemokines and growth factors and indeed are likely to play a role in the inflammatory response. Importantly, these structural cells are increased in number in severe disease and contribute to the remodelling process, which leads onto progressive disease and persistent airflow obstruction [51].
Corticosteroids are the mainstay of therapy for asthma and attenuate the inflammatory response. However, in refractory asthma by definition this effect is inadequate; therefore, alternative strategies are required. Cytokine or anti-cytokine therapy presents an important alternative or adjunct to current therapy. Whether or not the success of modulating the cytokine milieu in animal models can be translated in human disease is challenging, due to the complexity of severe asthma and the potential for redundancy when targeting single cytokines. In spite of these concerns, there remains the possibility that single cytokines play dominant roles in the development of specific features of disease within subgroups of asthmatics.
Clinical trials of anti-cytokine therapy
To date, clinical trials of anti-cytokine therapies in asthma have, in the majority, targeted the Th1 versus Th2 balance. Following the success of anti-TNF-α therapy in rheumatoid disease and inflammatory bowel disease, together with evidence supporting a role for TNF-α, particularly in severe asthma, has led to a number of trials targeting this axis (Tables 2 and 3).
Table 2.
Summary of anti-cytokine anti-interleukin (IL) therapy trials in asthma (excluding anti-tumour necrosis factor-α).
| Author | No/severity | Design | Treatment | Outcomes | Results |
|---|---|---|---|---|---|
| Haldar et al.[56] | 61/severe | Randomized, double-blind placebo-controlled parallel group | Mepolizumab 50 weeks Anti-IL-5 | 1.Exacerbations 2.Symptoms, FEV1, AQLQ, AHR, sputum and blood Eos | Reduced exacerbations Improved AQLQ, reduced Eos |
| Nair et al.[57] | 20/severe | Randomized, double-blind placebo-controlled parallel group | Mepolizumab 26 weeks Anti-IL-5 | 1.Exacerbations, oral steroid reduction 2.FEV1, ACQ, sputum and blood Eos | Reduced exacerbations and steroid sparing Improved FEV1 and ACQ, reduced Eos |
| Flood-Page et al.[55]] | 362/severe | Randomized, double blind placebo controlled parallel group | Mepolizumab 12 weeks Anti-IL-5 | 1.PEFR 2.Exacerbations, AQLQ, FEV1, sputum and blood eosinophil counts, safety | Improved PEFR Significantly reduced Eos, trend to reduced exacerbations, safe |
| Flood-Page et al.[53] | 24/mild | Randomized, double-blind placebo-controlled parallel group | Mepolizumab 20 weeks Anti-IL-5 | 1.Reduction in Eos in BAL, EBB, bone marrow and blood 2.PEFR, FEV1, AHR | Significant reduction in Eos Trend towards better PEFR |
| Leckie et al.[52] | 24/mild | Randomized, double-blind placebo-controlled parallel group | SB-240563 hmAb IL-5 single dose | 1.Sputum and blood Eos 2.Early and late asthma response, AHR | Reduced sputum and blood Eos Secondary outcomes insignificant |
| Kips et al.[54] | 26/severe | Randomized, double-blind placebo-controlled parallel group | SCH55700 12 weeks Anti-IL-5 | 1.Sputum and blood Eos, symptoms, FEV1 | Reduced blood Eos, safe No other significant outcomes |
| Wenzel et al.[62] | 24/mild 32/mild | Randomized, double-blind placebo-controlled parallel group | Pitrakinra (IL-4Rα inhibitor) 28 days | 1.FEV1 reduction post-allergen challenge 2.FENO, AHR, sputum Eos, blood IgE, skin prick | Significant attenuation to LAR Improved symptoms |
| Borish et al.[59,60] | 25/moderate 62/moderate | Randomized, double-blind placebo-controlled parallel group | SIL-4R Single dose (n 25) 12 weeks (n 62) | 1.FEV1, symptoms 2.Safety | Improved FEV1, symptoms Safe |
| O'Byrne et al.[67] | 10/mild + moderate | Randomized, double-blind placebo-controlled parallel group | MEDI-528 HmAb IL-9 Single dose | 1.Safety 2.PFTs | Safety established, no significant PFT changes |
FEV1, forced expiratory volume in 1 s; AQLQ, Asthma Quality of Life Questionnaire; AHR, airways hyperresponsiveness; Eos, eosinophils; ACQ, Asthma Control Questionnaire; PEFR, peak expiratory flow rate; PFT, pulmonary function test; BAL, brochoalveolar lavage; EBB, endobronchial biopsy; FENO, fractional exhaled nitric oxide; LAR, lifetime attributable risk; IL, interleukin; hmAb, humanized monoclonal antibody.
Table 3.
Summary of anti-tumour necrosis factor (TNF)-α, interferon and interleukin (IL) therapy trials in asthma.
| Author | No/ severity | Design | Treatment | Outcome | Result |
|---|---|---|---|---|---|
| Wenzel et al.[75] | 309/severe | Randomized, double-blinded, placebo-controlled, parallel group | Golimumab 24 weeks, anti-TNF-α | 1.FEV1, exacerbations 2.AQLQ, PEFR | 1.FEV1 unchanged, reduced exacerbations 2.No significant differences Unfavourable adverse effect profile |
| Howarth et al.[35] | 15/severe | Open-label, uncontrolled | Etanercept 12 weeks, anti-TNF-α | 1.ACQ 2.FEV1, AHR | 1.Significantly improved ACQ, FEV1, AHR |
| Berry et al.[36] | 30/moderate + severe | Randomized, double-blinded, placebo-controlled, cross-over | Etanercept 10 weeks, anti-TNF-α | 1.AHR, AQLQ | 1.Improved AQLQ, FEV1, AHR |
| Morjaria et al.[74] | 39/moderate + severe | Randomized, double-blinded, placebo-controlled, parallel group | Etanercept 12 weeks, anti-TNF-α | 1.AQLQ, ACQ 2.FEV1, AHR, PEFR | 1.Improved ACQ 2.Improved AHR |
| Erin et al.[73] | 38/moderate | Randomized, double-blinded, placebo-controlled, parallel group | Infliximab 6 weeks, anti-TNF-α | 1.PEFR 2.Exacerbations | 1.No change in PEFR 2.Trend towards reduced exacerbations, reduced TNF-α in sputum |
| Boguniewicz et al.[68] | 5/mild | Open-labelled uncontrolled | Interferon-γ | FEV1, PEF variability | No effect |
| Simon et al.[69] | 10/severe | Observational case report | Interferon-α | PFTs, leucocyte counts, safety | Improved PFTs Increased IL-10, Th1 cell differentiation Reduced steroid dose |
| Kroegel et al.[70] | 3/severe | Observational case report | Interferon alphacon-1 | Exacerbations, oral steroid use, safety | Reduced exacerbations, steroid withdrawal Improved PFTs Safety profile acceptable |
| Bryan SA et al.[71] | 39/mild | Randomized, double-blind placebo-controlled parallel group | IL-12 | 1.FEV1 reduction post-allergen challenge 2.AHR, blood and sputum Eos | Primary outcome non significant Trend towards improved AHR, significant reduction in Eos |
| Chernoff et al.[72] | 17/normal subjects | Observational placebo-controlled | IL-10 | Safety | Safety established. Significant reduction of TNF-α, IL-1β, T cell proliferation |
FEV1, forced expiratory volume in 1 s; AQLQ, Asthma Quality of Life Questionnaire; AHR, airways hyperresponsiveness; Eos, eosinophils; ACQ, Asthma Control Questionnaire; PEFR, peak expiratory flow rate; PFT, pulmonary function test; Th1, T helper type 1.
IL-5
Two humanized, human-IL-5-specific monoclonal antibodies (mAbs), Sch- 55, 700 and mepolizumab (SB-240, 563), and an IL-5R-specific mAb (MEDI-563) have been developed for the treatment of asthma. In a small double-blind trial, mepolizumab resulted in a rapid dose-dependent reduction in the number of circulating and sputum eosinophils but, surprisingly, this had no effect on either the late asthmatic response or on airway hyperresponsiveness [52]. A further study using mepolizumab confirmed the persistent suppression of eosinophilia in blood, bone marrow and airway lavage, but in airway biopsies there was only a 55% reduction in the number of tissue eosinophils [53]. In a group of 24 patients with severe persistent asthma, treatment with Sch- 55700 resulted in a decrease in the number of blood eosinophils, but over the course of 10 weeks it had no effect on symptoms or physiological outcomes [54]. This observation has been confirmed in a dose-ranging trial of 324 severe asthmatics with mepolizumab [55]. Interestingly, in this study there was a trend towards reduced risk of moderate/severe exacerbations in the high-dose mepolizumab arm by about 50%. However, this study was not powered sufficiently to show a difference in exacerbations.
Most recently, Haldar [56] and colleagues have studied 61 subjects with eosinophilic refractory asthma in a randomized double-blinded placebo-controlled study of mepolizumab for 1 year. The primary outcome measure was the number of severe exacerbations per subject, and secondary outcomes included eosinophil counts in blood and sputum, airway hyperresponsiveness, lung function, health status and symptoms. The study demonstrated significant reductions in severe asthma exacerbations (2·0 mepolizumab versus 3·4 placebo) (Fig. 2) and reductions in both blood and sputum eosinophilia. This reiterates findings from previous studies that mepolizumab has no significant effect on airways hyperresponsivemess (AHR), lung function or symptoms, while ameliorating eosinophilic inflammation. This beneficial effect of mepolizumab was also observed in a prednisolone withdrawal study in severe eosinophilic asthma [57]. In addition to its role in asthma exacerbations anti-IL-5 therapy may also attenuate airway remodelling as airway wall area [56], and immunostaining for tenascin, lumican and procollagen III in the bronchial mucosal subepithelial basal lamina [58] was reduced.
Fig. 2.
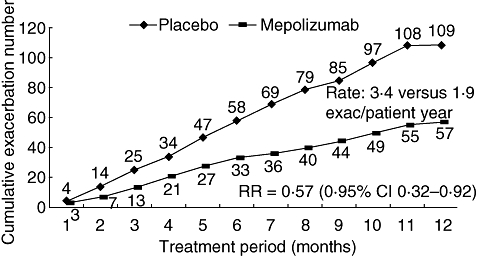
Severe asthma exacerbations were reduced in subjects treated with anti-interleukin-5 in a 1-year placebo-controlled parallel group study (adapted from [56]).
These more recent studies have rekindled enthusiasm for anti-IL-5 in asthma, and suggest that although eosinophilic inflammation and airway hyperresponsiveness are dissociated that the eosinophil does play a role in exacerbations and airway remodelling. To date, the side effect profile of anti-IL-5 has been favourable, and therefore further studies are under way to determine the efficacy of this therapy in severe asthma.
IL-4/IL-13
A small trial of nebulized inhaled altrakincept (soluble, recombinant human IL-4 receptor) for 12 weeks in patients with mild to moderate asthma indicated efficacy by allowing withdrawal from treatment with inhaled corticosteroids without relapse [59], and this result was confirmed subsequently in a larger trial [60]. However, a Phase III trial failed to confirm the efficacy of altrakincept for the treatment of asthma, although there were concerns over the bioavailability of altrakincept in this study. Further Phase II studies are in progress using humanized IL-4-specific and IL-4Rα-blocking antibodies such as pascolizumab (SB240, 683) [61].
Two recent placebo-controlled allergen challenge studies showed that an IL-4 variant (pitrakinra) administered subcutaneously or nebulized can inhibit the binding of IL-4 and IL-13 to the alpha subunit of the IL-4 receptor. Pitrakinra reduced the allergen-induced late-phase response and the need for rescue medication in asthmatic patients [62]. Trials are now under way using an inhaled preparation [63]. Similarly, a humanized IL-13 mAb, IMA-638, inhibited both the early and late allergen challenge response, but did not affect allergen-induced hyperresponsiveness to methacholine [64]. Several other mAbs against IL-13 have completed early safety trials in humans, including CAT-354 [65] and AMG 317 [66], and are undergoing clinical trials for asthma.
IL-9
Two phase I dose-escalation studies of an IL-9-specific mAb (MEDI- 528) in healthy volunteers have been completed [67]. Phase II trials are in progress for treating symptomatic, moderate–severe, persistent asthma.
Interferons
Subcutaneous administration of recombinant human IFN-γ in asthma has been disappointing [68]. By contrast, two small trials have shown that systemic administration of IFN-α for 18 months is effective for the treatment of severe corticosteroid refractory asthma, with one study showing reversal of the Th2-cell cytokine profile in blood mononuclear cells after treatment of patients with severe asthma [69,70]. These preliminary observations warrant further investigations in larger trials.
IL-12
Injection of recombinant human IL-12 in patients with mild asthma decreased the number of circulating blood eosinophils after allergen challenge (but not sputum eosinophilia, the late-phase response or airway hyperresponsiveness) [71] and this was accompanied by flu-like symptoms, abnormal liver-function tests and, most worryingly, cardiac arrhythmias.
IL-10
Administration of IL-10 to normal volunteers decreases the numbers of circulating CD4+ and CD8+ T cells [72]. Recombinant human IL-10 has been developed and is currently being tested in rheumatoid arthritis, inflammatory bowel disease, psoriasis, organ transplantation and chronic hepatitis C, but its effect in asthma has yet to be studied.
TNF-α
A number of strategies to block the TNF-α axis are available, including infliximab (a chimeric mouse/humanized mAb), etanercept (a soluble fusion protein combining two p75 TNF receptors with an Fc fragment of human IgG1), and fully human mAbs adalimumab and golimumab [9].
Enthusiasm for anti-TNF-α in severe asthma was first derived from an uncontrolled study of etanercept for 12 weeks in patients with severe asthma. Howarth et al.[35] reported a significant improvement in airway hyperresponsiveness, lung function and quality of life. These findings were replicated in another small randomized, placebo-controlled cross-over study [36]. One of the most striking aspects of this study was that the clinical response correlated closely with the expression of mTNF-α and TNF-α receptor 1 on monocytes. This suggests that measurement of TNF-α expression in monocytes might be a useful biomarker of responsiveness, but also suggests that anti-TNF-α approaches will be effective in only a subgroup of asthmatic patients. Another interesting aspect of the study was that there was no effect of etanercept therapy on the number of sputum eosinophils or neutrophils, but there was a reduction in sputum histamine concentration. One intriguing possible explanation for this apparent lack of effect on airway inflammation by anti-TNF-α in contrast to a marked effect on AHR is that TNF-α derived from mast cells within the airway smooth muscle (ASM) bundle might play a critical role in the development of AHR. Similar beneficial effects, albeit less profound, have been reported in patients with moderate asthma. Erin et al.[73] performed a randomized placebo-controlled study with infliximab in patients with moderate asthma. No improvement in morning peak flow occurred with infliximab, but there was an improvement in peak flow variability and a 50% reduction in the number of mild exacerbations encountered. Importantly, two further studies of etanercept in moderate–severe asthma, one unpublished, have failed to demonstrate efficacy [74].
Most recently, Wenzel and colleagues [75] published their study using golimumab. This is the largest published study to date on patients with severe asthma, with 309 subjects assigned to either placebo arm or three different dose arms of golimumab injections, monthly for 1 year in a randomized, double-blinded fashion. The primary end-points were an improvement from baseline forced expiratory volume in 1 s (FEV1) and also the number of severe asthma exacerbations. Secondary end-points were change from baseline in Asthma Quality of Life Questionnaire AQLQ) score, peak expiratory flow (PEF) and rescue medication use. The study was terminated early at 24 weeks due to a large number of serious adverse events in the golimumab arm and no demonstrable efficacy. Of greatest concern was the high number of serious adverse effects in all the golimumab arms. Pneumonia, sepsis, reactivation of tuberculosis (TB), increased rate of malignancy and one death were reported. A similar unfavourable safety profile for anti-TNF-α therapy has been reported in chronic obstructive pulmonary disease (COPD) [76]. Therefore, even if subgroups can be identified in which anti-TNF-α has efficacy, this will need to be achieved with a substantial reduction in treatment-related adverse outcomes.
Other potential cytokines
Several other cytokines have been implicated in severe asthma for which treatments are in development. Much interest has surrounded the identification of Th17 cells [77]. The expression of IL-17A and F has been reported recently to be elevated in severe asthma [78]. Modulation of these cytokines may affect neutrophil recruitment. Th2 responses are augmented by IL-25 (IL-17E), IL-31 and IL-33, thereby positioning these cytokines as potential targets [79,80]. It is likely that as our understanding of the cytokine networks in severe asthma develop, so the number of potential targets will increase.
Conclusion
Anti-cytokine therapy in severe asthma has underscored the importance to consider the heterogeneity of the disease and the relationship between airway inflammation, dysfunction and clinical expression of disease. Anti-IL-5 treatment demonstrates that eosinophilic inflammation is dissociated with airway responsiveness, but plays a central role in exacerbations. Whether the amelioration of eosinophilic inflammation in response to anti-IL-5 removes the cause of an exacerbation or reduces the risk of an exacerbation event in response to another trigger such as an infection is unknown. In contrast, TNF-α may be important in the development of airway hyperresponsiveness. Therefore, future clinical trials need to be cognizant of the most appropriate outcome measures for different cytokine therapies, or at least not to dismiss novel therapies without considering the impact upon several outcomes. Anti-TNF-α treatments have highlighted the importance of the risk–benefit ratio and how safety concerns need to be at the forefront in drug development. To date, the poor safety profile for anti-TNF-α in obstructive airways disease has undermined any potential efficacy that has been observed. It is likely that the development and validation of biomarkers will enable researchers to choose more accurately the most appropriate patients for these highly specific biological therapies. We are now moving towards a paradigm of targeted patient-specific therapy and the possibility for the need to treat patients with combination biological therapy. The increasing number of biological therapies in early clinical trials means that shortly we shall begin to unravel the complexity of severe asthma and are likely to have novel therapies for our patients. To date, as a consequence of our naive application of anti-cytokine therapy in asthma, or due perhaps to biological redundancy when targeting single cytokines, anti-cytokine therapy for severe asthma has promised much but delivered little. We are now at the threshold of understanding the value of biological therapy in severe asthma; the next few years promise to be exciting times for asthma research and may make cytokine and anti-cytokine therapy in the clinic a reality.
Acknowledgments
C. B. is funded by a Wellcome Senior Clinical Fellowship.
Disclosure
D.D. has no conflict of interest; C.B. has received research grant support in the last 3 years for £1.1 million from GSK, AstraZeneca and MedImmune and consultancy fees for less than £5000 per annum.
References
- 1.Braman SS. The Global Burden of asthma. Chest. 2006;130(1) Suppl.:4S–12S. doi: 10.1378/chest.130.1_suppl.4S. [DOI] [PubMed] [Google Scholar]
- 2.British Thoracic Society/Scottish Intercollegiate Guidelines Network. British Guideline on Management of Asthma. 05/08. Available at: http://www.brit-thoracic.org.uk (accessed 30 April 2009.
- 3.Global Initiative for Asthma Guidelines 11/06. Available at: http://www.ginasthma.com (accessed 30 April 2009.
- 4.Chanez P, Wenzel SE, Anderson GP, et al. Severe asthma: what are the important questions? J Allergy Clin Immunol. 2008;119:1337–48. doi: 10.1016/j.jaci.2006.11.702. [DOI] [PubMed] [Google Scholar]
- 5.Proceedings of the ATS Workshop on Refractory Asthma. Current understanding, recommendations, and unanswered questions. Am J Respir Crit Care Med. 2000;162:2341–51. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- 6.Haldar P, Pavord ID, Shaw DE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178:218–24. doi: 10.1164/rccm.200711-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green RH, Brightling CE, McKenna S, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–21. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 8.Jayaram L, Pizzichini MM, Cook RJ, et al. Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur Respir J. 2006;27:483–94. doi: 10.1183/09031936.06.00137704. [DOI] [PubMed] [Google Scholar]
- 9.Brightling CE, Berry M, Amrani Y. Anti-TNF-α in asthma. J Allergy Clin Immunol. 2008;121:5–10. doi: 10.1016/j.jaci.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarantini F, Baiardini I, Passalacqua G, Braido F, Canonica GW. Asthma treatment: ‘magic bullets which seek their own targets’. Allergy. 2007;62:605–10. doi: 10.1111/j.1398-9995.2007.01390.x. [DOI] [PubMed] [Google Scholar]
- 11.Walker S, Monteil M, Phelan K, Lasserson TJ, Walters EH. Anti-IgE for chronic asthma in adults and children. Cochrane Database of Systematic Reviews. 2006, Issue 2. Art. no.: CD003559. DOI: 10.1002/14651858.CD003559.pub3. [DOI] [PubMed]
- 12.Wardlaw AJ, Brightling C, Green R, Woltmann G, Pavord I. Eosinophils in asthma and other allergic diseases. Br Med Bull. 2000;56:985–1003. doi: 10.1258/0007142001903490. [DOI] [PubMed] [Google Scholar]
- 13.Humbles AA, Lloyd CM, McMillan SJ, et al. A critical role for eosinophils in allergic airways remodelling. Science. 2004;305:1776–9. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 14.Wardlaw AJ, Brightling CE, Green R, Woltmann G, Bradding P, Pavord ID. New insights into the relationship between airway inflammation and asthma. Clin Sci (Lond) 2002;103:201–11. doi: 10.1042/cs1030201. [DOI] [PubMed] [Google Scholar]
- 15.Brightling CE, Bradding P, Pavord ID, Wardlaw AJ. New insights into the role of the mast cell in asthma. Clin Exp Allergy. 2003;33:550–6. doi: 10.1046/j.1365-2222.2003.01636.x. [DOI] [PubMed] [Google Scholar]
- 16.Haldar P, Pavord ID. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol. 2007;119:1043–52. doi: 10.1016/j.jaci.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 17.Holgate ST. Novel targets of therapy in asthma. Curr Opin Pulm Med. 2009;15:63–71. doi: 10.1097/MCP.0b013e32831da867. [DOI] [PubMed] [Google Scholar]
- 18.Pepe C, Foley S, Shannon J, et al. Differences in airway remodelling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;166:544–9. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 19.Hammad H, Lambrecht BN. Recent progress in the biology of airway dendritic cells and implications for understanding the regulation of asthmatic inflammation. J Allergy Clin Immunol. 2006;118:331–6. doi: 10.1016/j.jaci.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 20.Medoff BD, Thomas SY, Luster AD. T cell trafficking in allergic asthma: the ins and outs. Annu Rev Immunol. 2008;26:205–32. doi: 10.1146/annurev.immunol.26.021607.090312. [DOI] [PubMed] [Google Scholar]
- 21.Larché M, Robinson DS, Kay AB. The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol. 2003;111:450–63. doi: 10.1067/mai.2003.169. [DOI] [PubMed] [Google Scholar]
- 22.Robinson DS, Hamid Q, Ying S, et al. Predominant TH-2 like bronchoalveolar T-lymphocyte populations in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 23.Brightling CE, Symon FA, Birring SS, Bradding P, Pavord ID, Wardlaw AJ. Th2 cytokine expression in bronchoalveolar lavage fluid T-lymphocytes and bronchial submucosa is a feature of asthma and eosinophilic bronchitis. J Allergy Clin Immunol. 2002;110:899–905. doi: 10.1067/mai.2002.129698. [DOI] [PubMed] [Google Scholar]
- 24.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Interleukin-4 and -13 expression is co-localized to mast cells within the airway smooth muscle in asthma. Clin Exp Allergy. 2003;33:1711–6. doi: 10.1111/j.1365-2222.2003.01827.x. [DOI] [PubMed] [Google Scholar]
- 25.Saha SK, Berry MA, Parker D, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121:685–91. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humbert M, Corrigan CJ, Durham SR, Kimmit P, Till SJ, Kay AB. Relationship between bronchial mucosal IL-4 and IL-5 mRNA expression and disease severity in atopic asthma. Am J Respir Crit Care Med. 1997;156:704–8. doi: 10.1164/ajrccm.156.3.9610033. [DOI] [PubMed] [Google Scholar]
- 27.Humbert M, Durham SR, Kimmitt P, et al. Elevated expression of messenger ribonucleic acid encoding IL-13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J Allergy Clin Immunol. 1997;99:657–65. doi: 10.1016/s0091-6749(97)70028-9. [DOI] [PubMed] [Google Scholar]
- 28.Broide DH, Firestein GS. Endobronchial allergen challenge in asthma. Demonstration of cellular source of granulocyte macrophage colony-stimulating factor by in situ hybridization. J Clin Invest. 1991;88:1048–53. doi: 10.1172/JCI115366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha SK, Doe C, Mistry V, et al. Granulocyte macrophage colony stimulating factor expression in induced sputum and bronchial mucosa in asthma and COPD. Thorax. doi: 10.1136/thx.2008.108290. 2009 Thorax Online First, published 12 February 2009 as 10.1136/thx.2008.108290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grunig G, Warnock M, Wakil AE, et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–3. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su YC, Rolph MS, Hansbro NG, Mackay CR, Sewell WA. Granulocyte–macrophage colony stimulating factor is required for bronchial eosinophilia in a murine model of allergic airway inflammation. J Immunol. 2008;180:2600–7. doi: 10.4049/jimmunol.180.4.2600. [DOI] [PubMed] [Google Scholar]
- 33.Ying S, Robinson DS, Varney V, et al. TNF alpha mRNA expression in allergic inflammation. Clin Exp Allergy. 1991;21:745–50. doi: 10.1111/j.1365-2222.1991.tb03205.x. [DOI] [PubMed] [Google Scholar]
- 34.Bradding P, Roberts JA, Britten KM, et al. Interleukin-4, -5, and -6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994;10:471–80. doi: 10.1165/ajrcmb.10.5.8179909. [DOI] [PubMed] [Google Scholar]
- 35.Howarth PH, Babu KS, Arshad HS, et al. Tumour necrosis factor (TNFα) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–8. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berry MA, Hargadon B, Shelley M, et al. Evidence of a role of tumour necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- 37.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346:1699–705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 38.Carroll NG, Mutavdzic S, James AL. Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur Respir J. 2002;19:879–85. doi: 10.1183/09031936.02.00275802. [DOI] [PubMed] [Google Scholar]
- 39.Berger P, Girodet PO, Begueret H, et al. Tryptase-stimulated human airway smooth muscle cells induce cytokine synthesis and mast cell chemotaxis. FASEB J. 2003;17:2139–41. doi: 10.1096/fj.03-0041fje. [DOI] [PubMed] [Google Scholar]
- 40.Slats AM, Janssen K, van Schadewijk A, et al. Bronchial inflammation and airway responses to deep inspiration in asthma and COPD. Am J Respir Crit Care Med. 2007;176:121–8. doi: 10.1164/rccm.200612-1814OC. [DOI] [PubMed] [Google Scholar]
- 41.Siddiqui S, Mistry V, Doe C, et al. Airway hyperresponsiveness is dissociated from airway wall structural remodeling. J Allergy Clin Immunol. 2008;122:335–41. doi: 10.1016/j.jaci.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amin K, Janson C, Boman G, Venge P. The extracellular deposition of mast cell products is increased in hypertrophic airways smooth muscles in allergic asthma but not in nonallergic asthma. Allergy. 2005;60:1241–7. doi: 10.1111/j.1398-9995.2005.00823.x. [DOI] [PubMed] [Google Scholar]
- 43.Siddiqui S, Brightling CE. Microlocalisation of inflammatory cells and structural cells: functional consequences in airways disease. Eur Respir J. 2007;30:1043–56. doi: 10.1183/09031936.00162506. [DOI] [PubMed] [Google Scholar]
- 44.Tliba O, Deshpande D, Chen H, et al. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. 2003;140:1159–62. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laporte JC, Moore PE, Baraldo S, et al. Direct effects of interleukin-13 on signalling pathways for physiological responses in cultured human airway smooth muscle cells. Am J Respir Crit Care Med. 2001;164:141–8. doi: 10.1164/ajrccm.164.1.2008060. [DOI] [PubMed] [Google Scholar]
- 46.Grunstein MM, Hakonarson H, Leiter J, et al. IL-13 dependent autocrine signalling mediates altered responsiveness of IgE-sensitized airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;282:520–8. doi: 10.1152/ajplung.00343.2001. [DOI] [PubMed] [Google Scholar]
- 47.Woodman L, Siddiqui S, Cruse G, et al. Mast cells promote airway smooth muscle cell differentiation via autocrine upregulation of TGF-β1. J Immunol. 2008;181:5001–7. doi: 10.4049/jimmunol.181.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wenzel S. Severe asthma in adults. Am J Resp Crit Care Med. 2005;172:149–60. doi: 10.1164/rccm.200409-1181PP. [DOI] [PubMed] [Google Scholar]
- 49.Kamath AV, Pavord AV, Ruparelia PR, Chilvers ER. Is the neutrophil the key effector cell in severe asthma? Thorax. 2005;60:529–30. doi: 10.1136/thx.2005.043182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Idris SF, Chilvers ER, Haworth C, McKeon D, Condliffe AM. Azithromycin therapy for neutrophilic airways disease: myth or magic? Thorax. 2009;64:186–9. doi: 10.1136/thx.2008.103192. [DOI] [PubMed] [Google Scholar]
- 51.Benayoun L, Druilhe A, Dombret M-C, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Resp Crit Care Med. 2003;167:1360–8. doi: 10.1164/rccm.200209-1030OC. [DOI] [PubMed] [Google Scholar]
- 52.Leckie MJ, ten Brinke A, Khan J, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–8. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 53.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil's role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 54.Kips J, O'Connor BJ, Langley SJ, et al. Effect of SCH55700, a humanized anti-human interleukin-5 antibody, in severe persistent asthma: a pilot study [abstract. Am J Respir Crit Care Med. 2003;167:1655–9. doi: 10.1164/rccm.200206-525OC. [DOI] [PubMed] [Google Scholar]
- 55.Flood-Page P, Swenson C, Faiferman I, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062–71. doi: 10.1164/rccm.200701-085OC. [DOI] [PubMed] [Google Scholar]
- 56.Haldar P, Brightling CE, Hargadon B, et al. Meoplizumab and exacerbations of eosinophilic refractory asthma. N Engl J Med. 2009;360:973–84. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–93. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 58.Flood-Page P, Menzies-Gow A, Phipps S, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–36. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borish LC, Nelson HS, Lanz MJ, et al. Interleukin-4 receptor in moderate atopic asthma. A phase I/II randomized, placebo-controlled trial. Am J Respir Crit Care Med. 1999;160:1816–23. doi: 10.1164/ajrccm.160.6.9808146. [DOI] [PubMed] [Google Scholar]
- 60.Borish LC, Nelson HS, Corren J, et al. Efficacy of soluble IL-4 receptor for the treatment of adults with asthma. J Allergy Clin Immunol. 2001;107:963–70. doi: 10.1067/mai.2001.115624. [DOI] [PubMed] [Google Scholar]
- 61.Hart TK, Blackburn MN, Brigham-Burke M, et al. Preclinical efficacy and safety of pascolizumab (SB240683): a humanised anti-interleukin-4 antibody with therapeutic potential in asthma. Clin Exp Immunol. 2002;130:93–100. doi: 10.1046/j.1365-2249.2002.01973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–31. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 63.Getz EB, Wilbraham D, Lalor C, Longphre M, Fuller R. Pharmacokinetics and local tolerance of an IL-4/IL-13 antagonist inhalation powder in humans. ERS Conference. 2008:545S. (abstract). Available at: http://www.ersnet.org. [Google Scholar]
- 64.Gauvreau GM, Boulet LP, Fitzgerald JM, et al. The effects of IMA-638 on allergen induced airway responses in subjects with mild atopic asthma. ERS Conference. 2008:827S. (abstract). Available at: http://www.ersnet.org. [Google Scholar]
- 65.Bhowmick B, Singh D, Molfino N, et al. A double-blind, placebo-controlled, study to assess the pharmacokinetics, safety and tolerability of multiple ascending intravenous doses of CAT 354, a recombinant human anti-IL13 antibody, in subjects with moderate asthma. ERS Conference. 2008:515S. (abstract). Available at: http://www.ersnet.org. [Google Scholar]
- 66.Banfield C, Vincent M, Kakkar T, et al. Multiple-dose study of AMG317 in adults with asthma: pharmacokinetics and safety. Am J Respir Crit Care Med. 2008;177:A568. [Google Scholar]
- 67.O'Byrne P, Boulet L-P, Gauvreau G, Leon F, Sari S, White B. A single dose of MEDI-528, a monoclonal antibody against interleukin-9, is well tolerated in mild and moderate asthmatics in the phase II trial MI-CP-138. Chest. 2007;132:478. [Google Scholar]
- 68.Boguniewicz M, Martin RJ, Martin D, et al. The effects of nebulised recombinant interferon-γ in asthmatic airways. J Allergy Clin Immunol. 1995;95:133–5. doi: 10.1016/s0091-6749(95)70162-1. [DOI] [PubMed] [Google Scholar]
- 69.Simon HU, Seelbach H, Ehmann R, Schmitz M. Clinical and immunological effects of low-dose IFNα treatment in patients with corticosteroid-resistant asthma. Allergy. 2003;58:1250–5. doi: 10.1046/j.1398-9995.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 70.Kroegel C, Bergmann N, Foerster M, et al. Interferon-alphacon-1 treatment of three patients with severe glucocorticoid-dependent asthma. Effect on disease control and systemic glucocorticoid dose. Respiration. 2006;73:566–70. doi: 10.1159/000088660. [DOI] [PubMed] [Google Scholar]
- 71.Bryan SA, O'Connor BJ, Matti S, et al. Effects of recombinant human interleukin-12 on eosinophils, airway hyperresponsiveness, and the late asthmatic response. Lancet. 2000;356:2149–53. doi: 10.1016/S0140-6736(00)03497-8. [DOI] [PubMed] [Google Scholar]
- 72.Chernoff AE, Granowitz EV, Shapiro L, et al. A randomised, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J Immunol. 1995;154:5492–9. [PubMed] [Google Scholar]
- 73.Erin EM, Leaker BR, Nicholson GC, et al. The effects of a monoclonal antibody directed against tumor necrosis factor-alpha in asthma. Am J Respir Crit Care Med. 2006;174:753–62. doi: 10.1164/rccm.200601-072OC. [DOI] [PubMed] [Google Scholar]
- 74.Morjaria JB, Chauhan AJ, Babu KS, Polosa R, Davies DE, Holgate ST. The role of a soluble TNFα receptor fusion protein (Etanercept) in corticosteroid refractory asthma: a double blind, randomised, placebo controlled trial. Thorax. 2008;63:584–91. doi: 10.1136/thx.2007.086314. [DOI] [PubMed] [Google Scholar]
- 75.Wenzel SE, Barnes PJ, Bleecker ER, et al. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-α blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009;179:549–58. doi: 10.1164/rccm.200809-1512OC. [DOI] [PubMed] [Google Scholar]
- 76.Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:926–34. doi: 10.1164/rccm.200607-995OC. [DOI] [PubMed] [Google Scholar]
- 77.Louten J, Boniface K, de Waal Malefyt R. Development and function of Th17 cells in health and disease. J Allergy. Clin Immunol. 2009;123:1004–11. doi: 10.1016/j.jaci.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 78.Al-Ramli W, Préfontaine D, Chouiali F, et al. Th-17 associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123:1185–7. doi: 10.1016/j.jaci.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 79.Ballantyne SJ, Barlow JL, Jolin HE, et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007;120:1234–1. doi: 10.1016/j.jaci.2007.07.051. [DOI] [PubMed] [Google Scholar]
- 80.Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST blocks interleukin-33 signalling in allergic airway inflammation. J Biol Chem. 2007;282:26369–80. doi: 10.1074/jbc.M704916200. [DOI] [PubMed] [Google Scholar]