

Abstract
Myeloperoxidase (MPO)-anti-neutrophil cytoplasmic autoantibody (ANCA)-associated necrotizing crescentic glomerulonephritis (NCGN) is characterized by abundant leucocyte infiltration. Chemokines are chemotactic cytokines involved in receptor-mediated recruitment of leucocytes. Our objective was to analyse spatiotemporal gene expression of chemokines and chemokine receptors in anti-MPO-mediated NCGN, to find potential targets for intervening with leucocyte influx. NCGN was induced in mice by co-administration of anti-MPO immunoglobulin (Ig)G and lipopolysaccharide. mRNA expression levels of chemokines and chemokine receptors were analysed in whole kidney lysates as well as in laser microdissected glomeruli and tubulo-interstitial tissue 1 and 7 day(s) after NCGN induction. Several chemokines and chemokine receptors were induced or up-regulated in anti-MPO-mediated NCGN, both on day 1 (chemokines CCL3, 5; CXCL2, 5, 13; receptor CXCR2) and on day 7 (chemokines CCL2, 5, 7, 8, 17, 20; CXCL1, 2, 5, 10; CX3CL1; receptors CCR2, 8; CX3CR1). The expression levels of most chemokines and receptors were higher in glomeruli than in the tubulo-interstitium. Because of the temporal induction of CXCR2 on day 1, we hypothesized CXCR2 as a potential target for treatment in anti-MPO-induced NCGN. Inhibition of CXCR2 using a goat-anti-CXCR2 serum prior to NCGN induction increased glomerular neutrophil influx but did not affect crescent formation and albuminuria. In conclusion, expression levels of various chemokines and chemokine receptors were increased in anti-MPO NCGN, and expressed particularly in glomeruli. These chemokines and receptors may serve as potential targets for treatment. Inhibition of a single target, CXCR2, did not attenuate anti-MPO NCGN. Combinatorial interventions may be necessary to avoid redundancy.
Keywords: ANCA, chemokines, crescentic glomerulonephritis, CXCR2, MPO
Introduction
Circulating anti-neutrophil cytoplasmic autoantibodies (ANCA) directed against myeloperoxidase (MPO) are associated with systemic small vessel vasculitis, often characterized by necrotizing crescentic glomerulonephritis (NCGN) [1]. In mice, administration of murine anti-MPO antibodies induces an acute glomerular inflammation that progresses to NCGN within days [2]. This process is aggravated severely upon co-administration of lipopolysaccharide (LPS) [3]. Pathologically, the model is characterized by an early glomerular accumulation of neutrophils that progresses to crescentic glomerulonephritis with abundant glomerular and interstitial macrophage infiltration. Moreover, neutrophils are the main effector cells in disease induction, as neutrophil depletion completely prevented disease development [4].
Recruitment of inflammatory cells to sites of inflammation is, to a large extent, regulated by chemokines. Chemokines are small chemotactic cytokines that are secreted by activated or injured cells and can be recognized by specific G-protein coupled receptors expressed on leucocytes [5,6]. Chemokines are classified into four families, -C, CC, CXC and CX3C-, according to the position of the first two cysteines in the conserved amino acid sequence. Most chemokines belong to the CXC chemokine family, recruiting neutrophils and T and B cells, or the CC chemokine family, recruiting multiple leucocyte subsets including monocytes and T cells but not neutrophils. CXC chemokines containing an ELR+ (glutamic acid–leucine–arginine) motif are particularly powerful chemoattractants for neutrophils. The most potent ELR+CXC chemokine is interleukin (IL)-8 (CXCL8), which binds and activates its receptors CXCR1 and CXCR2 with similar affinity. CXCR1 is specific for IL-8, whereas other ELR+CXC chemokines (CXCL1, 2, 3, 5 and 7) can act through CXCR2. MPO-ANCA can activate neutrophils to produce IL-8 [7,8], and IL-8 is present in crescentic lesions of ANCA-associated NCGN patients [7], suggesting a pathogenic role for IL-8 in MPO-ANCA-associated vasculitis. Because a murine orthologue of human IL-8 does not exist, the activities of other ELR+CXC chemokines, e.g. keratinocyte-derived chemokine (KC/CXCL1) and macrophage inflammatory protein-2 (MIP-2/CXCL2), are more prominent in mice. CXCR2 is expressed predominantly on neutrophils, but can also be detected on monocytes/macrophages and non-inflammatory cells, including microvascular endothelial cells [9–12].
Because of the diversity in chemokine receptors expressed by leucocytes, production of different chemokines during distinct phases of an inflammatory response determines the recruitment of specific leucocyte subsets in time and space. Spatiotemporal analysis of chemokine and chemokine receptor expression patterns during the course of glomerulonephritis may reveal potential targets for intervening in recruitment of specific leucocyte subsets. Chemokine and chemokine receptor expression patterns have been analysed in several glomerulonephritis models, such as nephrotoxic nephritis and immune complex glomerulonephritis (reviewed in [13] and [14]). No studies have been conducted, however, in experimental models of ANCA-associated vasculitis.
In this study, we analysed the renal gene expression levels of chemokines and chemokine receptors in the mouse model of anti-MPO-mediated NCGN, both in time and space, to identify potential targets for intervention. Based upon our initial results, we examined specifically the role of the chemokine receptor CXCR2 in experimental anti-MPO immunoglobulin (Ig)G-induced NCGN using a CXCR2-blocking serum.
Materials and methods
Animals
Mpo−/− mice were back-crossed to a C57BL/6 background seven times [15] and bred in-house. Female C57BL/6 wild-type mice were purchased from Harlan (Horst, the Netherlands). All animal experiments were performed according to national guidelines and upon approval of the Animal Care and Use Committee of Groningen University.
Production of polyclonal mouse anti-MPO IgG
Murine MPO was purified from WEHI-3 cells and used for immunization of Mpo−/− mice, as described previously [3]. Total IgG was isolated from pooled sera of immunized Mpo−/− mice and the anti-MPO titre was checked by enzyme-linked immunosorbent assay (ELISA), as reported previously [3].
Induction and evaluation of anti-MPO IgG-induced NCGN
Wild-type C57BL/6 mice (8–10 weeks) received 100 µg/g body weight of anti-MPO IgG intraperitoneally, followed by an intraperitoneal injection with 1500 EU/g (0·5 µg/g) LPS (Escherichia coli, serotype O26:B6; Sigma, St Louis, MO, USA) 1 h later. Mice were killed after 1 or 7 day(s), and kidneys were harvested, cut and partly snap-frozen for gene and protein analyses and partly embedded in paraffin for histopathological evaluation. Plasma and (17-h) urine were collected at both time-points. Urine samples were tested for haematuria (0–4+ score) by Combur-Test® strips (Roche Diagnostics BV, Almere, the Netherlands) and albuminuria by ELISA (Bethyl Laboratories, Montgomery, TX, USA). Periodic acid-Schiff staining was performed on paraffin sections and the number of glomerular crescents was counted in 100 consecutive glomerular cross-sections in a blinded fashion, as described previously [16]. Immunohistochemical staining for neutrophils was performed on acetone-fixed 5 µm cryosections using an anti-rabbit peroxidase-based Envision®+ system (DakoCytomation, Carpinteria, CA, USA), according to the manufacturer's protocol. Sections were incubated for 30 min with 10 µg/ml rat-anti-mouse-Ly6G (clone 1A8; BD Biosciences, Breda, the Netherlands) or isotype control antibody (IgG2a; Antigenix America, Huntington Station, NY, USA) followed by a 30-min incubation with 10 µg/ml unlabelled rabbit-anti-rat secondary antibody (Vector Laboratories, Burlingame, CA, USA). After detection of peroxidase activity with 3-amino-9-ethylcarbazole, sections were counterstained with Mayer's haematoxylin.
Chemokine expression analysis
For chemokine analysis, we studied kidneys from untreated mice (n = 12), mice subjected to LPS for 1 day (n = 9) and 7 days (n = 7) and mice subjected to anti-MPO IgG-induced NCGN for 1 day (n = 11; 1·88 ± 0·69 neutrophils/glomerular cross-section; albuminuria 59·0 ± 69·6 µg/16 h) and 7 days (n = 14; 19·9 ± 6·6% crescents; albuminuria 1203 ± 944 µg/16 h). For whole kidney gene expression analysis, RNA was isolated using the RNeasy mini kit (Qiagen Benelux BV, Venlo, the Netherlands) with DNase I treatment on the column. For analysis of microdissected material, 606 (range 410–873) glomeruli (equal to 2·72 ± 0·29 × 106 µm2) and surrounding tubulo-interstitial tissue (2·88 ± 0·25 × 106 µm2) were dissected using the Laser Robot Microbeam System (PALM Micro Laser Technology, Bernried, Germany), as described previously [17], and RNA was isolated using the RNeasy micro kit (Qiagen). Reverse transcription was carried out using Superscript III reverse transcriptase (Invitrogen, Breda, the Netherlands) and random hexamer primers (Promega, Leiden, the Netherlands). Gene expression was analysed with a chemokine-focused 384-well micro fluidic card, containing primer-probe sets for 48 different genes (Table 1) using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Nieuwerkerk a/d IJssel, the Netherlands). Relative mRNA levels were calculated as 2−ΔCT, in which ΔCT is CTgene of interest − CTgapdh. CT-values that were beyond detection level were set manually to 40. Plasma levels of CXCL1 and CXCL2 protein were detected on a Luminex 100-based analyser (Luminex Corporation, Austin, TX, USA), using a Fluorokine Mouse MultiAnalyte Profiling Base Kit, containing antibody-coated microparticles and biotin-conjugated detection antibodies (R&D Systems Europe, Abingdon, UK). Protein levels of CXCL1, CXCL2 and CXCL5 in renal homogenates were determined with specific DuoSet ELISA kits (R&D Systems) and corrected for total protein concentration as measured with Bradford protein assay (Bio-Rad Laboratories, Veenendaal, the Netherlands).
Table 1.
List of the genes that were analysed using a custom low-density array.
| Gene | Protein | Mouse synonym† | Human synonym† | Assay ID |
|---|---|---|---|---|
| CC chemokines and receptors | ||||
| Ccl1 | CCL1 | TCA-3 | I-309 | Mm00441236_m1 |
| Ccl2 | CCL2 | JE | MCP-1 | Mm00441242_m1 |
| Ccl3 | CCL3 | MIP-1α | MIP-1α | Mm00441258_m1 |
| Ccl4 | CCL4 | MIP-1β | MIP-1β | Mm00443111_m1 |
| Ccl5 | CCL5 | RANTES | RANTES | Mm01302428_m1 |
| Ccl7 | CCL7 | MARC | MCP-3 | Mm00443113_m1 |
| Ccl8 | CCL8 | MCP-2 | MCP-2 | Mm01297183_m1 |
| Ccl11 | CCL11 | Eotaxin | Eotaxin | Mm00441238_m1 |
| Ccl17 | CCL17 | TARC | TARC | Mm00516136_m1 |
| Ccl20 | CCL20 | MIP-3α | MIP-3α | Mm00444228_m1 |
| Ccl22 | CCL22 | ABCD-1 | MDC | Mm00436439_m1 |
| Ccr1 | CCR1 | Mm00438260_s1 | ||
| Ccr2 | CCR2 | Mm99999051_gH | ||
| Ccr3 | CCR3 | Mm00515543_s1 | ||
| Ccr4 | CCR4 | Mm00438271_m1 | ||
| Ccr5 | CCR5 | Mm01216171_m1 | ||
| Ccr6 | CCR6 | Mm99999114_s1 | ||
| Ccr8 | CCR8 | Mm99999115_s1 | ||
| CXC chemokines and receptors | ||||
| Cxcl1 | CXCL1 | KC | GROα | Mm00433859_m1 |
| Cxcl2 | CXCL2 | MIP-2 | GROβ | Mm00436450_m1 |
| Pf4 | CXCL4 | PF-4 | PF-4 | Mm00451315_g1 |
| Cxcl5 | CXCL5 | LIX | ENA-78 | Mm00436451_g1 |
| Cxcl9 | CXCL9 | Mig | Mig | Mm00434946_m1 |
| Cxcl10 | CXCL10 | IP-10 | IP-10 | Mm00445235_m1 |
| Cxcl11 | CXCL11 | I-TAC | I-TAC | Mm00444662_m1 |
| Cxcl12 | CXCL12 | SDF-1 | SDF-1 | Mm00445552_m1 |
| Cxcl13 | CXCL13 | BLC | BCA-1 | Mm00444533_m1 |
| Il8ra | CXCR1 | Mm00731329_s1 | ||
| Il8rb | CXCR2 | Mm00438258_m1 | ||
| Cxcr3 | CXCR3 | Mm00438259_m1 | ||
| Cxcr4 | CXCR4 | Mm99999055_m1 | ||
| Cxcr5 | CXCR5 | Mm00432086_m1 | ||
| CX3C and C chemokines and receptors | ||||
| Cx3cl1 | CX3CL1 | Neurotactin | Fractalkine | Mm00436454_m1 |
| Cx3cr1 | CX3CR1 | Mm00438354_m1 | ||
| Xcl1 | XCL1 | Lymphotactin | Lymphotactin | Mm00434772_m1 |
| Xcr1 | XCR1 | Mm00442206_s1 | ||
| Endothelial and angiogenic | ||||
| Pecam1 | CD31 | Mm00476702_m1 | ||
| Sele | E-selectin | Mm00441278_m1 | ||
| Selp | P-selectin | Mm00441295_m1 | ||
| Angpt1 | Angiopoietin 1 | Mm00456503_m1 | ||
| Angpt2 | Angiopoietin 2 | Mm00545822_m1 | ||
| Tek | Tie-2 | Mm00443242_m1 | ||
| Housekeeping and other | ||||
| Gapdh | GAPDH | Mm99999915_g1 | ||
| 18S | 18S | Hs99999901_s1 | ||
| Nphs1 | Nephrin | Mm00497828_m1 | ||
| Nphs2 | Podocin | Mm00499929_m1 | ||
| Aqp2 | Aquaporin 2 | Mm00437575_m1 | ||
| Lrp2 | Megalin | Mm01328171_m1 |
BCA-1, B cell-activating chemokine-1; BLC, B lymphocyte chemoattractant; ENA-78, epithelial cell-derived neutrophil-activating factor, 78 amino acids; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GROα/β, growth-related oncogene α/β; IP-10, interferon-inducible protein-10; I-TAC, interferon-inducible T cell α-chemoattractant; KC, keratinocyte-derived chemokine; LIX, lipopolysaccharide-induced CXC chemokine; MARC, mast cell activation-related chemokine; MCP-#, monocyte chemoattractant protein-#; MDC, macrophage-derived chemokine; Mig, monokine induced by interferon-γ; MIP-#, macrophage inflammatory protein-#; PF-4, platelet factor-4; RANTES, regulated on activation normal T cell expressed and secreted; SDF-1, stromal cell-derived factor-1; TARC, thymus- and activation-related chemokine; TCA-3, T-cell activation protein-3.
Cell culture
Human conditionally immortalized glomerular endothelial cells (CiGEnC) [18] were cultured in endothelial growth medium 2-microvascular (EGM2-MV; Cambrex-Lonza, Breda, the Netherlands) containing fetal calf serum (5%) and growth factors as supplied, without vascular endothelial growth factor (VEGF). CiGEnC up to passage 40 were propagated at 33°C (when cells have a proliferative phenotype), whereas experiments were carried out after 5–7 days of incubation at 37°C (non-proliferative/quiescent phenotype).
Gene expression analysis of human glomerular endothelial cells and neutrophils
CiGEnC were seeded in 12-well plates (90 000 cells/well) and incubated at 33°C for 1 day before thermoswitching to 37°C. Neutrophils were isolated from heparinized venous blood of healthy donors by density gradient centrifugation on Lymphoprep (Axis-Shield, Oslo, Norway). Erythrocytes were lysed with ice-cold ammonium chloride buffer, and neutrophils were washed in Hanks's balanced salt solution without Ca2+/Mg2+ (HBSS−/−; Gibco/Life Technologies, Breda, the Netherlands). RNA was isolated using the RNeasy Plus Mini kit (Qiagen), and cDNA was prepared and individual gene real-time polymerase chain reaction (PCR) analyses were carried out as described in the ‘Chemokine expression analysis’ section. Primer-probe sets specific for human CXCR1 (Hs00174146_m1), CXCR2 (Hs00174304_m1) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs99999905_m1) (Applied Biosystems) were used.
Intracellular calcium measurements
For single-cell calcium imaging, CiGEnC were grown on glass coverslips (30 mm ∅) in six-well plates (60 000 cells/well) at 33°C for 1 day before thermoswitching to 37°C. Cells were washed briefly in loading buffer (150 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES/NaOH, 10 mM glucose, pH ∼ 7·35) and loaded with 5 µM Fura-2(AM) (TEF Laboratories, Austin, TX, USA) in loading buffer in the presence of 0·08% Pluronic® F-127 (Molecular Probes, Invitrogen) at 37°C for 30 min. After an additional washing step in loading buffer for 30 min in the presence or absence of 15 µg/ml anti-CXCR1 and/or anti-CXCR2 blocking antibodies (R&D systems), the coverslips were fixed in a perfusion chamber (37°C) and attached to an inverted microscope (Axiovert 35M; Zeiss, Sliedrecht, the Netherlands) equipped with a 12-bit Sensicam CCD camera (PCO, Kelheim, Germany) supported by Imaging Workbench 5·0 software (INDEC BioSystems, Santa Clara, CA, USA). Using a 16× plan-neofluar objective, digital images were taken at an emission wavelength of 510 nm using paired exposure at 340 nm and 380 nm excitation wavelengths at a frequency of 1Hz. Changes in intracellular calcium levels upon IL-8 treatment (100 ng/ml; R&D systems) were detected as changes in the ratio of the 340 and 380 nm excitation wavelengths in time (seconds).
In vivo anti-CXCR2 treatment
The inhibitory goat anti-murine CXCR2 serum was raised against a peptide (MGEFKVDKFNIEDFFSG) of the ligand-binding region of CXCR2. In previous studies, this anti-serum (0·5–1·0 ml) has been shown to abrogate neutrophil influx in lung inflammatory mouse models without affecting circulating neutrophil numbers [19,20]. Mice (n = 6/group) received 0·8 ml anti-CXCR2 or normal goat serum (AbD Serotec, Düsseldorf, Germany) intraperitoneally every other day, starting with the first treatment 24 h before anti-MPO IgG administration. In a separate experiment, mice (n = 3/group) received a daily dose (30 mg/kg) of the CXCR2-inhibitor repertaxin (Sigma) subcutaneously, starting with the first treatment 1 h before anti-MPO IgG administration, and were killed after 7 days.
Statistical analysis
Statistical significance was determined using one-way analysis of variance (anova) with Bonferroni's multiple comparison test (whole kidney mRNA analysis, intracellular calcium measurements and CXCL1/CXCL2/CXCL5 protein measurements) or two-tailed Student's t-test (mRNA analysis of dissected renal compartments and quantification of glomerular neutrophils).
Results
Chemokine and chemokine receptor expression in the acute inflammatory phase of anti-MPO IgG-induced NCGN
Gene expression analysis of chemokines and chemokine receptors in renal tissue 1 day after induction of experimental anti-MPO IgG-mediated NCGN revealed induction or up-regulation of several chemokines when compared to expression levels in non-treated and LPS-treated mice (Fig. 1a–e). These chemokines belonged to the CC-family of chemokines, CCL3 and CCL5, and the CXC-family, CXCL2, CXCL5 and CXCL13. Interestingly, CXCR2 was the only chemokine receptor with a significantly higher expression level in the acute phase of disease development. Subsequently, we determined the localization of chemokine expression by comparing expression levels between laser microdissected glomeruli and the tubulo-interstitial area. We confirmed accurate separation of the compartments with laser microdissection by analysing expression of glomerulus-specific (podocin and nephrin) and tubulus-restricted (megalin and aquaporin-2) genes (data not shown). We found that gene expression of most of the chemokines and chemokine receptors was localized predominantly in the glomerular compartment (Fig. 1f–j).
Fig. 1.
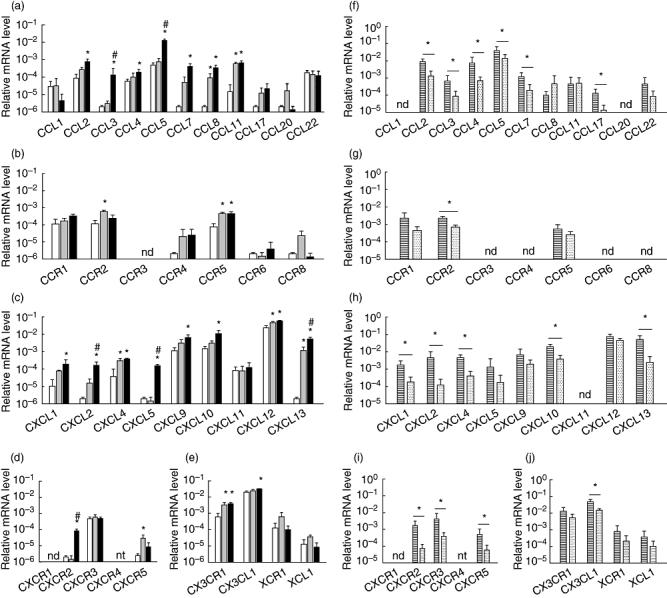
Relative gene expression of chemokines and chemokine receptors in mouse kidney during the acute inflammation phase of anti-myeloperoxidase (MPO) immunoglobulin (Ig)G-induced necrotizing crescentic glomerulonephritis (NCGN). (a–e) Relative mRNA levels of (a) CC-chemokines, (b) CC-chemokine receptors, (c) CXC-chemokines, (d) CXC-chemokine receptors and (e) CX3C- and XC-chemokines and their receptors in kidneys from healthy control mice (white bars), mice that received lipopolysaccharide (LPS) only (grey bars) and mice that received anti-MPO IgG and LPS (black bars) 1 day after administration. Bars represent mean ± standard deviation (s.d.). *P < 0·05 versus control mice; #P < 0·05 versus LPS-treated mice. (f–j) Relative mRNA levels in laser-microdissected glomeruli (striped bars) and tubulo-interstitial areas (dotted bars) from mice that received anti-MPO IgG and LPS 1 day after administration. (f) CC-chemokines, (g) CC-chemokine receptors, (h) CXC-chemokines, (i) CXC-chemokine receptors and (j) CX3C- and XC-chemokines and their receptors. Bars represent mean ± s.d. *P < 0·05; n.d.: not detected; n.t.: not tested – CXCR4 could not be analysed due to insufficient amplification reactions.
Chemokine and chemokine receptor expression in the crescentic phase of anti-MPO IgG-induced NCGN
Gene expression analysis in renal tissue obtained from mice 7 days after induction of anti-MPO IgG-mediated glomerulonephritis demonstrated the induction or up-regulation of various chemokines and chemokine receptors compared to non-treated and LPS-treated mice (Fig. 2a–e). Most of the chemokines with increased expression at day 7 belonged to the CC-chemokines: CCL2, CCL5, CCL7, CCL8, CCL17 and CCL20. In addition, increased expression was observed for the CC-chemokine receptors CCR2 and CCR8 and CXC-chemokine CXCL10. In contrast to day 1, no increased expression of CCL3 and CXCL13 was found at day 7. CXCL1, CXCL2 and CXCL5, the ligands for CXCR2, were also up-regulated in the crescentic phase, although the receptor itself was expressed at a level similar to control mice. Furthermore, CX3CL1 and its receptor CX3CR1 were expressed to an increased extent. Analysis of dissected renal tissue showed that expression of the majority of chemokines and receptors was also localized in glomeruli at this later time-point (Fig. 2f–j). However, for some chemokines the pattern changed. CXCL2 was expressed highly in glomeruli but less so in the tubulo-interstitium on day 1, while on day 7 no significant difference was found.
Fig. 2.
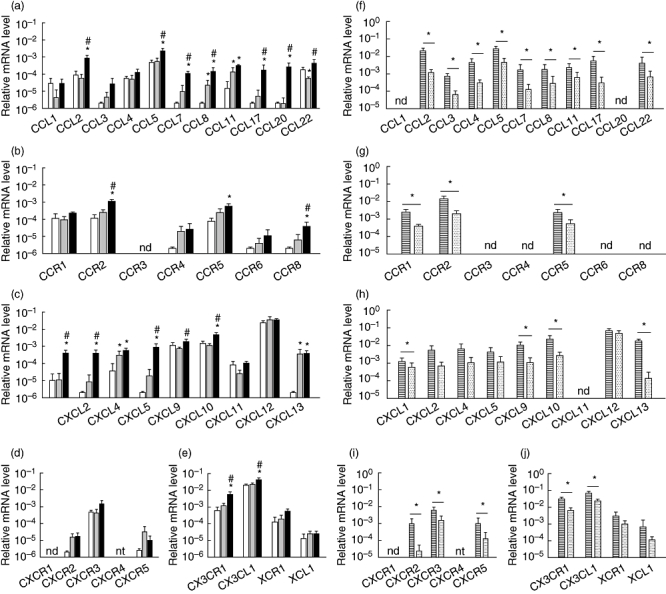
Relative gene expression of chemokines and chemokine receptors in mouse kidney during the crescentic phase of anti- myeloperoxidase (MPO) immunoglobulin (Ig)G-induced necrotizing crescentic glomerulonephritis (NCGN). (a–e) Relative mRNA levels of (a) CC-chemokines, (b) CC-chemokine receptors, (c) CXC-chemokines, (d) CXC-chemokine receptors and (e) CX3C- and XC-chemokines and their receptors in kidneys from healthy control mice (white bars), mice that received lipopolysaccharide (LPS) only (grey bars) and mice that received anti-MPO IgG and LPS (black bars) 7 days after administration. Bars represent mean ± standard deviation (s.d.). *P < 0·05 versus control mice; #P < 0·05 versus LPS-treated mice. (f–j) Relative mRNA levels in laser-microdissected glomeruli (striped bars) and tubulo-interstitial areas (dotted bars) from mice that received anti-MPO IgG and LPS 7 days after administration. (f) CC-chemokines, (g) CC-chemokine receptors, (h) CXC-chemokines, (i) CXC-chemokine receptors and (j) CX3C- and XC-chemokines and their receptors. Bars represent mean ± s.d. *P < 0·05; n.d.: not detected; n.t.: not tested – CXCR4 could not be analysed due to insufficient amplification reactions.
Protein expression of CXCR2 and CXCR2 ligands
Because a temporal induction of CXCR2 was detected in the acute phase and induction of its ligands in both the acute and the crescentic phases, we aimed to investigate further the role of CXCR2 and its ligands in anti-MPO IgG-induced NCGN. Analysis of circulating CXCL1 and CXCL2 protein levels demonstrated increased CXCL1 levels in the crescentic phase of anti-MPO IgG-induced NCGN (Table 2). In contrast, CXCL1, CXCL2 and CXCL5 protein levels in renal homogenates did not change during the course of anti-MPO IgG-induced NCGN (Table 2). In addition, we wanted to determine to what extent intrinsic glomerular cells contribute to glomerular CXCR2 expression. Recent data indicate that expression of CXCR2 is not restricted to inflammatory cells, but can also be detected on microvascular endothelial cells. Furthermore, endothelial CXCR2 has been demonstrated to be involved in LPS-induced neutrophil infiltration in the lungs [21]. We speculated that CXCR2 was expressed on glomerular endothelial cells and that endothelial CXCR2 could contribute to the early neutrophil accumulation, as observed in anti-MPO/LPS-induced glomerulonephritis. We studied whether human CiGEnC expressed CXCR2. CXCR2 mRNA was detected in CiGEnC, but its expression was much lower compared to expression in human neutrophils (Fig. 3a). CXCR1 mRNA was not detected in CiGEnC. Treatment of CiGEnC with IL-8 induced an intracellular calcium flux in approximately 70% of the cells, which was mediated by CXCR2 but not by CXCR1 (Fig. 3b). These results indicate that glomerular endothelial cells express a functional CXCR2 receptor.
Table 2.
Plasma and renal tissue protein levels of CXCL1, CXCL2 and CXCL5 in mice suffering from anti-MPO IgG-induced NCGN.
| Plasma (pg/ml) |
Renal homogenate (ng/mg total protein) |
|||||
|---|---|---|---|---|---|---|
| Group | CXCL1 | CXCL2 | CXCL5 | CXCL1 | CXCL2 | CXCL5 |
| Control | 166·1 ± 80·9 | n.d. | n.t. | 0·80 ± 0·15 | 1·94 ± 0·30 | 5·30 ± 0·93 |
| LPS day 1 | 601·6 ± 210·4 | 78·3 ± 32·7 | n.t. | 0·86 ± 0·23 | 1·83 ± 0·46 | 4·81 ± 0·97 |
| Anti-MPO IgG + LPS day 1 | 432·5 ± 47·5 | 60·1 ± 22·8 | n.t. | 0·78 ± 0·10 | 1·80 ± 0·14 | 5·65 ± 0·26 |
| LPS day 7 | 172·1 ± 22·9 | n.d. | n.t. | 0·62 ± 0·15 | 1·41 ± 0·29 | 4·64 ± 1·09 |
| Anti-MPO IgG + LPS day 7 | 1075·6 ± 1030·5* | n.d. | n.t. | 0·59 ± 0·05 | 1·34 ± 0·17 | 3·73 ± 0·76 |
P < 0·05 compared to both control and lipopolysaccharide (LPS) day 7; mean ± standard deviation; n.d., not detected; n.t., not tested. Ig, immunoglobulin; MPO, myeloperoxidase; NCGN, necrotizing crescentic glomerulonephritis.
Fig. 3.
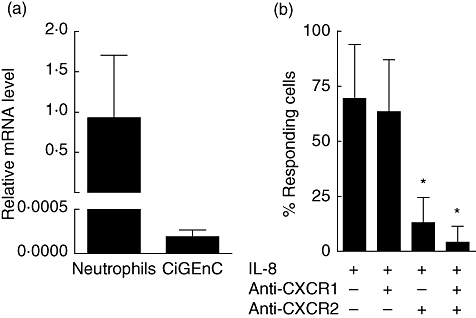
Human glomerular endothelial cells functionally express CXCR2 in vitro. (a) Relative CXCR2 mRNA levels in conditionally immortalized glomerular endothelial cells (CiGEnC) and human neutrophils. Bars represent mean ± standard deviation (s.d.) of measurements in two independent cell preparations (CiGEnC) or three donors (neutrophils). (b) Percentage of CiGEnC responding to interleukin (IL)-8 treatment by intracellular calcium flux in the presence or absence of CXCR1- and CXCR2-blocking antibodies. Bars represent the mean percentage of cells responding to IL-8 in three independent experiments (n = 34–142 cells/condition). *P < 0·01.
In vivo inhibition of CXCR2 does not attenuate renal injury in anti-MPO IgG-induced NCGN
To study the functional role of the CXCR2–CXCR2 ligand axis in anti-MPO IgG/LPS-mediated glomerulonephritis, we blocked the receptor employing a CXCR2 blocking serum. Compared to control-treated mice, mice that had received anti-CXCR2 serum showed an increase in glomerular neutrophil accumulation 1 day after anti-MPO IgG/LPS administration (Fig. 4a and b). Moreover, inhibition of CXCR2 did not change the percentage of glomeruli positive for neutrophils (control 65·7 ± 5·0 and anti-CXCR2 66·3 ± 6·3%) but rather increased the mean number of neutrophils per positive glomerulus (control 1·75 ± 0·30 and anti-CXCR2 2·59 ± 0·50, P < 0·05). Treatment of mice suffering from anti-MPO IgG-induced NCGN with the anti-CXCR2 blocking serum did not reduce haematuria and albuminuria (Fig. 4c and d). In addition, CXCR2 inhibition did not influence glomerular crescent formation, as a similar percentage of crescentic glomeruli was detected in control and anti-CXCR2-treated mice (Fig. 4e). To confirm the effects of the CXCR2 blocking serum, a group of anti-MPO IgG-induced NCGN mice were treated with the CXCR2 inhibitor repertaxin. No differences were found between vehicle- and repertaxin-treated animals with respect to albuminuria on day 1 (vehicle 28·9 ± 16·0 and repertaxin 21·7 ± 0·8 µg/17 h, mean ± standard deviation, n = 3) and day 7 (110·8 ± 119·7 and 119·1 ± 52·2 µg/17 h) and the extent of glomerular crescent formation on day 7 (11·0 ± 3·6 and 9·3 ± 1·5%).
Fig. 4.
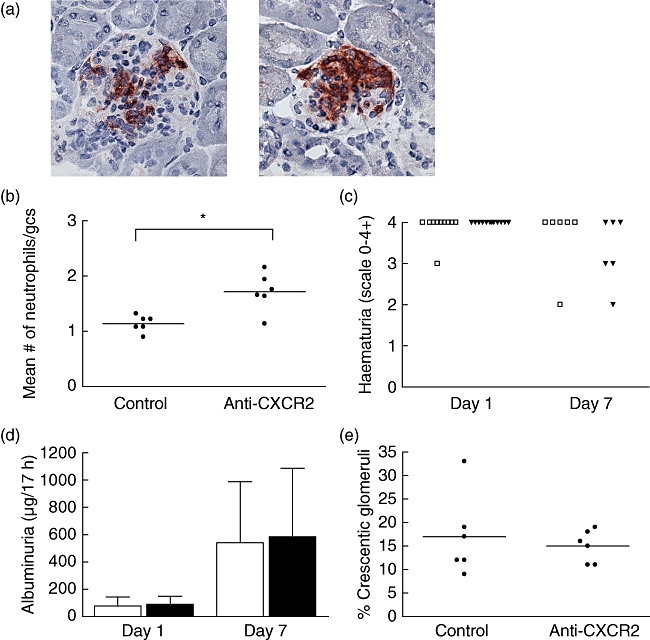
Inhibition of CXCR2 in anti-myeloperoxidase (MPO) immunoglobulin (Ig)G-induced necrotizing crescentic glomerulonephritis (NCGN) enhanced early glomerular neutrophil influx but did not affect urinary abnormalities and crescent formation. (a) Immunohistochemical staining of neutrophils (Ly6G) on kidney cryosections of mice treated with control serum (left) or CXCR2 blocking serum (right) 1 day after anti-MPO IgG/lipopolysaccharide (LPS) administration. Original magnification 400×. (b) Quantification of neutrophil influx at day 1. gcs: glomerular cross-section. *P < 0·01. (c) Administration of anti-MPO IgG/LPS caused marked haematuria after 1 and 7 day(s). No differences were observed between mice treated with control serum (□) and mice treated with CXCR2-blocking serum (▾). (d) Albuminuria at 1 and 7 days after anti-MPO IgG/LPS administration. Treatment with CXCR2-blocking serum did not prevent albuminuria. Open bars: control treatment; filled bars: anti-CXCR2 treatment. Bars represent mean ± standard deviation (s.d.). The average level of albumin in urine of untreated mice was 40·7 ± 24·0 µg/17 h. (e) Quantification of glomerular crescents revealed no difference between control-treated and anti-CXCR2-treated mice. Bars represent mean ± s.d.
Discussion
In the present study, we demonstrate spatiotemporal differences in chemokine and chemokine receptor gene expression levels in a mouse model of anti-MPO IgG-induced NCGN. Our objective was to identify those chemokines and chemokine receptors whose expression patterns suggest involvement in the recruitment of specific leucocyte subsets.
Induction of CXCR2 was restricted to the acute inflammation phase. Its ligands, CXCL1, CXCL2 and CXCL5, were up-regulated in both the acute and crescentic phases of the disease. Our findings suggest that CXCL2 expression is restricted to glomeruli in the acute phase of anti-MPO IgG-induced NCGN, whereas in the crescentic phase CXCL2 is expressed in both the glomerular and the tubulo-interstitial compartment, which correlates with the presence of both glomerular and interstitial inflammatory infiltrates at this time-point. We chose CXCR2 as an initial target for intervention because the temporal induction of CXCR2 suggested involvement of CXCR2 in recruitment of neutrophils, which are pivotal effector cells in anti-MPO IgG-induced NCGN [4]. Furthermore, several studies have demonstrated reduced glomerular neutrophil influx and albuminuria in models of glomerulonephritis upon inhibition of CXCR2 or its ligands [22–25]. The measurements of CXCL1, CXCL2 and CXCL5 protein in kidney tissue did not confirm the increase in tissue-specific expression of these chemokines suggested by analysis of mRNA levels. In plasma, we observed a higher CXCL2 level at day 7. Interestingly, increased circulating levels of IL-8 have been observed in ANCA-associated vasculitis patients [26].
Interestingly, blocking CXCR2 in our model of anti-MPO IgG-induced NCGN did not prevent early glomerular neutrophil influx but increased glomerular accumulation of neutrophils. In vitro flow assays have revealed that inhibition of neutrophil CXCR2 reduces transendothelial migration of ANCA-activated neutrophils [27]. We speculate that in our experiment neutrophils were recruited to glomeruli via chemoattractants other than CXCR2 ligands but, due to inhibition of CXCR2-mediated transendothelial migration, were retained within the vascular compartment. In line with this, Cockwell et al. hypothesized that frustrated neutrophil transmigration due to high levels of intravascular IL-8 contributes to glomerular injury in ANCA-associated glomerulonephritis [7]. The observation that inhibition of CXCR2-accelerated glomerular neutrophil accumulation would also imply a worsening of other disease parameters, but we did not observe increased urinary abnormalities or crescent formation, which was supported by the repertaxin experiment. Based on the experiments conducted, we cannot explain this discrepancy, although our data suggest that the extent of kidney injury is determined primarily by the number of affected glomeruli. A probable neutrophil chemoattractant involved in the neutrophil recruitment in anti-MPO IgG-induced NCGN is C5a. Recently, genetic ablation or inhibition of complement factor C5, and consequently C5-derived C5a, was found to abrogate disease development completely in this model [16,28]. Moreover, neutrophils from CXCR2−/− mice retain chemotactic activity towards C5a [29]. An additional possibility is that the activity of other chemokines and chemokine receptors becomes more important, as there is redundancy in the chemokine system [30].
The observed increase in CXCR2 mRNA at day 1 in our disease model is caused most probably by the glomerular influx of neutrophils and not by up-regulation of endothelial CXCR2, as neutrophils express relatively high levels of CXCR2. Reutershan and colleagues demonstrated that endothelial CXCR2 was involved in LPS-induced neutrophil transmigration in the lungs [21]. Although lung tissue has a higher expression of CXCR2 compared to kidney, we cannot exclude the possibility that inhibition of endothelial CXCR2 in glomeruli contributed to neutrophil transmigration impairment in our CXCR2-intervention experiment.
Other chemokines that had increased expression in the acute phase were CCL3 and CCL5, suggesting their involvement in leucocyte influx. Increased protein levels of CCL3 and CCL5 have been found in vasculitic lesions in lungs of Wegener's granulomatosis [31]. None of the receptors for CCL3 and CCL5 (e.g. CCR1, CCR5), however, were up-regulated in our model.
In the crescentic phase of anti-MPO IgG-induced NCGN, CCR2 was up-regulated and expressed predominantly in glomeruli. The CCR2 ligands CCL2 and CCL7 were also expressed to an increased extent and mainly in glomeruli. Up-regulation of CCR2 and its ligands suggests involvement in the monocyte/macrophage influx observed in the crescentic phase of anti-MPO IgG-induced NCGN. Neutralization studies have demonstrated important roles for CCR2 and CCL2 in monocyte/macrophage influx and, in some cases, crescent formation in other models of crescentic glomerulonephritis [32–34]. Furthermore, CCL2 protein has been detected in glomerular and interstitial cells in ANCA-associated vasculitis patients with renal involvement [35].
In addition, we observed up-regulation of CX3CL1 and its receptor CX3CR1 in the crescentic phase. CX3CL1 is a transmembrane domain-containing chemokine with monocyte attracting properties. CX3CL1 mRNA expression has been observed in glomerular lesions of vasculitic patients [36], whereas inhibition of CX3CR1 in a rat model of crescentic glomerulonephritis attenuated glomerular leucocyte influx and reduced crescent formation [37]. These data suggest that CX3CL1 may be involved in the pathogenesis of anti-MPO IgG-induced NCGN.
Our finding that several chemokines were up-regulated during anti-MPO IgG-induced glomerulonephritis raises questions on the cellular source of chemokine production. We speculate that intrinsic renal (predominantly glomerular) cells are, in part, responsible for the production of chemokines, as are infiltrating inflammatory cells. The relative contributions of the different intrinsic glomerular cells – glomerular endothelial cells, podocytes and mesangial cells – in chemokine production could be investigated in vitro in future studies.
In this study, we have used murine anti-MPO IgG-induced glomerulonephritis as a model for human ANCA-associated vasculitis. For the translation of our data to the human situation, it is important to realize that the mouse model mimics ANCA-induced effects on neutrophils (and other cells) but does not involve a genuine autoimmune response. In addition, the model encompasses a proinflammatory stimulus, which may have modulated expression of chemokines and receptors in a way that does not occur in human ANCA-associated vasculitis.
In conclusion, various chemokine receptors and chemokines, including CXCR2, CCR2 and CX3CR1 and their ligands, are up-regulated during the course of anti-MPO IgG-induced NCGN, and they probably contribute to shaping the inflammatory response. These chemokines and chemokine receptors can be tested further as potential targets in anti-MPO IgG-induced glomerulonephritis. Inhibition of one potential target, CXCR2, did not diminish anti-MPO IgG-induced NCGN. On the contrary, CXCR2 inhibition may, hypothetically, accelerate the vasculitic process by retaining neutrophils in the vascular compartment. Intervening with multiple chemokine targets is probably necessary to avoid redundancy of the chemokine system.
Acknowledgments
Peter Heeringa is supported by the Dutch Organization of Scientific Research (ZonMW VIDI 917·066·341). We thank Hester I. Bakker for excellent technical assistance, Tjerk Feenstra and Peter J. Zwiers for help with laser microdissection and Attje S. Hoekstra and Hilmar R. J. van Weering for help with intracellular calcium measurements.
Disclosure
The authors declare no conflict of interest.
References
- 1.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318:1651–7. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 2.Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–63. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huugen D, Xiao H, van Esch A, et al. Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: role of tumor necrosis factor-alpha. Am J Pathol. 2005;167:47–58. doi: 10.1016/s0002-9440(10)62952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao H, Heeringa P, Liu Z, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167:39–45. doi: 10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95:3032–43. [PubMed] [Google Scholar]
- 6.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 7.Cockwell P, Brooks CJ, Adu D, Savage CO. Interleukin-8: a pathogenetic role in antineutrophil cytoplasmic autoantibody-associated glomerulonephritis. Kidney Int. 1999;55:852–63. doi: 10.1046/j.1523-1755.1999.055003852.x. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh SC, Yu HS, Cheng SH, et al. Anti-myeloperoxidase antibodies enhance phagocytosis, IL-8 production, and glucose uptake of polymorphonuclear neutrophils rather than anti-proteinase 3 antibodies leading to activation-induced cell death of the neutrophils. Clin Rheumatol. 2007;26:216–24. doi: 10.1007/s10067-006-0285-3. [DOI] [PubMed] [Google Scholar]
- 9.Salcedo R, Resau JH, Halverson D, et al. Differential expression and responsiveness of chemokine receptors (CXCR1-3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J. 2000;14:2055–64. doi: 10.1096/fj.99-0963com. [DOI] [PubMed] [Google Scholar]
- 10.Addison CL, Daniel TO, Burdick MD, et al. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165:5269–77. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 11.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–76. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 12.Heidemann J, Ogawa H, Dwinell MB, et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–15. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- 13.Panzer U, Steinmetz OM, Stahl RA, Wolf G. Kidney diseases and chemokines. Curr Drug Targets. 2006;7:65–80. doi: 10.2174/138945006775270213. [DOI] [PubMed] [Google Scholar]
- 14.Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol. 2000;11:152–76. doi: 10.1681/ASN.V111152. [DOI] [PubMed] [Google Scholar]
- 15.Aratani Y, Koyama H, Nyui S, Suzuki K, Kura F, Maeda N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect Immun. 1999;67:1828–36. doi: 10.1128/iai.67.4.1828-1836.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huugen D, van Esch A, Xiao H, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–54. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- 17.Asgeirsdottir SA, Kamps JA, Bakker HI, et al. Site-specific inhibition of glomerulonephritis progression by targeted delivery of dexamethasone to glomerular endothelium. Mol Pharmacol. 2007;72:121–31. doi: 10.1124/mol.107.034140. [DOI] [PubMed] [Google Scholar]
- 18.Satchell SC, Tasman CH, Singh A, et al. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006;69:1633–40. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 19.Mehrad B, Strieter RM, Moore TA, Tsai WC, Lira SA, Standiford TJ. CXC chemokine receptor-2 ligands are necessary components of neutrophil-mediated host defense in invasive pulmonary aspergillosis. J Immunol. 1999;163:6086–94. [PubMed] [Google Scholar]
- 20.Moore TA, Newstead MW, Strieter RM, Mehrad B, Beaman BL, Standiford TJ. Bacterial clearance and survival are dependent on CXC chemokine receptor-2 ligands in a murine model of pulmonary Nocardia asteroides infection. J Immunol. 2000;164:908–15. doi: 10.4049/jimmunol.164.2.908. [DOI] [PubMed] [Google Scholar]
- 21.Reutershan J, Morris MA, Burcin TL, et al. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wada T, Tomosugi N, Naito T, et al. Prevention of proteinuria by the administration of anti-interleukin 8 antibody in experimental acute immune complex-induced glomerulonephritis. J Exp Med. 1994;180:1135–40. doi: 10.1084/jem.180.3.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Wittwer AJ, Carr LS, Crippes BA, DeLarco JE, Lefkowith JB. Cytokine-induced neutrophil chemoattractant mediates neutrophil influx in immune complex glomerulonephritis in rat. J Clin Invest. 1994;94:337–44. doi: 10.1172/JCI117326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng L, Xia Y, Yoshimura T, Wilson CB. Modulation of neutrophil influx in glomerulonephritis in the rat with anti-macrophage inflammatory protein-2 (MIP-2) antibody. J Clin Invest. 1995;95:1009–17. doi: 10.1172/JCI117745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, Dolecki GJ, Sherry B, Zagorski J, Lefkowith JB. Chemokines are expressed in a myeloid cell-dependent fashion and mediate distinct functions in immune complex glomerulonephritis in rat. J Immunol. 1997;158:3917–24. [PubMed] [Google Scholar]
- 26.Ohlsson S, Wieslander J, Segelmark M. Circulating cytokine profile in anti-neutrophilic cytoplasmatic autoantibody-associated vasculitis: prediction of outcome? Mediat Inflamm. 2004;13:275–83. doi: 10.1080/09629350400003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderwood JW, Williams JM, Morgan MD, Nash GB, Savage CO. ANCA induces beta2 integrin and CXC chemokine-dependent neutrophil–endothelial cell interactions that mimic those of highly cytokine-activated endothelium. J Leukoc Biol. 2005;77:33–43. doi: 10.1189/jlb.0104054. [DOI] [PubMed] [Google Scholar]
- 28.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155:2158–64. [PubMed] [Google Scholar]
- 30.Mantovani A. The chemokine system: redundancy for robust outputs. Immunol Today. 1999;20:254–7. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y, Huang D, Farver C, Hoffman GS. Relative importance of CCR5 and antineutrophil cytoplasmic antibodies in patients with Wegener's granulomatosis. J Rheumatol. 2003;30:1541–7. [PubMed] [Google Scholar]
- 32.Bird JE, Giancarli MR, Kurihara T, et al. Increased severity of glomerulonephritis in C-C chemokine receptor 2 knockout mice. Kidney Int. 2000;57:129–36. doi: 10.1046/j.1523-1755.2000.00848.x. [DOI] [PubMed] [Google Scholar]
- 33.Lloyd CM, Minto AW, Dorf ME, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185:1371–80. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panzer U, Thaiss F, Zahner G, et al. Monocyte chemoattractant protein-1 and osteopontin differentially regulate monocytes recruitment in experimental glomerulonephritis. Kidney Int. 2001;59:1762–9. doi: 10.1046/j.1523-1755.2001.0590051762.x. [DOI] [PubMed] [Google Scholar]
- 35.Tam FW, Sanders JS, George A, et al. Urinary monocyte chemoattractant protein-1 (MCP-1) is a marker of active renal vasculitis. Nephrol Dial Transpl. 2004;19:2761–8. doi: 10.1093/ndt/gfh487. [DOI] [PubMed] [Google Scholar]
- 36.Cockwell P, Chakravorty SJ, Girdlestone J, Savage CO. Fractalkine expression in human renal inflammation. J Pathol. 2002;196:85–90. doi: 10.1002/path.1010. [DOI] [PubMed] [Google Scholar]
- 37.Feng L, Chen S, Garcia GE, et al. Prevention of crescentic glomerulonephritis by immunoneutralization of the fractalkine receptor CX3CR1 rapid communication. Kidney Int. 1999;56:612–20. doi: 10.1046/j.1523-1755.1999.00604.x. [DOI] [PubMed] [Google Scholar]