

Abstract
Studies of patients with active tuberculosis (TB) and infected healthy individuals have shown that interferon (IFN)-γ is present in sites of Mycobacterium tuberculosis infection in comparable levels. This suggests that there is a deficiency in the macrophage response to IFN-γ in TB patients. We used recombinant human IFN-γ to stimulate adherent monocyte-derived macrophages from three groups of people: patients with active tuberculosis (TBP), their healthy household contacts (HHC) and healthy uninfected controls from the community (CC). We then evaluated the ability of the macrophages to inhibit the growth of M. tuberculosis H37Rv as well as their cytokine profile at early in infection (48 h). After IFN-γ treatment, macrophages of healthy individuals (HHC and CC) controlled M. tuberculosis growth and produced mainly nitric oxide (NO) and interleukin (IL)-12p70, whereas TBP macrophages did not kill M. tuberculosis. Additionally, TBP macrophages produced low levels of NO and IL-12p70 and high levels of tumour necrosis factor (TNF)-α and IL-10. Transforming growth factor (TGF)-β levels were similar among all three groups. M. tuberculosis infection had little effect on the cytokine response after IFN-γ stimulus, but infection alone induced more IL-10 and TGF-β in TBP macrophages. There were no differences in Stat1 nuclear translocation and DNA binding between the groups. However, the phosphorylated Stat1 and c-Jun (AP-1) in nuclear protein extracts was diminished in TBP macrophages compared to macrophages of healthy individuals. These results indicate an impairment of Stat1-dependent and Stat1-independent IFN-γ signalling in macrophages of people with active tuberculosis, suggesting a different molecular regulation that could impact macrophage functionality and disease outcome.
Keywords: healthy household contacts, IFN-γ signalling, macrophage, TB control, tuberculosis
Introduction
One-third of the total human population is infected with Mycobacterium tuberculosis. This bacterium causes illness in up to 9 million people annually and is responsible for three deaths every minute worldwide [1,2]. Tuberculosis (TB) is a disease which infects people with apparently normal immune systems [3]. Of all the population infected with M. tuberculosis, approximately 10% progress to disease [2]. Characterization of immunity from infected but healthy people (latency) has shown that M. tuberculosis persists within professional phagocytes in granulomas organizing the immune cell response to restrain bacterial growth [4]. This is due in part to the interaction of macrophages with antigen-dependent interferon (IFN)-γ-producing T cells [5–7] that mediate macrophage activation [5,8,9]. Activation of the macrophage with IFN-γ prior to infection with M. tuberculosis enables the host cell to overcome the block in phagosome maturation, which leads to bacterial lysis through the production of nitric oxide (NO) and reactive oxygen intermediates (ROI) [10], thus preventing bacterial dispersion and disease [11]. IFN-γ also transforms the macrophage into a potent antigen-presenting cell that induces the adaptive immune response with a dominant TH1 profile by releasing cytokines such as interleukin (IL)-12 [12].
Binding of IFN-γ to its receptor induces transphosphorylation of the receptor-associated Janus kinase (Jak)-1 and Jak2. The activated Jaks phosphorylate the intracellular domain of the IFN-γ receptor (IFNGR), which serves as a docking site for Stat1. Stat1 is phosphorylated at tyrosine 701 (Tyr 701) by Jak2. Phosphorylated Stat1 then forms a homodimer, translocates to the nucleus and regulates gene expression by binding to gamma-activated sequence (GAS) elements in the promoters of IFN-γ-regulated genes. In response to IFN-γ, IFNGR activates other kinases in a Stat1-independent manner, including mitogen-activated protein (MAP)-kinases such as c-jun N-terminal kinase (JNK), phosphatidylinositol 3-kinase-AKT, Pyk2 and extracellular-signal-regulated kinase 1/2 (ERK1/2) [13,14]. These kinases, which are also integrated in macrophage Toll-like receptor (TLR) signalling pathways, have important physiological consequences during M. tuberculosis infection [15,16].
Examining the immune response in patients with active tuberculosis has confirmed that IFN-γ is compartmentalized in infected lungs as well as in lymph nodes [17–23]. Therefore, the basis for the failure to control M. tuberculosis despite the action of IFN-γ remains unknown. The present study aimed to evaluate the phenotype and signalling cascades of IFN-γ-stimulated macrophages from patients with active tuberculosis and healthy individuals.
Materials and methods
Patients and control subjects
A total of 14 tuberculosis patients (TBP), 15 infected but healthy household contacts (HHC) and 14 naive controls from the community (CC) were recruited for the donation of whole blood to isolate and culture monocytes. Pulmonary tuberculosis was diagnosed in all TBP by acid-fast screening of a sputum smear, by being M. tuberculosis culture-positive and by chest radiography. Additionally, they all reacted within 72 h to an intradermal injection of 5 tuberculin units of purified protein derivative from M. tuberculosis with an average induration size above 17 mm in diameter (tuberculin-positive; PPD+). None of the donors had a previous history of tuberculosis, nor were they under anti-tuberculosis chemotherapy for longer than 1 week. Latently infected healthy household contacts (HHC) [19] were people who lived with a tuberculosis patient for more than 5 months, had both acid-fast sputum smear and M. tuberculosis culture negatives and were PPD+. Naive controls (CC) were people who had no known contact with any tuberculosis patients and were tuberculin-negative (<5 mm induration size; PPD−) by testing twice with an interval of a month. Subjects with any chronic or immunosuppressive disease [human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS), vascular hypertension, diabetes, hepatitis, asthma] or taking immunosuppressive medication (corticosteroids) were excluded. The median age was 41 years (range 19–50) for the TBP group, 37 years (range 23–50) for the HHC group and 27 years (range 23–34) for the CC group. All subjects provided informed consent from protocols approved by the institutional review boards of the National Institute of Respiratory Diseases (INER) and National Autonomous University of Mexico (UNAM).
Culture of human monocyte-derived macrophages and IFN-γ stimulation
PBMCs obtained from the whole venous blood of each donor were layered over Ficoll-Hypaque (Pharmacia, Uppsala, Sweden) and centrifuged for 25 min at 1200 g at room temperature. After washing four times with phosphate-buffered saline (PBS) containing 10% autologous plasma, peripheral blood mononuclear cells (PBMCs) were incubated at 37°C in RPMI-1640 culture medium supplemented with 5% fetal bovine serum (FBS) in 100 mm plastic Petri dishes. After 3 h the non-adherent cells were washed with four changes of warmed RPMI-1640 medium. Adherent cells were >95% monocytes as determined by flow cytometry with anti-CD14 [phycoerythrin (PE)-conjugated] and anti-CD11b (Alexa 488-conjugated) antibodies (Beckton Dickinson, San Diego, CA, USA). Human monocyte-derived macrophages (referred to hereafter as macrophages) were prepared by culturing monocytes at 37°C in a humidified 5% CO2 atmosphere for 7 days in the presence of 20% FBS plus 2% human AB pooled serum. Once differentiated, macrophages were stimulated to induce cell size enlargement, membrane protrusions and nitric oxide production by treating cells for 22 h in serum-free RPMI-1640 medium containing human recombinant IFN-γ (500 U/ml; Peprotech, Rocky Hill, NJ, USA). Macrophages were certified as uninfected prior to our experiments based on three assays: (i) inspection of adherent monocyte-derived macrophages of each subject by viewing 100 microscope fields after auramine–rhodamine staining, (ii) plating serial dilutions of cell lysates onto Middlebrook 7H10 agar and (iii) polymerase chain reaction (PCR) and DNA-hybridization of the CFP-10 gene of M. tuberculosis (sensitivity limit of 5 fg of genomic DNA) in DNA extracts of 1 × 106 macrophages.
Measurement of mycobactericidal response of human macrophages
Our protocol was adopted from a previous study by Wong et al.[24]. We quantified the M. tuberculosis growth index in human macrophages. Briefly, we used live virulent M. tuberculosis H37Rv strain (ATCC 25618) grown to mid-log phase in Middlebrook 7H9 broth (Difco, Detroit, MI, USA) supplemented with 10% ADC (albumin, dextrose, catalase; Becton & Dickinson, San Jose, CA, USA) and 0·05% Tween 80 (Sigma, St Louis, MO, USA). Bacteria were washed and suspended in RPMI-1640 medium. Suspensions of mycobacteria were sonicated to disperse clumps as described previously [25] and were added to triplicate batches of adherent human macrophages at a multiplicity of infection (MOI) of 10 viable bacilli [colony-forming units (CFU)]. After 4 h of incubation, cells were washed three times with pre-warmed PBS to remove extracellular bacteria. This was considered time 0. At times 0 and 48 h, infected macrophages were lysed by incubating with PBS containing 0·1% Tween 20 for 10 min followed by pipetting. The organisms were then processed for the CFU assay as described previously [25]. Serial 10-fold dilutions of the bacterial suspension were made and plated on Middlebrook 7H10 agar plates supplemented with 10% of OADC (ADC plus oleic acid). After 21 days, dilutions containing 10–200 colonies, each arising from a single mycobacterium, were counted. As a control, equal inocula of mycobacteria were added to wells without macrophages and cultured under the same conditions as infected macrophages. The growth index was defined as the number of CFU at 48 h of culture with macrophages divided by the number of CFU at 0 h of culture with macrophages. A growth index greater than 1 indicated that the strain multiplied inside human macrophages, whereas a growth index of less than 1 indicated that the bacteria were engulfed and gradually killed by macrophages [24].
Measurement of cytokines and nitric oxide
Supernatants from triplicate cultures of macrophages were collected at 48 h post-infection. All supernatants were immediately snap-frozen at −70°C before being tested. Cytokine assays were performed for IL-10, IL-12p70, tumour necrosis factor alpha (TNF-α) and transforming growth factor (TGF)-β using a kit according to the manufacturer's instructions (DuoSet ELISA development system; R&D Systems, Inc., Minneapolis, MN, USA). Nitrite levels, as a stable product of NO, were determined by the Griess assay as published elsewhere [26]. The amount of NO was calculated from a standard curve of NaNO2.
Preparation of nuclear and cytoplasmic extracts
Macrophage monolayers were washed and incubated for 10 min with ice-cold PBS, scraped into PBS with 1 mM phenymethylsulphonyfluoride (PMSF) and pelleted by centrifugation (500 g) at 4°C for 10 min. Cell pellets were resuspended in buffer A (20 mM HEPES, pH 7·9, 10 mM KCl, 0·1 mM ethylenediamine tetraacetic acid (EDTA), 1 mM ethyleneglycol tetraacetic acid (EGTA), 1 mM sodium orthovanadate, 50 mM NaF, 1 mM PMSF, 20 mg/ml aprotinin, 20 mg/ml antipain, 20 mg/ml leupeptin and 10 mg/ml pepstatin A) and incubated on ice for 10 min. Nonidet P-40 was added to a final concentration of 0·2% and the cell suspension was passed through a 26-gauge needle to break open the cells. After centrifugation (12 000 g, in a microcentrifuge) at 4°C for 1 min, supernatants were collected as cytoplasmic extracts, and the pellets (crude nuclei) were resuspended in buffer B (20 mM HEPES, pH 7·9, 0·4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol (DTT), 1 mM sodium orthovanadate, 50 mM NaF and the same protease inhibitors as in buffer A). After incubating for 60 min on ice, insoluble materials were pelleted by centrifugation (15 000 g) at 4°C for 10 min and supernatants were collected as nuclear extracts.
Western blot and electrophoretic mobility shift assay (EMSA)
Western blots using cytoplasmic and nuclear lysates, and EMSAs with nuclear lysates were performed to determine signal activation. Cells treated with 500 U/ml of IFN-γ for 45 min were processed for cytoplasmic and nuclear extraction as mentioned above. Equal amounts of protein samples were run on a 10% sodium dodecyl sulphate (SDS)-polyacrylamide gel in reducing conditions followed by transfer onto a polyvinylidene difluoride (PVDF) membrane. The blots were blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) with 0·1% Tween 20. Western blots were then probed with antibodies recognizing phosphorylated forms of Stat1 (Tyr701) (1/200; Santa Cruz Biotechnologies, Santa Cruz, CA, USA), AKT (1/1000; Cell Signaling Technology, Danvers, MA, USA) and c-Jun (Cell Signaling Technology; 1/1000). The secondary antibodies used were horseradish peroxidase (HRP)-conjugated anti-mouse (1/10 000; Jackson, West Grove, PA, USA) or anti-rabbit (1/5000; Zymed, San Francisco, CA, USA) and were detected by using the Inmobilon Western system (Millipore, Billerica, MA, USA). Membranes were subsequently stripped (100 mM β-mercaptoethanol, 2% SDS, 62·5 mM Tris-HCl; at 50°C for 30 min) and reprobed with antibodies that bound non-phosphorylated forms of Stat1, AKT and c-Jun (all at 1/200; all from Santa Cruz Biotechnologies). EMSA was performed with 10 µg of nuclear protein incubated with a 32P-end-labelled double-stranded DNA oligonucleotide (5′-CATGTTATGCATATTCCTGTAAGTG-3′; underlining indicates GAS binding site for Stat1; Santa Cruz Biotechnology). Binding reactions were performed for 10 min at room temperature in 1× binding buffer (50 mM Tris-HCl, 20% glycerol, 250 mM NaCl, 5 mM MgCl2, 2·5 mM EDTA, 1 mM DTT, 1 mM PMSF), with 1 µg of poly(dI-dC) and 50- or 100-fold molar excess unlabelled competitor or Supershift antibodies for Stat1 and phospho-Stat1 (Tyr 701) (1 µl; Santa Cruz Biotechnology) when needed. Protein complexes were separated from the free probe by electrophoresis in native 5% polyacrylamide gels prepared with 0·5× Tris–borate–EDTA (TBE) buffer and radiographed on X-ray film.
Statistical analysis
Data were analysed by Wilcoxon signed-rank test for paired comparisons (resting versus activated), and Kruskal–Wallis test for differences between groups using spss software (version 10).
Results
Mycobacterium tuberculosis survives well in IFN-γ-stimulated macrophages of patients with active tuberculosis
We observed a higher number of M. tuberculosis H37Rv CFUs in macrophages of patients with active tuberculosis (TBP) with and without prior IFN-γ stimulus both immediately after removal of non-phagocytosed bacteria (Fig. 1a), and after 48 h of growth than M. tuberculosis CFUs obtained of macrophages from healthy subjects (Fig. 1b). Even though TBP macrophages had more mycobacterial binding and phagocytic activity, they were unable to control mycobacterial growth when compared to macrophages from healthy subjects, as indicated by the marked difference in mycobacterial load reduction by these cells pretreated with IFN-γ (P < 0·005, Wilcoxon signed-rank test; Fig. 1c). Indeed, in some cases M. tuberculosis grew better in IFN-γ-stimulated TBP macrophages (Fig. 1c). These results suggest that IFN-γ treatment in TBP macrophages did not induce the mycobactericidal mechanisms which presumably occurred in macrophages of healthy infected (HHC) and uninfected (CC) people. Interestingly, IFN-γ-induced inhibition of mycobacterial growth was more marked in macrophages of subjects who were naive to infection (CC) (P < 0·001, Kruskal–Wallis test).
Fig. 1.
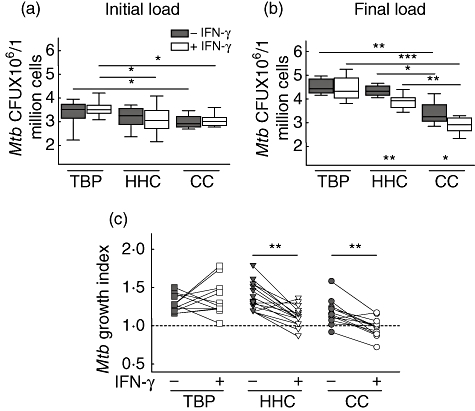
Interferon (IFN)-γ allows the growth of Mycobacterium tuberculosis in macrophages of patients with active tuberculosis. Boxes of median-quartile range and whiskers of min to max were performed with the 10 viable bacilli [colony-forming units (CFU)] mean of triplicates of macrophage batches of each subject at 0 h (a) and 48 h (b) post-infection and grouped in tuberculosis patients (TBP, n = 14), healthy household contacts (HHC, n = 15) and healthy uninfected community controls (CC, n = 14). Significant differences are at *P < 0·05, **P < 0·01 and ***P < 0·001 with Wilcoxon signed-rank test for comparisons within groups and with Kruskal–Wallis test for comparisons between groups. M. tuberculosis growth index (c) is expressed as the fold change over time of bacterial growth in macrophages cultured in media alone (solid symbols) or with IFN-γ (empty symbols) and was calculated by dividing mean CFU at 48 h (final load) by mean CFU at 0 h (initial load) for each donor.
IFN-γ induced low bactericidal mechanisms and a dominant anti-inflammatory profile in macrophages of TB patients
NO, which is produced from L-arginine in a reaction catalyzed by inducible NO synthase (iNOS) in macrophages, is an IFN-γ induced endogenous free radical that mediates mycobacterial killing, inflammation and cell signalling which, together with cytokines such as IL-12p70 and TNF-α, participates in the control and apparent inactivation of M. tuberculosis. We observed that continuous stimulus with IFN-γ (500 U/ml for 22 h) induced macrophages of the TBP group to produce low levels of NO and IL-12p70, but high levels of TNF-α and IL-10 at 48 h after IFN-γ removal. This is contrary to what was observed for both healthy infected (HHC) and uninfected (CC) groups, where macrophages responded to IFN-γ by producing higher levels of NO, IL-12p70 and TGF-β and low levels of IL-10 and TNF-α (Fig. 2). Interestingly, M. tuberculosis infection alone induced TBP cells to produce more IL-10 and TGF-β than macrophages of healthy subjects. However, M. tuberculosis did not alter the overall responsiveness to IFN-γ because NO and cytokines were produced similarly in IFN-γ-prestimulated macrophages of all groups after M. tuberculosis infection. This reflects the macrophage bactericidal responsiveness to IFN-γ of each group; the more NO produced, the greater the inhibition of M. tuberculosis growth by macrophages.
Fig. 2.
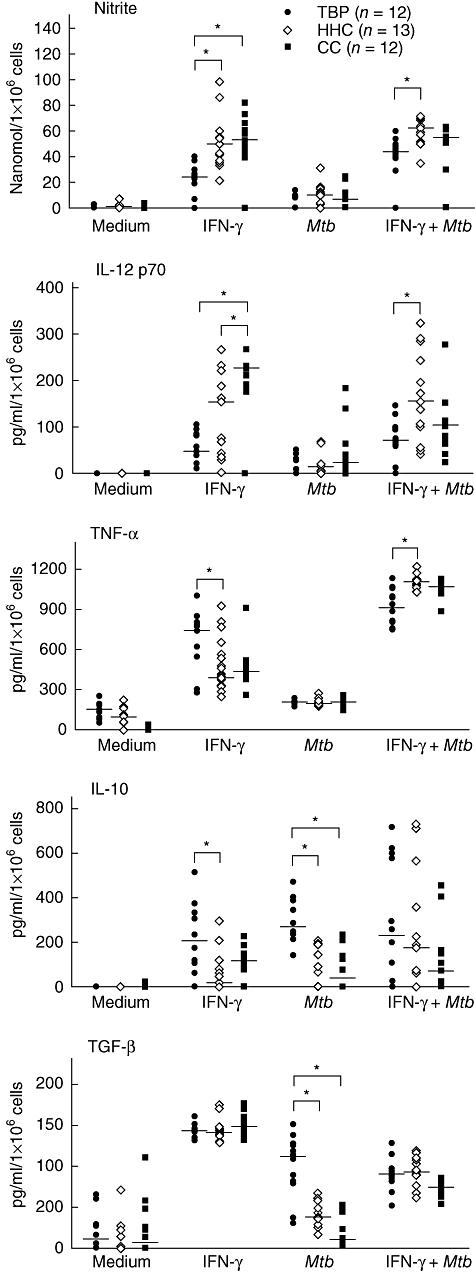
Anti-inflammatory response to interferon (IFN)-γ and Mycobacterium tuberculosis in macrophages of patients with active tunerculosis. Plots are the mean of two replicate values of two independent experiments of each subject. Lanes at median values of each group. *p < 0·05 with the Kruskal–Wallis test.
IFN-γ-signalling is down-regulated in macrophages from TB patients
Because we observed that TBP macrophages respond to IFN-γ in an opposite manner to HHC and CC macrophages, we analysed their Stat1-dependent and -independent signalling pathways. Once it was shown that Stat1 signalling is activated rapidly in an IFN-γ-dependent manner (Fig. 3a and b), we observed at the half of its signalling intensity (45 min of exposure to IFN-γ) (Fig. 3c) that there were no disruptions of such signal pathway in TBP macrophages, nor were there any differences in DNA-binding ability of Stat1 within the groups. These data indicate that the different functionalities of those cells were independent of the ability of Stat1 to translocate to the host cell nucleus and bind to the consensus DNA sequence (GAS). Therefore, to determine the role of IFN-γ induced Stat1-dependent and -independent signalling events in the phenotype of these macrophages, we analysed the intensity of activation of Stat1, AKT and c-Jun by Western blotting for phosphorylated proteins in cytoplasmic and nuclear extracts. As shown in Fig. 4, Stat1 and c-Jun are activated concomitantly by IFN-γ with low and prominent strength in TBP and HHC macrophages, respectively, whereas in CC macrophages both proteins are activated dissimilarly. The AKT activation was relatively similar between groups, confirming that macrophage-specific signalling pathways are regulated differentially in relation to donor status.
Fig. 3.
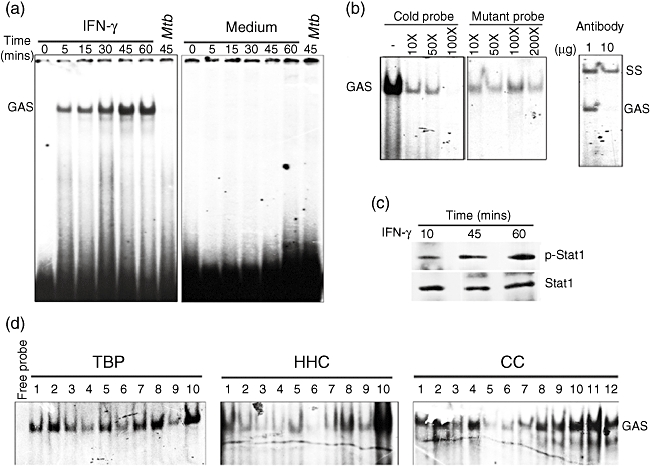
Stat1-dependent interferon (IFN)-γ signalling is accomplished in Mycobacterium tuberculosis-infected macrophages of patients with active tuberculosis (TB) as healthy normal subjects. Ten micrograms of nuclear extracts were incubated with 32P-labelled DNA probe containing the gamma interferon activating sequence (GAS) and then assayed for electrophoretic mobility shift assay (EMSA) (a). Stat1-GAS specific binding was corroborated by competing with the same unlabelled probe (cold probe) or with a probe mutated for GAS site (mutant probe) or by using an anti-Stat1 antibody to produce a supershift (SS) in EMSAs of nuclear extracts of macrophages incubated 45 min with IFN-γ (b). Stat1 phosphorylation (Tyr701) was corroborated by Western blotting of nuclear extracts (c). EMSAs were performed with 10 µg of nuclear extracts of M. tuberculosis-infected macrophages (48 h) incubated for 45 min with IFN-γ in patients with active TB (TBP, n = 10), healthy household contacts (HHC, n = 10) and healthy uninfected subjects (CC, n = 12) (d).
Fig. 4.
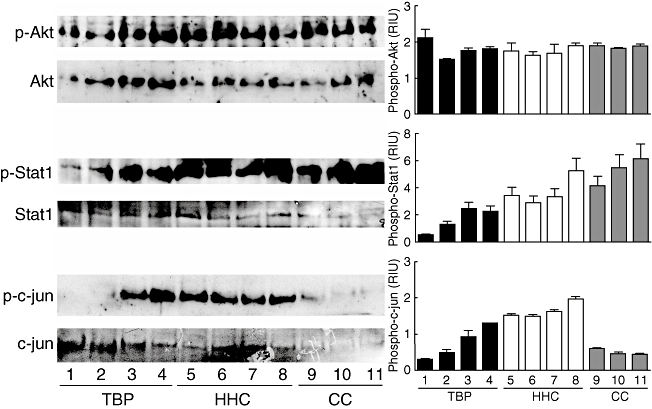
Down-activation of Stat1 and c-Jun in interferon (IFN)-γ-treated macrophages from patients with active tuberculosis (TB). Uninfected macrophages of patients with active tuberculosis (TBP), healthy household contacts (HHC) and healthy uninfected subjects (CC) were incubated with IFN-γ (500 U/ml) for 45 min and processed for cytoplasmic and nuclear extraction (see Material and methods). Western blots were assayed with 10 µg of cytoplasmic protein for phosphorylated AKT (p-Akt) (Thr308) and 10 µg of nuclear protein for phosphorylated Stat1 (p-Stat1)(Tyr701) and phosphorylated c-Jun (p-c-jun) (Ser73). The blots were stripped and re-probed with antibodies against total AKT, Stat1 and c-Jun, respectively. Densitometry analysis was performed in blots and relative intensity units (RIU) were calculated by dividing values of phosphorylated protein in values of total protein.
Discussion
The data presented here show that IFN-γ and M. tuberculosis stimuli induce a different response on human monocyte-derived macrophages of patients with active tuberculosis than is induced on macrophages from healthy but M. tuberculosis-infected (HHC) or uninfected (CC) human controls. The comparisons between patients and controls were focused upon three points: the capability of their macrophages to limit the growth of the pathogen, their cytokine response at early evaluation times and the intensity of intracellular signals activated by IFN-γ.
Macrophages and IFN-γ-producing lymphocytes are considered the main effectors of the protective immune response against tuberculosis. These cells respond by containing mycobacteria in granulomatous lesions, resulting in a lack of clinical symptoms, otherwise known as latency [4]. However, in less than 10% of infected individuals, M. tuberculosis infection progresses to active disease in the face of a prominent TH1 immune response with the presence of IFN-γ, indicating a deficiency in cell responsiveness to IFN-γ.
Because IFN-γ augments the bactericidal function of macrophages by stimulating the synthesis of reactive oxygen intermediates and nitric oxide, the level of intracellular mycobacterial growth has been correlated with the intensity of the immune response under physiological conditions [25]. In our study, treatment with IFN-γ led rapidly to a decrease in growth of M. tuberculosis in macrophages from both control groups but not in TBP macrophages (Fig. 1). This effect was not due to phagocytosis, because IFN-γ treatment did not reduce the number of phagocytosed bacteria. However, phagocytic activity was different between groups. Macrophages of uninfected subjects (CC) with or without IFN-γ treatment phagocytosed fewer mycobacteria than TBP, but not HHC macrophages. TBP and HHC macrophages showed differences in phagocytosis only with IFN-γ treatment. Moreover, M. tuberculosis displayed better growth in TBP macrophages than in HHC and CC macrophages, which were more effective at controlling growth. Indeed, when treated with IFN-γ, TBP macrophages were highly permissive to M. tuberculosis growth, while the opposite was true for CC macrophages. These results indicate that macrophage bactericidal response to IFN-γ is reduced in subjects with active TB.
Differences in macrophage phagocytosis between patients and controls may be related to the possibility of differences in density of surface phagocytic receptors for M. tuberculosis, in particular of those associated with its intracellular growth permissiveness [27,28], like the TBP macrophage response shown in our study. The human mannose receptor directs mycobacterial–phagosome biogenesis [29] to M. tuberculosis tolerance by inhibiting ligand-induced proinflammatory effects on macrophages [30,31]. A reduction in surface density of this receptor in macrophages reduces M. tuberculosis intake [27], thus an increase in M. tuberculosis phagocytosis, as observed for TBP macrophages, suggests an increase in this receptor. Moreover, the highest levels of M. tuberculosis-induced IL-10 and TGF-β produced by TBP macrophages (Fig. 2) are in agreement with the possibility that differences in receptors are accounting for the differences in cell response at the ligand level [32]. Although we did not evaluate density or functionality of any phagocytic or innate immune receptor, it is an important factor that needs future attention.
The improvement of M. tuberculosis killing by macrophages of healthy subjects in our study disagrees with the prior notion that incubation of monocyte-derived human macrophages with IFN-γ did not improve M. tuberculosis killing [33,34]. Differences between these studies and our study may be related to differences in the time used to evaluate mycobactericidal activity. The reason that other studies show a slight reduction, or even an increase, in viability of M. tuberculosis is due to IFN-γ-induced death of M. tuberculosis-infected cells [7,35]. This macrophage death results in an increase in the number of free-cell mycobacteria and therefore in an increase in the MOI, as reported by themselves [33]. The increase in intracellular growth was probably caused by increasing phagocytosis rates due to the higher availability of bacteria, rather than a deficiency in controlling its growth. Moreover, in our conditions of IFN-γ with mycobacterial stimulation, cells began to detach after 3 days, affecting their functionality. Therefore we decided not to evaluate times beyond 48 h post-infection.
The mycobacterial growth control found in our study led us to examine bactericidal mechanisms used by the macrophages. Macrophages with low NO production harboured the highest mycobacterial loads, while high NO producers had the lowest counts for living mycobacteria. Concordantly, TBP macrophages are low NO producers and HHC and CC macrophages are high NO producers. These data indicate that activation or the strength of oxidative burst in TBP macrophages is reduced compared to cells from healthy hosts. The data also indicate that there is not a lack of response to IFN-γ or impairment of its signal pathway in TBP cells, because IFN-γ treatment increased their TNF-α and IL-10 production further with significant prominence above HHC and CC macrophages (Fig. 2). This suggests a dissimilar regulation of IFN-γ response. Park et al. did not find any differences in the rate in which TNF-α production increased between IFN-γ-treated macrophages of patients with clinical TB and controls with clear chest radiography prior to lipopolysaccharide (LPS) stimulation [36]. However, it is difficult to determine whether or not a subtle change in the IFN-γ response in TBP macrophages is reflected in TNF-α production, because of the LPS stimulus. Moreover, the sample size of groups they used was too small to determine a functional difference.
Due to the different functional responses of macrophages observed in our study, we decided to evaluate M. tuberculosis-infected macrophages from all groups for their Stat1-dependent IFN-γ signal transduction pathway by means of nuclear translocation and DNA binding. We did not find substantial differences among individuals, indicating that there was no disruption of signal transduction or dysfunction of this transcription activator in TBP cells (Fig. 3). These results confirm that impairment of the IFN-γ signalling pathway does not account for cases of clinical TB. Condos et al.[37] showed that the refractory response to IFN-γ in alveolar macrophages from multi-drug resistance tuberculosis patients reverted with aerosol treatment of recombinant IFN-γ that induced Stat1 activation and improved the clinical course of patients. These data confirm that the IFN-γ signal transduction system is functional, and suggests that a modification in the response to IFN-γ, beyond the deleterious effects caused by M. tuberculosis[28,38,39], could be responsible for the development of clinical tuberculosis, while the onset and establishment of infection depends upon the nature of the pathogen.
In fact, our observations describe a non-classical response to IFN-γ in TBP macrophages. How this response to IFN-γ relates to its well-documented influence on macrophage activation, which involves functional up-regulation of several proinflammatory response genes, is not clear. One possible explanation is through the regulation of signalling pathways. Among the most important endogenous regulatory factors of signalling pathways is the signal transduction cross-talk that results in the downstream activation of pro- or anti-inflammatory genes [40]. Stat1 is considered one of the cross-talk regulators that influence the activation or repression of genes through synergism or antagonism with transcription factors belonging to other signalling pathways [41]. We found that although IFN-γ-dependent Stat1 activation was reduced in TBP macrophages (Fig. 4), this resulted in turn in less IFN-γ-dependent gene activation of IL-12 and iNOS, as suggested by the low levels of their final products (Fig. 3). This correlates the quality of the signal activation with both the phenotype of the cell and the infection/inflammatory status of the donor. It is well documented that a sustained and strong activation of Stat1 leads to increased IFN-γ-dependent activation of genes related to M. tuberculosis protection [13]. Besides this, activation of other transcription factors downstream of several signalling pathways may enhance the response, inhibit it or be necessary for cells to respond fully to the essential Stat1 IFN-γ-dependent signalling [14,42]. In addition to the well known Jak2–Stat1 signalling pathway, IFN-γ activates Pyk2, PI3-K/Akt, mitogen-activated protein kinases (MAPK) including extracellular signal-regulated kinases (ERK1/2), p38 MAPK and c-Jun N-terminal kinases/stress-activated protein kinases (JNKs/SAPKs) [14,42]. Activation of c-Jun is required for the activation of IFN-γ response genes through AP-1, but is independent of Stat1 signalling [43]. Here we present evidence that c-Jun is activated differentially through IFN-γ in macrophages of patients with active disease, and in individuals with precedent of infection but without clinical disease (HHC). Moreover, c-Jun is activated strongly in HHC cells and minimally in cells of diseased individuals and naive controls. These results show that activation of c-Jun plays a key role in regulating IFN-γ signalling in a Stat1-independent manner. Interestingly, the activation of other Stat1-independent signalling pathways such as PI3-K/AKT are similar between groups, indicating that functional modification involves only particular signalling pathways, and reinforcing the notion that a regulatory mechanism is related to a precedent of infection. Under physiological circumstances (no tuberculosis infection or disease), the mechanism of action of IFN-γ on the macrophage results in IL-10 gene down-regulation by GSK3 activation which, in turn, contributes to the inhibition of AP-1–DNA binding by reducing AP-1 expression [44]. The elevated production of IL-10 together with the diminished c-Jun activation induced by IFN-γ suggests that this mechanism is altered in TBP macrophages.
Our results provide evidence that dysfunction of macrophages in response to IFN-γ is dependent not only on the Stat1 signalling pathway, but on AP-1 activation. There has not been a reported dysfunction of c-Jun proteins, referred to as gene mutations or polymorphisms, associated with susceptibility to mycobacterial diseases [45]. However, alterations in the regulation of AP-1 proteins (c-jun/c-fos) are well described in human pathologies of chronic inflammation, resulting in modification of macrophage functionality in response to inflammatory stimulus or cytokines [46]. Macrophage heterogeneity has been altered in chronic inflammatory diseases such as asthma, organ tissue fibrosis, arthritis, obesity and even in granulomatous diseases [47–49]. As a result of loss of equilibrium in host mechanisms of inflammation, macrophages could be polarized into functional populations that respond differently to the same stimulus. Polarized human macrophage phenotypes have been described previously and are named M1 and M2 macrophages [by transposition of T helper type 1 (Th1) and Th2 phenotypes]. M1 and M2 populations are derived by culturing with granulocyte–macrophage colony-stimulating factor (GM–CSF) or M-CSF and produce mainly IL-12 or IL-10, respectively [50]. Such phenotypes are preserved even after their activation with different stimulus: LPS, zymosan, IFN-γ or CD40L. Macrophage polarization has been identified previously in human populations by defining monocyte subsets through cell surface markers with specific differential properties observed in vitro as well as in vivo[51,52]. Such macrophage polarization could be present in individuals with active tuberculosis, thus affecting the response of cells to a protective signal such as IFN-γ. Our results show a non-classical IFN-γ-response in monocyte-derived macrophages from TB patients, derived from an altered regulatory mechanism in Stat1-independent and -dependent signalling pathways. Whether this is the reason for tuberculosis susceptibility or is a consequence of it still remains unknown.
Acknowledgments
We thank Gabriel Jesús Moreno González and Gabriela Rodríguez Rodríguez for technical assistance with EMSA. Also we thank Programa de Doctorado en Ciencias Biomédicas. This work was funded by grant DGAPA PAPIIT-IN225506 and -IN209708 from the Universidad Nacional Autónoma de México (UNAM), Doctoral scholarship grant CONACyT-158472 from Consejo Nacional de Ciencia y Tecnología, Mexico; and the Programa Transdiciplinario en Investigación y Desarrollo para Facultades y Escuelas del Macroproyecto ‘Nuevas Estrategias Epidemiológicas, Genómicas y Proteómicas en Salud Pública UNAMSDEI.PTID.05·04.
Disclosure
The authors do not have any conflict of interests to declare.
References
- 1.Raviglione MC. The TB epidemic from 1992 to 2002. Tuberculosis (Edinb) 2003;83:4–14. doi: 10.1016/s1472-9792(02)00071-9. [DOI] [PubMed] [Google Scholar]
- 2.Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat Rev Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- 3.Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. Immunol J Clin. 2007;27:347–62. doi: 10.1007/s10875-007-9084-0. [DOI] [PubMed] [Google Scholar]
- 4.Ulrichs T, Kosmiadi GA, Jorg S, et al. Differential organization of the local immune response in patients with active cavitary tuberculosis or with nonprogressive tuberculoma. J Infect Dis. 2005;192:89–97. doi: 10.1086/430621. [DOI] [PubMed] [Google Scholar]
- 5.Carranza C, Juarez E, Torres M, Ellner JJ, Sada E, Schwander SK. Mycobacterium tuberculosis growth control by lung macrophages and CD8 cells from patient contacts. Am J Respir Crit Care Med. 2006;173:238–45. doi: 10.1164/rccm.200503-411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufmann SH. Protection against tuberculosis: cytokines, T cells, and macrophages. Ann Rheum Dis. 2002;61(Suppl 2):ii54–8. doi: 10.1136/ard.61.suppl_2.ii54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonecini-Almeida MG, Chitale S, Boutsikakis I, et al. Induction of in vitro human macrophage anti-Mycobacterium tuberculosis activity: requirement for IFN-gamma and primed lymphocytes. J Immunol. 1998;160:4490–9. [PubMed] [Google Scholar]
- 8.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to infection. J Exp Med. 1993;178:2249–54. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorman SE, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364:2113–21. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 10.Macmicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–8. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–7. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zissel G, Ernst M, Schlaak M, Muller-Quernheim J. Pharmacological modulation of the IFNgamma-induced accessory function of alveolar macrophages and peripheral blood monocytes. Inflamm Res. 1999;48:662–8. doi: 10.1007/s000110050519. [DOI] [PubMed] [Google Scholar]
- 13.Hu X, Herrero C, Li WP, et al. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3:859–66. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 14.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 15.Yang CS, Lee JS, Song CH, et al. Protein kinase C zeta plays an essential role for Mycobacterium tuberculosis-induced extracellular signal-regulated kinase 1/2 activation in monocytes/macrophages via Toll-like receptor 2. Cell Microbiol. 2007;9:382–96. doi: 10.1111/j.1462-5822.2006.00797.x. [DOI] [PubMed] [Google Scholar]
- 16.Yang CS, Shin DM, Lee HM, et al. ASK1-p38 MAPK-p47phox activation is essential for inflammatory responses during tuberculosis via TLR2-ROS signalling. Cell Microbiol. 2008;10:741–54. doi: 10.1111/j.1462-5822.2007.01081.x. [DOI] [PubMed] [Google Scholar]
- 17.Barnes PF, Fong SJ, Brennan PJ, Twomey PE, Mazumder A, Modlin RL. Local production of tumor necrosis factor and IFN-gamma in tuberculous pleuritis. J Immunol. 1990;145:149–54. [PubMed] [Google Scholar]
- 18.Bai X, Wilson SE, Chmura K, Feldman NE, Chan ED. Morphometric analysis of Th(1) and Th(2) cytokine expression in human pulmonary tuberculosis. Tuberculosis (Edinb) 2004;84:375–85. doi: 10.1016/j.tube.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Schwander SK, Torres M, Carranza CC, et al. Pulmonary mononuclear cell responses to antigens of Mycobacterium tuberculosis in healthy household contacts of patients with active tuberculosis and healthy controls from the community. J Immunol. 2000;165:1479–85. doi: 10.4049/jimmunol.165.3.1479. [DOI] [PubMed] [Google Scholar]
- 20.Ribera E, Ocana I, Martinez-Vazquez JM, Rossell M, Espanol T, Ruibal A. High level of interferon gamma in tuberculous pleural effusion. Chest. 1988;93:308–11. doi: 10.1378/chest.93.2.308. [DOI] [PubMed] [Google Scholar]
- 21.Lin Y, Zhang M, Hofman FM, Gong J, Barnes PF. Absence of a prominent Th2 cytokine response in human tuberculosis. Infect Immun. 1996;64:1351–6. doi: 10.1128/iai.64.4.1351-1356.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson DS, Ying S, Taylor IK, et al. Evidence for a Th1-like bronchoalveolar T-cell subset and predominance of interferon-gamma gene activation in pulmonary tuberculosis. Am J Respir Crit Care Med. 1994;149:989–93. doi: 10.1164/ajrccm.149.4.8143065. [DOI] [PubMed] [Google Scholar]
- 23.Barnes PF, Lu S, Abrams JS, Wang E, Yamamura M, Modlin RL. Cytokine production at the site of disease in human tuberculosis. Infect Immun. 1993;61:3482–9. doi: 10.1128/iai.61.8.3482-3489.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong KC, Leong WM, Law HK, et al. Molecular characterization of clinical isolates of Mycobacterium tuberculosis and their association with phenotypic virulence in human macrophages. Clin Vaccine Immunol. 2007;14:1279–84. doi: 10.1128/CVI.00190-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laochumroonvorapong P, Paul S, Manca C, Freedman VH, Kaplan G. Mycobacterial growth and sensitivity to H2O2 killing in human monocytes in vitro. Infect Immun. 1997;65:4850–7. doi: 10.1128/iai.65.11.4850-4857.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–8. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 27.Schlesinger LS. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol. 1993;150:2920–30. [PubMed] [Google Scholar]
- 28.Banaiee N, Kincaid EZ, Buchwald U, Jacobs WR, Jr, Ernst JD. Potent inhibition of macrophage responses to IFN-gamma by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. J Immunol. 2006;176:3019–27. doi: 10.4049/jimmunol.176.5.3019. [DOI] [PubMed] [Google Scholar]
- 29.Kang PB, Azad AK, Torrelles JB, et al. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. 2005;202:987–99. doi: 10.1084/jem.20051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang BK, Schlesinger LS. Characterization of mannose receptor-dependent phagocytosis mediated by Mycobacterium tuberculosis lipoarabinomannan. Infect Immun. 1998;66:2769–77. doi: 10.1128/iai.66.6.2769-2777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nadesalingam J, Dodds AW, Reid KB, Palaniyar N. Mannose-binding lectin recognizes peptidoglycan via the N-acetyl glucosamine moiety, and inhibits ligand-induced proinflammatory effect and promotes chemokine production by macrophages. J Immunol. 2005;175:1785–94. doi: 10.4049/jimmunol.175.3.1785. [DOI] [PubMed] [Google Scholar]
- 32.Chang JS, Huggett JF, Dheda K, Kim LU, Zumla A, Rook GA. Myobacterium tuberculosis induces selective up-regulation of TLRs in the mononuclear leukocytes of patients with active pulmonary tuberculosis. J Immunol. 2006;176:3010–18. doi: 10.4049/jimmunol.176.5.3010. [DOI] [PubMed] [Google Scholar]
- 33.Douvas GS, Looker DL, Vatter AE, Crowle AJ. Gamma interferon activates human macrophages to become tumoricidal and leishmanicidal but enhances replication of macrophage-associated mycobacteria. Infect Immun. 1985;50:1–8. doi: 10.1128/iai.50.1.1-8.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rook GA, Steele J, Ainsworth M, Champion BR. Activation of macrophages to inhibit proliferation of Mycobacterium tuberculosis: comparison of the effects of recombinant gamma-interferon on human monocytes and murine peritoneal macrophages. Immunology. 1986;59:333–8. [PMC free article] [PubMed] [Google Scholar]
- 35.Placido R, Mancino G, Amendola A, et al. Apoptosis of human monocytes/macrophages in Mycobacterium tuberculosis infection. J Pathol. 1997;181:31–8. doi: 10.1002/(SICI)1096-9896(199701)181:1<31::AID-PATH722>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 36.Park GY, Im YH, Ahn CH, et al. Functional and genetic assessment of IFN-gamma receptor in patients with clinical tuberculosis. Int J Tuberc Lung Dis. 2004;8:1221–7. [PubMed] [Google Scholar]
- 37.Condos R, Raju B, Canova A, et al. Recombinant gamma interferon stimulates signal transduction and gene expression in alveolar macrophages in vitro and in tuberculosis patients. Infect Immun. 2003;71:2058–64. doi: 10.1128/IAI.71.4.2058-2064.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fortune SM, Solache A, Jaeger A, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J Immunol. 2004;172:6272–80. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- 39.Ting LM, Kim AC, Cattamanchi A, Ernst JD. Mycobacterium tuberculosis inhibits IFN-gamma transcriptional responses without inhibiting activation of STAT1. J Immunol. 1999;163:3898–906. [PubMed] [Google Scholar]
- 40.Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol. 2007;82:237–43. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- 41.Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex roles of Stat1 in regulating gene expression. Oncogene. 2000;19:2619–27. doi: 10.1038/sj.onc.1203525. [DOI] [PubMed] [Google Scholar]
- 42.Ramana CV, Gil MP, Han Y, Ransohoff RM, Schreiber RD, Stark GR. Stat1-independent regulation of gene expression in response to IFN-gamma. Proc Natl Acad Sci USA. 2001;98:6674–9. doi: 10.1073/pnas.111164198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gough DJ, Sabapathy K, Ko EY, et al. A novel c-Jun-dependent signal transduction pathway necessary for the transcriptional activation of interferon gamma response genes. J Biol Chem. 2007;282:938–46. doi: 10.1074/jbc.M607674200. [DOI] [PubMed] [Google Scholar]
- 44.Hu X, Paik PK, Chen J, et al. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–74. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 45.Fortin A, Abel L, Casanova JL, Gros P. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genomics Hum Genet. 2007;8:163–92. doi: 10.1146/annurev.genom.8.080706.092315. [DOI] [PubMed] [Google Scholar]
- 46.Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005;208:126–40. doi: 10.1111/j.0105-2896.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 47.Gil A, Maria AC, Gil-Campos M, Canete R. Altered signalling and gene expression associated with the immune system and the inflammatory response in obesity. Br J Nutr. 2007;98(Suppl. 1):S121–6. doi: 10.1017/S0007114507838050. [DOI] [PubMed] [Google Scholar]
- 48.Kawanaka N, Yamamura M, Aita T, et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–86. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- 49.Scherberich JE, Nockher WA. CD14++ monocytes, CD14+/CD16+ subset and soluble CD14 as biological markers of inflammatory systemic diseases and monitoring immunosuppressive therapy. Clin Chem Lab Med. 1999;37:209–13. doi: 10.1515/CCLM.1999.039. [DOI] [PubMed] [Google Scholar]
- 50.Verreck FA, de Boer T, Langenberg DM, van der ZL, Ottenhoff TH. Phenotypic and functional profiling of human proinflammatory type-1 and anti-inflammatory type-2 macrophages in response to microbial antigens and IFN-gamma- and CD40L-mediated costimulation. J Leukoc Biol. 2006;79:285–93. doi: 10.1189/jlb.0105015. [DOI] [PubMed] [Google Scholar]
- 51.Moreno-Altamirano MM, Aguilar-Carmona I, Sanchez-Garcia FJ. Expression of GM1, a marker of lipid rafts, defines two subsets of human monocytes with differential endocytic capacity and lipopolysaccharide responsiveness. Immunology. 2007;120:536–43. doi: 10.1111/j.1365-2567.2006.02531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]