

Abstract
Telomerase activity is over-expressed in nearly all pancreatic carcinomas, but not in chronic pancreatitis. Here, we investigated various protocols for expansion of telomerase-specific T cells for adoptive cell transfer and their use in a syngeneic pancreatic carcinoma mouse model. Telomerase-specific T cells were generated by stimulation of splenocytes from peptide-immunized donor mice with either interleukin (IL)-2, IL-15, artificial antigen-presenting cells, anti-signalling lymphocyte activation molecule (SLAM) microbeads or allogeneic dendritic cells in combination with a limited dilution assay. T cells were tested for antigen specificity in vitro and for anti-tumour activity in syngeneic mice with orthotopically implanted tumours pretreated with cyclophosphamide. The immune cells from recipients were immunophenotyped. During a period of 2 weeks, the expansion approach using IL-2 was very successful in generating a high number of telomerase-specific CD8+ T cells without losing their function after adoptive cell transfer. Significantly slower tumour growth rate and less metastasis were observed after adoptively transferring telomerase specific CD8+ T cells, expanded using IL-2. Further investigations showed that anti-tumour efficacy was associated with a significant shift from naive CD8+ T cells to CD8+ central memory T cells, as well as recruitment of a high number of dendritic cells. Remarkable amounts of telomerase-specific T cells were detectable in the tumour. Generation of telomerase-specific T cells is feasible, whereat IL-2-based protocols seemed to be most effective and efficient. Antigen-specific T cells showed significant cytotoxic activity in a syngeneic, orthotopic mouse model, whereas central memory T cells but not effector memory T cells appear to be of high importance.
Keywords: adoptive cell transfer, antigen-specific T cells, non-myeloablative chemotherapy, pancreatic carcinoma, telomerase
Introduction
Carcinoma of the exocrine pancreas has a particularly poor prognosis. The 5-year survival rate is < 1%, with a median survival of 4–6 months. Even after surgical intervention with curative intention, the 2-year survival rate in specialized centres is, at best, 15% [1]. New therapies are needed urgently and immunotherapy might be an option [2,3]. Adoptive cell transfer (ACT) immunotherapy is based on the ex vivo selection of tumour-reactive lymphocytes, and their activation and expansion before reinfusion into the tumour-bearing host [4,5]. Murine models of ACT have established the ability of this approach to mediate regression of established cancers and have provided important principles to guide human studies [6]. In murine models, prior host immunosuppression can improve dramatically the anti-tumour effects of ACT therapy [7,8]. Lymphodepleting but non-myeloablative chemotherapy prior to ACT provides room for transferred lymphocytes and allows their clonal repopulation in the host. Lymphodepletion may also decrease the number of regulatory T cells, which are strongly suspected to disturb the balance between activating and suppressing immune cells.
Telomerase is a ribonucleoprotein enzyme that plays a key role in maintaining chromosomal stability and cellular life span. Three components of human telomerase, human telomerase RNA component (hTERC), human telomerase protein 1 (hTEP1) and human telomerase reverse transcriptase (hTERT), have been identified. hTERT, the catalytic subunit of human telomerase, is expressed in 85% of human cancers but not usually in normal cells. Therefore, it seems to be an ideal candidate for a ubiquitous, specific tumour-associated antigen. Different antigenic epitopes from hTERT inducing tumour-specific cytotoxic T lymphocytes have been described [9–13]. Pancreatic carcinomas have been described to over-express telomerase [14]; therefore, telomerase might be a good candidate for the generation of antigen-specific T cells.
Here, we tested the feasibility of ex vivo generation and expansion of telomerase-specific T cells derived from human peripheral blood lymphocytes and checked their functional status in cytotoxicity assays against telomerase-positive pancreatic tumour cells. In a mouse model, mice were immunized with telomerase peptide and their T cells were expanded ex vivo. The expanded T cells were then administered intravenously (i.v.) into syngeneic tumour-bearing mice after non-myeloablative chemotherapy and survival of the mice was determined.
Materials and methods
Subjects
Tissue samples were drawn from subjects suffering from advanced pancreatic carcinoma or chronic pancreatitis. After giving informed consent, samples were drawn (ethics votes S359/2007) and stored in liquid nitrogen. Benign and malignant samples were collected from the same patients whenever possible.
Determination of telomerase activity
The telomerase-polymerase chain reaction (PCR) enzyme-linked immunosorbent assay (ELISA) kit (Roche Diagnostics, Mannheim, Germany) was used according to the manufacturer's instructions.
Immunization of donor mice
Immunization was performed as described previously [15]. In brief, C5BL/6 (H-2b) male mice (6–8 weeks of age) were immunized to serve as donors for obtaining murine spleen-derived telomerase-specific T cells using 100 µg H2b telomerase peptide emulsified in 1:1 in incomplete Freund's adjuvant (IFA), with the addition of macrophage-activating lipopeptide-2 (MALP-2, a Toll-like receptor 2/6 agonist) [16] to enhance the immune response. Seven days after a booster injection, the spleens were harvested and T cells were obtained by density gradient centrifugation.
Limited dilution assay and expansion
Spleen-derived feeder cells were recovered from female Balb/c mice. Splenocytes were irradiated with 60 Gy shortly before being processed for the limited dilution analysis (LDA). The purified T cells were then plated in 96-well plates using a limited dilution analysis, starting with a concentration of 2 × 104 cells/ml and ending with up to 1 cell/well. T cells were stimulated with 30 ng/ml of mouse anti-CD3 antibody, 300 U/ml of mIL-2, 1 µg/ml of mouse telomerase peptide and 5 × 104 irradiated feeder cells/well. Cells were stimulated every third day with mIL-2 at 300 U/ml. Clones were expanded with 1 × 106 irradiated feeder cells in six-well plates. For IL-2-based expansion, 30 ng/ml of mouse anti-CD3 antibody and 300 U/ml of murine IL-2 were added. Expansion via signalling lymphocyte activation molecule (SLAM) microbeads was performed with 100 µl mouse anti-SLAM-coated beads, 30 ng/ml of mouse anti-CD3 antibody and 300 U/ml of IL-2. For expansion with artificial antigen presenting cells (aAPC), mouse anti-CD3/CD28-conjugated beads, 2 µl aliquots/ml of aAPC, 30 ng/ml of mouse anti-CD3 antibody and 300 U/ml of mIL-2 were added. In the case of IL-15-based expansion, 30 ng/ml of mouse anti-CD3 antibody and 10 ng/ml of mouse IL-15 were supplemented. In the first three approaches, expanded clones were stimulated every third day with 300 U/ml IL-2, whereas for the fourth approach expanded clones were stimulated every third day with 10 ng/ml IL-15. The clones were allowed to expand over a period of 14 days before performing the ACT. Generation and expansion using telomerase peptide-pulsed dendritic cells (DCs) was performed as described previously [15].
Animal model
Syngeneic pancreatic carcinoma cells were injected orthotopically in C57Bl/6 mice, as described elsewhere [17].
Flow cytometric analysis
Telomerase specificity of both in vitro-expanded cloned T cells and tumour-derived T cells was determined using telomerase peptide-loaded dimer (dimer XI: recombinant soluble dimeric mouse H-2Db : immunoglobulin). A dimer loaded with an irrelevant peptide was used as control. Cells were characterized further for CD8 (clone 53–67), CD4 (clone RM4-5) and T cell differentiation stages (naive T cells CD8+ CD62Lhigh CD45RBhigh; central memory T cells CD8+ CD62Llow CD45RBhigh; and effector memory T cells CD8+ CD62Llow CD45RBlow). All antibodies were purchased from BD Biosciences, Heidelberg, Germany (CD62L clone MEL-14, CD45RB clone 16A).
Statistical analysis
The Mann–Whitney U-test, Pearson's correlation and the paired t-test on spss version 11·5 were used to analyse statistical significance where appropriate. A P-value < 0·05 was considered significant.
Results
Detection of telomerase activity in pancreatic ductal adenocarcinoma cell lines
Telomerase activity was over-expressed significantly in murine and human pancreatic carcinoma cell lines and in tissue samples from pancreatic carcinoma patients, whereas chronic pancreatitis and benign pancreas showed no over-expression (Fig. 1a and b).
Fig. 1.
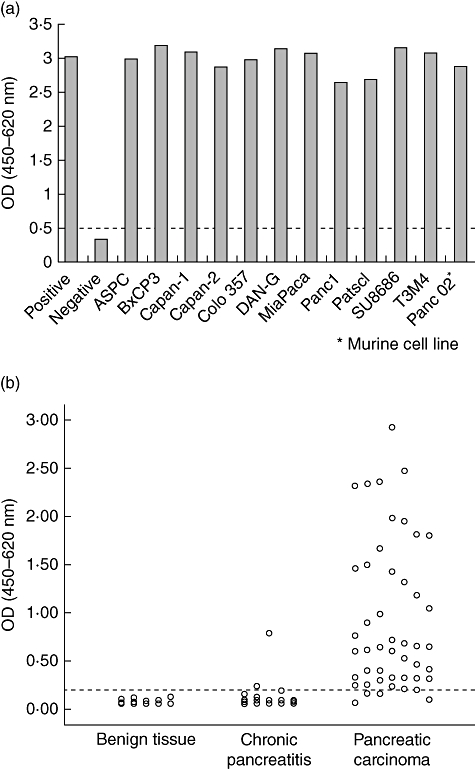
Telomerase activity in pancreatic cancer. Using a telomerase polymerase chain reaction (PCR) enzyme-linked immunosorbent assay (ELISA) technique, telomerase activity was investigated (a) in one murine pancreatic carcinoma cell line (Panc02) and 11 human pancreatic carcinoma cell lines (listed in the figure) and (b) in 50 pancreatic carcinoma samples, 25 tissue samples from patients suffering from chronic pancreatitis and 25 samples from healthy pancreas. The dotted line depicts the cut-off.
Optimization of in vitro generation and expansion of telomerase-specific T cells
Various expansion protocols were tested and compared regarding the achieved cell numbers, antigen specificity and anti-tumour activity. IL-2-based expansion led to the highest number of expanded cells (5 × 106 cells/ml) in comparison to expansion approaches using artificial APC, IL-15 and SLAM microbeads, which expanded the cells to a maximum of 2 × 106, 3 × 106 and 4 × 106, respectively. Prolonged expansion yielded further increased cell numbers, from 5 × 106 cells/ml to 40 × 106 cells/ml (Fig. 2). Telomerase specificity of in vitro-induced T cells was determined using telomerase peptide-loaded dimer and an irrelevant A3-loaded dimer used as control. IL-2-based expanded cells showed the highest telomerase specificity of 87% (range 64–92%). The anti-SLAM, aAPC and IL-15 expansion approach showed lower telomerase specificity of 42% (range 25–53%), 35% (range 26–41%) and 54% (range 12–63%), respectively. Prolongation of the expansion period of IL-2-based expanded cells led to a 53% (range 32–59%) decrease in telomerase specificity in comparison to the normal expansion period 87% (Fig. 3).
Fig. 2.
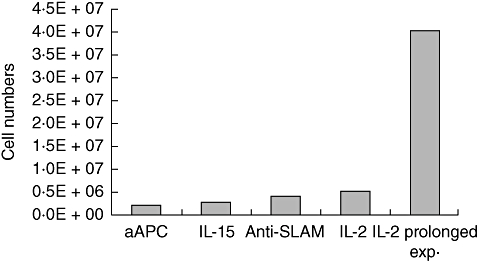
Expansion approach versus number of cells/ml. T cells were isolated from preimmunized mice and were then plated for 14 days in 96-well plates for limited dilution analysis (LDA), after which T cells were expanded for a further 14 days using artificial antigen presenting cells (aAPC), interleukin (IL)-15, signalling lymphocyte activation molecule (SLAM) microbeads or IL-2 for expansion. A set of T cells was allowed to expand for a prolonged expansion period using IL-2. Shortly before adoptive cell transfer (ACT), the cell count achieved in each expansion approach was determined. Data are shown from at least two independent experiments.
Fig. 3.
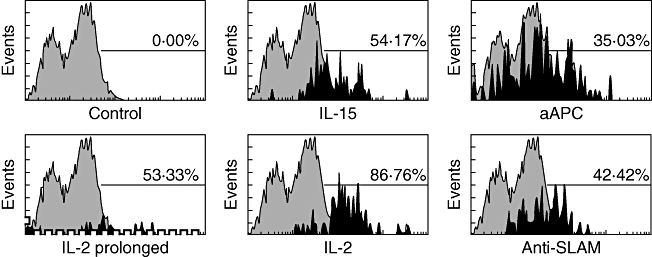
Telomerase specificity. In vitro-induced T cells – expanded via interleukin (IL)-2 (either normal or prolonged expansion), IL-15, artificial antigen presenting cells (aAPC) and signalling lymphocyte activation molecule (SLAM) microbeads – were incubated with either murine telomerase peptide-loaded dimer or with human A3 peptide-loaded dimer as control. After being stained with a dimer-specific antibody, cells were processed for specificity analysis using the flow cytometer. The figure depicts representative histograms from at least three independent experiments.
Anti-tumour activity of telomerase-specific T cells
Animals that received adoptively transferred T cells expanded using IL-2 developed significantly smaller tumours (0·016 cm3, IQR 0·014–0·018) in comparison to the control mice (0·167 cm3, IQR 0·067–0·258, P < 0·05). Mice treated with cells expanded via SLAM, aAPC and IL-15 developed larger tumour volume (0·078 cm3, IQR 0·069–0·087; 0·032 cm3, IQR 0·02–0·044; 0·226 cm3, IQR 0·134–0·318, respectively), in comparison to the IL-2 approach. Meanwhile, mice transferred adoptively with cells expanded with IL-2 but for a prolonged expansion period, or generated with modified protocol (expansion followed by LDA), showed similar effects (0·071 cm3, IQR 0·059–0·083; 0·081 cm3, IQR 0·047–0·116). However, this was not significant (Fig. 4). There appeared to be no difference concerning the metastases between the control and the other treated groups. However, animals treated with IL-15 expanded cells appeared to have a higher incidence of metastases.
Fig. 4.
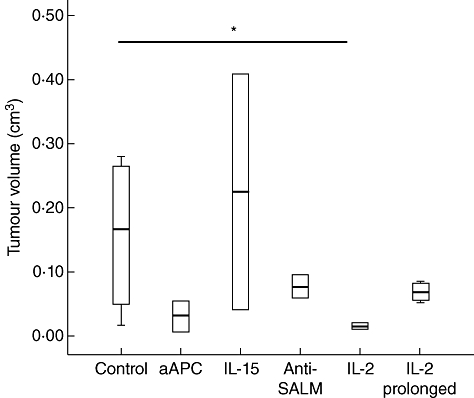
Anti-tumour activity of adoptively transferred cells. Tumour-bearing mice were treated with adoptively transferred T cells expanded via artificial antigen presenting cells (aAPC), interleukin (IL)-15, signalling lymphocyte activation molecule (SLAM) microbeads and two different IL-2-based protocols. On the day of killing, the tumour volume of all treated mice groups as well as the control was calculated. Data are shown as median and interquartile range (IQR). An asterisk indicates statistical significance. Data are shown from at least three independent experiments.
Phenotypic characterization of spleen from ACT-treated animals
Splenocytes obtained from mice transferred adoptively with IL-2-based expanded cells differed significantly from those derived either from control animals or from mice treated with cells expanded by aAPC, anti-SLAM or IL-15. Mice receiving ACT with IL-2 propagated cells had roughly 40% CD4 and CD8 cells in their spleen compared to fewer than 10% in control mice and those treated with differently expanded cells (P < 0·05, Fig. 4a). Most interestingly, composition regarding the T cell differentiation stage also differed significantly. Control animals and animals treated with non-IL-2-based expansion protocols had roughly 40% naive T cells compared to 1·0% in mice treated with IL-2 expanded cells (P < 0·05). For most parameters determined, no differences between animals treated with either IL-2 or IL-2 prolonged expansion cells could be observed. Nevertheless, this was different for effector and central memory T cells. Mice treated with IL-2 prolonged expanded cells had significantly more effector memory T cells (68%, IQR 58–78; P < 0·05) than the other groups (control 10%, IQR 9–11; aAPC/anti-SLAM/IL-15 18%, IQR 14–22; and IL-2 25%, IQR 13–38; P < 0·05). Differences were more prominent for central memory T cells where, after therapy with IL-2 expanded cells, 73% (IQR 64–87) of T cells had a central memory phenotype, but only 3% (IQR 2–4) after treatment with IL-2 prolonged expanded cells (P < 0·05). Control animals and mice treated with aAPC/anti-SLAM/IL-15 also showed significantly fewer central memory T cells (26%, IQR 18–35; 18%, IQR 14–22; P < 0·05). Analysis of antigen-presenting cells showed a similar pattern: single-digit percentages of CD40+, CD80+ and 33D1+ cells in control and aAPC/anti-SLAM/IL-15 mice and significantly more in IL-2-based protocol-treated mice (Fig. 5b; P < 0·05). There was a tendency towards IL-2 compared to IL-2 prolonged expansion. However, this was not significant (IL-2: 68% CD40+ cells versus 42% after prolonged IL-2 expansion; IL-2: 37% CD80+ cells versus 21% after prolonged IL-2 expansion; IL-2: 42% 33D1+ cells versus 29% after prolonged IL-2 expansion). Mice treated with IL-2 expanded cells accrued the highest percentage of CD11c+ cells (86%; IQR 78–93), whereas all other groups had significantly fewer CD11c+ cells (control 12%, IQR 11–14; aAPC/anti-SLAM/IL-15 12%, IQR 12–12; and IL-2 prolonged 47%, IQR 46–49; P < 0·05).
Fig. 5.
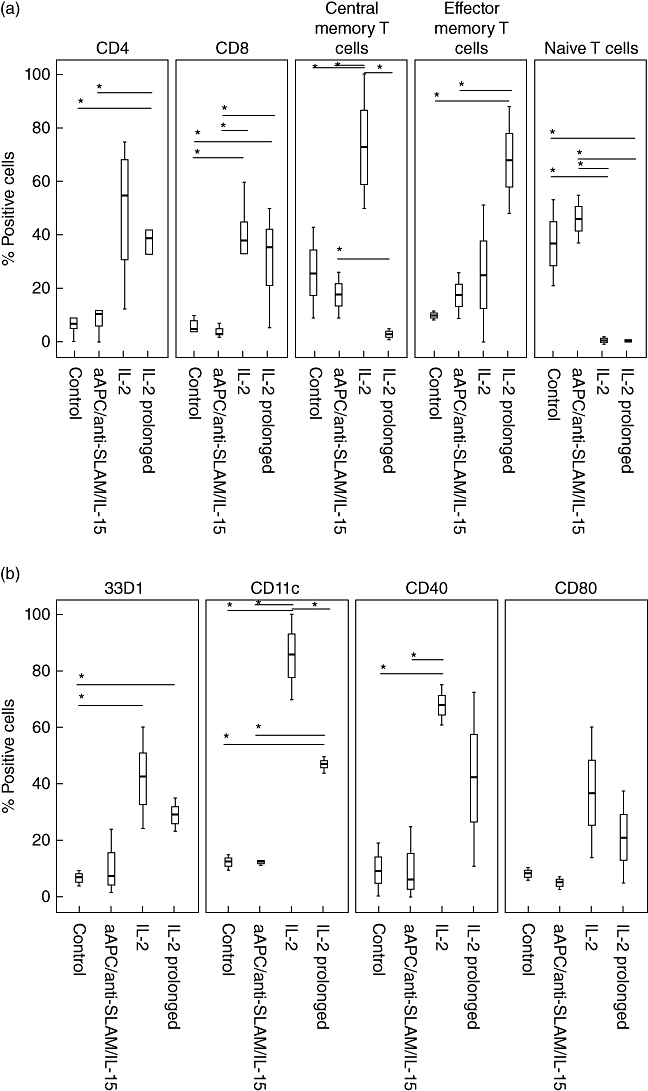
(a,b) Immunophenotyping of splenocytes. Tumour-bearing mice were treated as described. Splenocytes from killed animals were investigated by flow cytometry. Data are shown as median and interquartile range (IQR). An asterisk indicates statistical significance. Data are shown from at least three independent experiments.
Specificity and anti-tumour activity of T cells co-cultured with telomerase-pulsed DCs
Next, a direct comparison of IL-2 propagated cells with cells co-cultured with syngeneic DCs pulsed with telomerase peptide was performed. Animals that received adoptively transferred cells co-cultured with DCs developed significantly lower median tumour volume (0·004 cm3, IQR 0·002–0·007) than the IL-2 expanded cells (0·02 cm3, IQR 0·01–0·038) and control mice (0·06 cm3, IQR 0·01–0·09; Fig. 6a). Control animals had few telomerase-specific tumour-infiltrating T cells, whereas in tumours of ACT-treated mice 40% of the T cells were positive for telomerase recognizing T cell receptor (Fig. 6b). Because two tumours/group could be analysed, no clear difference was detectable between the two protocols.
Fig. 6.
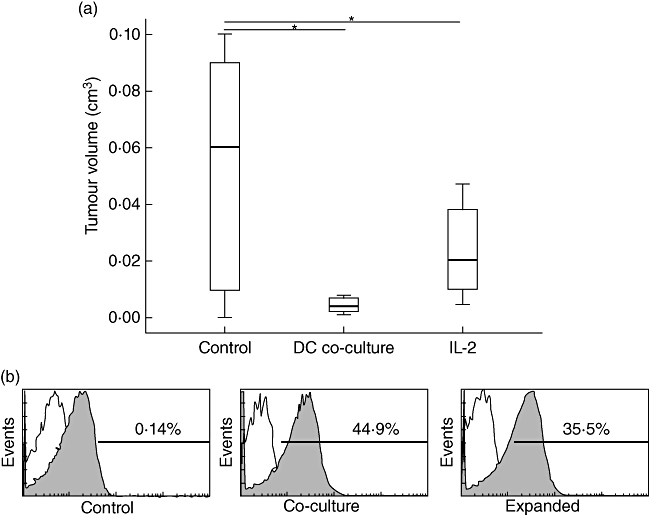
(a,b) Comparison of T cell activity generated either by dendritic cell (DC) co-culture or interleukin (IL)-2 expansion. (a) Both cells co-cultured with telomerase-pulsed DCs and those expanded using IL-2 were transferred adoptively into mice. On the day of killing, tumour volume of all treated mice groups as well as the control was calculated. Data are shown as median and interquartile range (IQR). An asterisk indicates statistical significance. (b) On the day of killing, T cells were isolated from the tumours and analysed by dimer stain and subsequent flow cytometric analysis for telomerase-specificity. The figure depicts representative histograms.
Discussion
The increased level, specificity and frequency of telomerase activity in cancers when compared with normal cells makes telomerase an extremely attractive target for anti-cancer strategies, particularly as it should be possible to use telomerase-based therapeutics over a broad range of malignancies [18–20]. Recently, enhancement of anti-tumour immune responses has aroused considerable interest as a therapeutic approach and has been applied to telomerase [9,11,21–25]. Previous work from our group has successfully generated and expanded telomerase-specific T cells which revealed cytotoxic effects when transferred adoptively into a subcutaneous pancreatic adenocarcinoma mouse model [15].
Among the various factors that appear to control the expansion and viability of antigen-activated T lymphocytes, IL-2 and IL-15 are perhaps the most extensively studied. Although IL-2 and IL-15 share two of three receptor subunits on the T lymphocyte surface, their biological function can be quite different, depending on the activation state and the specific subset of T lymphocytes that is targeted by these lymphokines [26,27]. Moreover, common ways to stimulate T cells include exposing them to other cells or antigens that provide an actual or mimicked interaction between the T cell antigen receptor (TCR) and peptide–major histocompatibility complexes (MHC) that reside on the surface of APCs. A simple way to imitate this in vivo interaction is to use aAPCa, which are coated with monoclonal antibodies against CD3 and CD28 molecules. Another molecule known as SLAM, or CD150, is involved in T cell stimulation and might be used for further optimization [28,29].
The IL-2-based expansion showed the highest increase in cell numbers, especially when the propagation period was prolonged. Nevertheless, antigen specificity diminished during the prolonged expansion period. Only the standard IL-2 protocol allowed us to generate reasonably pure T cell clones. Subsequently, the cells were tested for anti-tumour activity in immunocompetent mice which were pretreated with cyclophosphamide to promote the anti-tumour effects of the adoptively transferred T cells. There is evidence for numerous effects, including killing of host T regulatory cells that suppress anti-tumour immune responses; creating ‘space’ in the host so that the adoptively transferred T cells can engraft [30]; and perhaps enhancing cross-priming of tumour antigens. The tumour volume measured after 21 days post-implantation showed that telomerase-specific T cells expanded using IL-2 over a 2-week expansion period had the greatest effect. T cells expanded using IL-2 over a prolonged period had a lesser anti-tumour effect. When correlating both telomerase specificity results before ACT and tumour volumes recorded after ACT, there appears to be a correlation between the expansion approach used and the cytotoxic outcome of the ACT. It seems that T cells expanded using IL-2 were activated efficiently and had enough telomerase-specific cells to exhibit a significant anti-tumour effect presented by the lowest tumour volume. Meanwhile, cells expanded using the same approach but for a prolonged expansion period seem to have lost their efficacy, functionality and to some extent their telomerase specificity over the long expansion period.
This observation emphasizes that anti-tumour efficacy is more complex than having enough telomerase-specific T cell numbers. It actually exceeds the main requirement of recruiting enough effector cells or any other phenotypes of T cells that warrant an anti-tumour effect. Exposure to tumour antigens directs naive T cells to clonal expansion and differentiation into two antigen-specific subsets: short-lived effector T cells and long-lived memory T cells. Memory T cells can be divided further into two subsets: central memory T cells, which are a clonally expanded antigen-specific group anchoring in lymph nodes which can exert efficient stimulation to DCs and shift to effective T cell in the case of rechallenge with the same antigen; and the second subset comprises antigen-specific effective memory T cells homing in tissue and performing a rapid response to antigens [31,32].
Flow cytometric analysis indicated that there were significantly more CD8+ T cells infiltrating the spleens of mice treated with the IL-2 expanded T cells compared to control mice and those treated with T cells expanded using other approaches. A significant shift from CD8+ naive T cells to CD8+ central memory T cells was observed in the same group; furthermore, a significant increase of activated DCs migrating into the spleens of mice treated with IL-2 expanded T cells was also observed. The increased quantity of activated DCs in spleens of mice treated with IL-2 expanded T cells displayed an up-regulated recognition of telomerase antigen by DCs. Following the improved activation of DCs, the significant shift from naive T cells to telomerase-specific central memory T cells in the same mice group demonstrated more effective T cell recruitment. The most striking observation was the difference in central and effector memory T cells and in DCs between the IL-2-based protocols. Standard IL-2-propagated T cells, which had a greater anti-tumour effect, induced more central memory T cells and DC than prolonged expanded T cells which, in turn, induced more effector memory T cells. This is in line with reports that tumour-reactive CD8+ T cell populations with the phenotypic and functional attributes of central memory T cells may be superior to effector memory T cells for adoptive immunotherapies [33]. In summary, IL-2-expanded telomerase-specific T cells seem to have the highest anti-tumour response after ACT. Recruiting high numbers of activated DCs and shifting from CD8+ naive T cells to telomerase-specific CD8+ central memory T cells might be an explanation for the anti-tumour efficacy of this approach.
Priming T cells with peptide-pulsed DCs is a further, but more laborious, approach. Nevertheless, comparing DC co-culture with IL-2 expansion proved the efficacy of T cells primed by co-culture with DCs. However, although a tendency towards smaller tumours could be observed in mice treated with co-cultured DCs, this effect was not significant. Both approaches resulted in remarkable amounts of telomerase-specific T cells infiltrating the tumour.
In summary, standard expansion of antigen-specific T cells with IL-2 resulted in relevant cell numbers with high specificity with proper anti-tumour activity. Adoptive cell transfer in vivo induced a shift towards cytotoxic T cells, maturated DCs and differentiation to central memory T cells. The latter appear to be particularly important for the induction of tumour regression.
Acknowledgments
This work was supported by the H. W. & J. Hector foundation, Mannheim, Germany (grant M38).
Disclosure
None.
References
- 1.Raraty MG, Magee CJ, Ghaneh P, Neoptolemos JP. New techniques and agents in the adjuvant therapy of pancreatic cancer. Acta Oncol. 2002;41:582–95. doi: 10.1080/028418602321028184. [DOI] [PubMed] [Google Scholar]
- 2.Picozzi VJ, Kozarek RA, Traverso LW. Interferon-based adjuvant chemoradiation therapy after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am J Surg. 2003;185:476–80. doi: 10.1016/s0002-9610(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 3.Jaffee EM, Hruban RH, Biedrzycki B, et al. Novel allogeneic granulocyte–macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–56. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 4.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–75. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–66. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 7.Dudley ME, Wunderlich JR, Yang JC, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–51. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley ME, Wunderlich J, Nishimura MI, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–73. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Vonderheide RH, Hahn WC, Schultze JL, Nadler LM. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity. 1999;10:673–9. doi: 10.1016/s1074-7613(00)80066-7. [DOI] [PubMed] [Google Scholar]
- 10.Vonderheide RH, Anderson KS, Hahn WC, Butler MO, Schultze JL, Nadler LM. Characterization of HLA-A3-restricted cytotoxic T lymphocytes reactive against the widely expressed tumor antigen telomerase. Clin Cancer Res. 2001;7:3343–8. [PubMed] [Google Scholar]
- 11.Arai J, Yasukawa M, Ohminami H, Kakimoto M, Hasegawa A, Fujita S. Identification of human telomerase reverse transcriptase-derived peptides that induce HLA-A24-restricted antileukemia cytotoxic T lymphocytes. Blood. 2001;97:2903–7. doi: 10.1182/blood.v97.9.2903. [DOI] [PubMed] [Google Scholar]
- 12.Frolkis M, Fischer MB, Wang Z, Lebkowski JS, Chiu CP, Majumdar AS. Dendritic cells reconstituted with human telomerase gene induce potent cytotoxic T-cell response against different types of tumors. Cancer Gene Ther. 2003;10:239–49. doi: 10.1038/sj.cgt.7700563. [DOI] [PubMed] [Google Scholar]
- 13.Sievers E, Albers P, Schmidt-Wolf IG, Marten A. Telomerase pulsed dendritic cells for immunotherapy for renal cell carcinoma. J Urol. 2004;171:114–19. doi: 10.1097/01.ju.0000094803.60928.d7. [DOI] [PubMed] [Google Scholar]
- 14.Mizumoto K, Tanaka M. Detection of telomerase activity in patients with pancreatic cancer. Methods Mol Med. 2004;103:199–206. doi: 10.1385/1-59259-780-7:199. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt J, Ryschich E, Sievers E, Schmidt-Wolf IG, Büchler MW, Märten A. Telomerase-specific T-cells kill pancreatic tumor cells in vitro and in vivo. Cancer. 2006;106:759–64. doi: 10.1002/cncr.21655. [DOI] [PubMed] [Google Scholar]
- 16.Mühlradt PF, Kiess M, Meyer H, Sussmuth R, Jung G. Isolation, structure elucidation, and synthesis of a macrophage stimulatory lipopeptide from Mycoplasma fermentans acting at picomolar concentration. J Exp Med. 1997;185:1951–8. doi: 10.1084/jem.185.11.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serba S, Schmidt J, Wentzensen N, Ryschich E, Märten A. Transfection with CD40L induces tumour suppression by dendritic cell activation in an orthotopic mouse model of pancreatic adenocarcinoma. Gut. 2008;57:344–51. doi: 10.1136/gut.2007.130252. [DOI] [PubMed] [Google Scholar]
- 18.Holt SE, Shay JW. Role of telomerase in cellular proliferation and cancer. J Cell Physiol. 1999;180:10–18. doi: 10.1002/(SICI)1097-4652(199907)180:1<10::AID-JCP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 19.Keith WN, Jeffry Evans TR, Glasspool RM. Telomerase and cancer: time to move from a promising target to a clinical reality. J Pathol. 2001;195:404–14. doi: 10.1002/path.1001. [DOI] [PubMed] [Google Scholar]
- 20.White LK, Wright WE, Shay JW. Telomerase inhibitors. Trends Biotechnol. 2001;19:114–20. doi: 10.1016/s0167-7799(00)01541-9. [DOI] [PubMed] [Google Scholar]
- 21.Ayyoub M, Migliaccio M, Guillaume P, et al. Lack of tumor recognition by hTERT peptide 540-548-specific CD8(+) T cells from melanoma patients reveals inefficient antigen processing. Eur J Immunol. 2001;31:2642–51. doi: 10.1002/1521-4141(200109)31:9<2642::aid-immu2642>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 22.Heiser A, Maurice MA, Yancey DR, Coleman DM, Dahm P, Vieweg J. Human dendritic cells transfected with renal tumor RNA stimulate polyclonal T-cell responses against antigens expressed by primary and metastatic tumors. Cancer Res. 2001;61:3388–93. [PubMed] [Google Scholar]
- 23.Minev B, Hipp J, Firat H, Schmidt JD, Langlade-Demoyen P, Zanetti M. Cytotoxic T cell immunity against telomerase reverse transcriptase in humans. Proc Natl Acad Sci USA. 2000;97:4796–801. doi: 10.1073/pnas.070560797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minev BR, Chavez FL, Mitchell MS. Cancer vaccines: novel approaches and new promise. Pharmacol Ther. 1999;81:121–39. doi: 10.1016/s0163-7258(98)00039-4. [DOI] [PubMed] [Google Scholar]
- 25.Nair SK, Heiser A, Boczkowski D, et al. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med. 2000;6:1011–17. doi: 10.1038/79519. [DOI] [PubMed] [Google Scholar]
- 26.Ma A, Boone DL, Lodolce JP. The pleiotropic functions of interleukin 15: not so interleukin 2-like after all. J Exp Med. 2000;191:753–6. doi: 10.1084/jem.191.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 2001;14:105–10. [PubMed] [Google Scholar]
- 28.Mehrle S, Frank S, Schmidt J, Schmidt-Wolf IG, Märten A. SAP and SLAM expression in anti-CD3 activated lymphocytes correlates with cytotoxic activity. Immunol Cell Biol. 2005;83:33–9. doi: 10.1111/j.1440-1711.2004.01302.x. [DOI] [PubMed] [Google Scholar]
- 29.Mehrle S, Schmidt J, Buchler MW, Watzl C, Märten A. Enhancement of anti-tumor activity in vitro and in vivo by CD150 and SAP. Mol Immunol. 2008;45:796–804. doi: 10.1016/j.molimm.2007.06.361. [DOI] [PubMed] [Google Scholar]
- 30.Anasetti C, Mule JJ. To ablate or not to ablate? HSCs in the T cell driver's seat. J Clin Invest. 2007;117:306–10. doi: 10.1172/JCI30973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta S, Bi R, Su K, Yel L, Chiplunkar S, Gollapudi S. Characterization of naive, memory and effector CD8+ T cells: effect of age. Exp Gerontol. 2004;39:545–50. doi: 10.1016/j.exger.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 33.Klebanoff CA, Gattinoni L, Torabi-Parizi P, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. 2005;102:9571–6. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]