

Abstract
Folate deficiency may affect gene expression by disrupting DNA methylation patterns or by inducing base substitution, DNA breaks, gene deletions and gene amplification. Changes in expression may explain the inverse relationship observed between folate status and risk of colorectal cancer. Three cell lines derived from the normal human colon, HCEC, NCM356 and NCM460, were grown for 32–34 days in media containing 25, 50, 75 or 150 nM folic acid, and the expression of genes involved in cell-cycle checkpoints, intracellular signaling, folate uptake and cell adhesion and migration was determined. Expression of Folate Receptor 1 was increased with decreasing media folate in all cell lines, as was p53, p21, p16 and β-catenin. With decreasing folate, the expression of both E-cadherin and SMAD-4 was decreased in NCM356. APC was elevated in NCM356 but unchanged in the other lines. No changes in global methylation were detected. A significant increase in p53 exon 7–8 strand breaks was observed with decreasing folate in NCM460 cells. The changes observed are consistent with DNA damage-induced activation of cell-cycle checkpoints and cellular adaptation to folate depletion. Folate-depletion-induced changes in the Wnt/APC pathway as well as in genes involved in cell adhesion, migration and invasion may underlie observed relationships between folate status and cancer risk.
Keywords: Gene expression, p53, Strand break, Epithelial cell
1. Introduction
Human population studies show that a high dietary folate intake is associated with a diminished risk of colorectal cancer [1–3] and, possibly, of cancers of the breast [4–6], uterine cervix [7–9], lung [10,11], esophagus and pancreas [12] as well. The inverse association between folate status and the risk for carcinogenesis is thought to be explained by the role of folate coenzymes in providing one-carbon moieties for nucleotide and S-adenosylmethionine (via methionine) synthesis [13].
In cell culture, folate depletion increases the intracellular dUMP:dTMP ratio by up to 10-fold [14,15]. In vitro [15–18] and in vivo [19–21] studies reveal that this nucleotide imbalance promotes the misincorporation of uracil into DNA and the subsequent formation of double-stranded DNA breaks (DSB) through the excision of closely spaced uracil residues on opposing strands during repair [22]. Low dietary folate intake promotes genomic strand breakage in the liver of rats [21,23,24] and micropigs [25]. In addition, folate-depletion-induced DNA breakage has been observed in primary human lymphocytes [17,26] and rodent cell lines [15,27] in vitro. Folate-deficiency-induced chromosome breakage is relevant because the accumulation of chromosome aberrations, such as double-stranded breaks, is an established risk factor for the development of cancer [28].
In addition to disturbing nucleotide metabolism, folate depletion is reported to cause genomic hypomethylation in humans [19] and in cell culture [29] and p53 gene-specific hypomethylation in rats [23,24]. Although a depletion of the universal methyl donor S-adenosylmethionine is observed during folate depletion, the accumulation of its precursor S-adenosylhomocysteine, an inhibitor of DNA methyltransferases, appears to be a stronger predictor of hypomethylation [30]. Interestingly, under folate-depleted conditions, specific sequences may also become hypermethylated, as is the case with the H-cadherin gene [31]. DNA methylation is an important mechanism of gene silencing because the majority of tissue-specific genes exhibit a strong correlation between hypomethylation of the promoter region and gene expression [32].
These deleterious genetic and epigenetic changes likely elicit a change in cellular gene expression profiles, whether as a response to, or as a consequence of, a modified template. Several groups, including our own, have begun characterizing the effect of low folate status on the transcriptome using both rodent and cell culture models.
Previous studies have shown that Folate Receptor 1 (FOLR1) [33–36] is up-regulated due to folate depletion in vitro, while it is the reduced folate carrier (RFC) that is upregulated in the rat intestine [37,38], with the FOLR1 remaining undetectable [38]. Increased FOLR1 in vitro and the increased RFC in vivo reflect the abundance of available forms in the particular model and the affinity of each protein for them. FOLR1 has a higher affinity than RFC for folic acid (pteroylglutamic acid), the primary folate form in culture media. Conversely, the predominant folate form in plasma is 5-methyltetrahydrofolate, a form for which RFC has a greater affinity [39]. Nevertheless, these changes appear to be an adaptive response as cells attempt to take up more folate from their surroundings.
In addition to these adaptive changes, an array of interesting genes seem to be responsive to folate status. Among these are genes involved in implementing the cell-cycle checkpoints and DNA repair as well as genes involved in cell adhesion, motility and invasion. These two categories of genes are of clear relevance to carcinogenesis as they both include tumor suppressors and genes up-regulated in tumors.
To date, the reported response of cell-cycle checkpoint genes such as p21 and p53 to folate status has been varied. Folate depletion resulted in reduced p53 expression in the rat colon [40,41], which was associated with increased p53 strand breaks [40]. Similarly, p53 was decreased in HCT116, HT29 and LS513 but increased in Caco-2 colon cancer cells [42]. We recently observed an increase in p53 transcript and protein, despite substantial elevations in strand breaks, in primary human lymphocytes [43]. Immediately downstream of p53, p21 was also studied in colon cancer cells and was reported to be elevated in HCT116 and Caco-2 cells but down-regulated in HT29 and LS513 cells [42].
In the second category, Jhaveri et al. [31] noted the hypermethylation-related down-regulation of H-cadherin, a cell adhesion molecule, in folate-depleted KB cells. Similarly, we observed a reduction in the protocadherin-4 adhesion molecule in the colon of folate-depleted rats. This was accompanied by a concomitant reduction in attachment factors; integrin α5, glypican 1, nidogen, involucrin and proteases; MMP16, TIMP-2, metalloendo-peptidase, metallothionein and subtilisin and an elevation of urokinase and chymase [44]. Others report an elevation of metallothionein in Chinese hamster lung fibroblasts selected for growth in low folate [45]. The elevation of urokinase, as well as its receptor, under folate depletion, was observed by others in HCT116 but not in Caco-2 cells [42].
Emerging data also indicate that the APC/Wnt pathway, which is commonly disrupted in colon carcinogenesis [46], is sensitive to folate status. Novakovic et al. [42] report an elevation of β-catenin transcript in three of four colon cancer cell lines. Furthermore, we have recently observed that multiple but mild B-vitamin depletion (folate, B2, B6 and B12) induced an elevation of β-catenin protein and a decrease in APC gene expression, which was associated with elevated DNA breaks in the APC mutation cluster region (Liu et al., in preparation).
In light of these findings, we sought to further characterize the impact of folate status on the expression of genes involved in cell-cycle checkpoints, cell adhesion and migration and in the APC/Wnt pathway. Furthermore, since it remains unclear in the instance of one gene (p53), we sought to determine whether the induction of folate-dependent strand breaks in the coding region of the gene is related to diminished expression. Such studies may help us understand the mechanisms underlying the observed inverse relationship between cancer risk and folate status. Folate status has previously been observed to have a differential effect on tumorigenesis in animals, with supplementation being protective if started before the appearance of precancerous lesions while promoting tumor growth after initiation has already occurred [47,48]. For this reason, we have chosen to perform our experiments in cells derived from the normal colon, which, to the best of our knowledge, are among the most ‘normal’ colonic cells currently available. We cultured HCEC, NCM460 and NCM356 cell in media containing 25–150 nM folic acid for 30 days and studied the expression of genes involved in cell-cycle checkpoints (p16, p21, p53), adhesion and migration (urokinase, E-cadherin), the Wnt pathway (β-catenin, APC), folate uptake (RFC, folate receptor) and signaling (SMAD-4).
2. Methods
Three colonic epithelial cell lines derived from the normal adult human colon were cultured for 30–32 days in media containing varying concentrations of folic acid. Human colonic epithelial cells (HCECs) were a gift from Dr. A. Pfeifer (Nestle, Lausanne, Switzerland). HCECs were obtained from a scratch biopsy of a 69-year-old female, are SV-40 transformed and do not express cytokeratin 19. NCM356 and NCM460 cells were obtained from INCELL (San Antonio, TX). NCM cells are not transfected with any exogenous genetic material and do express cytokeratin 19 (data not shown). NCM356 and NCM460 are derived from the normal mucosa of 65- and 68-year-old males, respectively. Cells were grown in T75 flasks in 10 ml of A52 media supplemented with 30 µg/ml bovine pituitary extract, 100 nM retinoic acid (Biofluids, Rockville, MD, now Invitrogen, Carlsbad, CA), 8 µg/ml vitamin C, 2 mM l-glutamine and 1 nM dexamethasone. A52 was custom made without folic acid, which was added at concentrations of 25, 50, 75 and 150 nM before use. HCECs require the precoating of flasks with Matrigel extracellular matrix solution (BD Biosciences, San Jose, CA).
DNA was extracted from cells using phenol:chloroform via standard procedures and quantified using Pico green dye (Invitrogen). Total cellular RNA was isolated from cells using Trizol reagent, and cDNA was synthesized using Superscript II reverse transcriptase (both from Invitrogen).
The relative expression of selected transcripts was determined using Taqman gene expression assays and PCR mastermix with an ABI7300 thermal cycler according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA). Gene expression was normalized to that of GAPDH (ΔCt) and is reported as relative to the control group by using the following equation: relative expression=2−ΔΔCt, where ΔΔCt is the ΔCt of the test group (25, 50 and 75 nM folate) minus the ΔCt of the control group (150 nM).
The integrity of exons 7–8 of the p53 tumor suppressor gene was studied using a PCR-based assay [43] whereby damage such as double-stranded DNA breaks inhibits the progression of Taq polymerase and thereby decreases amplification rate. Briefly, 100 ng of DNA was subjected to PCR for p53ex7–8 and GAPDH. An increase in ΔCt (Ctp53−CtGAPDH) indicates a reduction in amplifiable template or increase in DNA breakage.
Genomic DNA methylation was determined in 1 µg DNA by enzymatic hydrolysis of DNA followed by LC-MS using the method of Friso et al. [49].
Data for each folic acid concentration were compared using one-way ANOVA with Tukey’s post hoc tests, and significance was accepted when P≤.05. In addition, linear regression was used to test the relationship between ΔCt and medium folate concentrations. Data are expressed as mean±S.E.M. All calculations were performed using SPSS Systat v10 (Point Richmond, CA). For gene expression, ΔCt values were analyzed; however, relative expression values are reported here for brevity and clarity.
3. Results
Decreasing media folate concentration caused an incremental reduction in cell growth for HCEC and NCM356 cells, while in NCM460, cell growth was similar for 50–150 nM and reduced only at 25 nM (Fig. 1).
Fig. 1.

Growth curves for colonic epithelial cells cultured with varying concentrations of folic acid. (A) HCEC, (B) NCM460, (C) NCM356. Data are expressed as mean±S.E.M.
Gene expression data are reported in Table 1 as relative expression values.
Table 1.
Effect of media folic acid concentration on relative gene expression in colon epithelial cell lines
| Gene | Cell line | Media folic acid (nM) | ANOVA | Trend | |||
|---|---|---|---|---|---|---|---|
| 25 | 50 | 75 | 150 | P | |||
| Reduced Folate Carrier | HCEC | 1.78a | 0.83 | 0.56a | 1.00 | .002 | 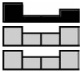 |
| SLC19A1 | NCM460 | 1.03 | 0.76 | 0.60 | 1.00 | .106 | |
| NM_003056.2 | NCM356 | 1.11 | 0.49 | 0.56 | 1.00 | .061 | |
| Folate Receptor 1 | HCEC | 5.17 | 4.55 | 2.57 | 1.00 | .101b | |
| FOLR1 | NCM460 | 9.59a | 0.93 | 1.11 | 1.00 | <.001 | |
| NM_016724.1 | NCM356 | 132.51a | 49.92a | 15.82a | 1.00 | <.001b | |
| Adenamatosis Polyposis Coli | HCEC | 1.21 | 1.50 | 1.06 | 1.00 | .660 | 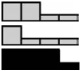 |
| APC | NCM460 | 1.52 | 1.01 | 1.05 | 1.00 | .310 | |
| NM_000038.3 | NCM356 | 1.34a | 1.23 | 1.37a | 1.00 | .018b | |
| β-Catenin | HCEC | 1.30 | 1.48 | 1.14 | 1.00 | .150b |  |
| CTNNB1 | NCM460 | 1.46a | 0.89 | 0.89 | 1.00 | .003 | |
| NM_001904.2 | NCM356 | 1.97a | 2.89a | 2.38a | 1.00 | <.001b | |
| E-Cadherin | HCEC | 1.88 | 1.62 | 0.98 | 1.00 | .550 | 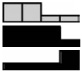 |
| CDH1 | NCM460 | 1.43 | 1.57a | 1.42 | 1.00 | .048b | |
| NM_004360.2 | NCM356 | 0.25a | 0.28a | 0.45a | 1.00 | <.001b | |
| Urokinase | HCEC | 3.23a | 2.79a | 1.88a | 1.00 | .001b | 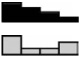 |
| PLAU | NCM460 | 2.29a | 1.26 | 1.00 | 1.00 | <.001b | |
| NM_002658.2 | NCM356 | 1.16 | 0.74 | 0.85 | 1.00 | .062 | |
| p53 | HCEC | 2.06a | 1.87a | 1.73a | 1.00 | .003b | |
| TP53 | NCM460 | 1.23a | 1.20a | 1.16a | 1.00 | .007b | |
| NM_000546.2 | NCM356 | 5.20a | 4.23a | 3.03a | 1.00 | <.001b | |
| p21 | HCEC | 3.28a | 1.70 | 1.28 | 1.00 | .004b | 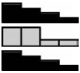 |
| CDKN1A | NCM460 | 1.34 | 1.27 | 0.90 | 1.00 | .117 | |
| NM_078467.1 | NCM356 | 7.06a | 4.92a | 3.23a | 1.00 | <.001b | |
| p16 | HCEC | 1.66 | 1.15 | 0.89 | 1.00 | .051 | 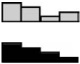 |
| CDKN2A | NCM460 | 1.37 | 1.14 | 1.11 | 1.00 | .073b | |
| NM_058195.2 | NCM356 | 3.20a | 2.18a | 1.56a | 1.00 | <.001b | |
| SMAD-4 | HCEC | 2.19a | 2.17a | 1.54 | 1.00 | .013b | 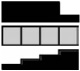 |
| SMAD-4 | NCM460 | 1.12 | 1.24 | 1.14 | 1.00 | .070 | |
| NM_005359.3 | NCM356 | 0.15a | 0.11a | 0.34a | 1.00 | <.001b | |
Relative expression=2−ΔΔCt. ΔΔCt=ΔCttest group−ΔCtcontrol group. ΔCt=Cttarget gene−CtGAPDH. Statistics were performed on ΔCt values.
P≤.05 vs. 150 nM Tukey).
P trend (regression)<.05. Gene symbols and accession numbers are listed below the gene name. Trends for gene expression change are illustrated with black graphs indicating significant trends.
Decreasing media folate caused a substantial elevation in the expression of FOLR1 in all three cell lines, especially in NCM356, which displayed a 132-fold elevation in FOLR1 at 25 nM, as compared with 150 nM. Interestingly, there is a tendency for the RFC to be slightly suppressed at the middle folate deficiencies and then normalize or slightly elevated at 25 nM, a trend that is significant in HCEC.
Decreasing media folate caused an incremental up-regulation of the DNA damage and cell-cycle-checkpoint-related genes p53, p16 and p21. This response is especially robust for p53, which was significantly up-regulated in all cell lines for even the mildest deficiency concentration of 75 nM, a concentration that could support maximal cell proliferation in NCM460. The trend for up-regulation of p21 is significant in HCEC and NCM356 but equivocal in NCM460. Similarly, p16 is significantly up-regulated in the folate-deficient NCM cell lines, but the response in HCEC is marginal.
Both APC and β-catenin are incrementally up-regulated during folate depletion in NCM356 cells (P<.05), a trend that does not reach significance in the other cell lines. E-cadherin, another member of the Wnt pathway, is down-regulated in NCM356 cells (P<.05) but marginally elevated in NCM460 and HCEC. Possibly related to these changes is SMAD-4, an inducer of E-cadherin, which is also significantly down-regulated in NCM356 but marginally elevated in NCM460 and HCEC under folate-deficient conditions.
Urokinase expression was elevated at the three deficiency folate concentrations in HCEC and at 25 nM in NCM460 but was unchanged in NCM356.
Genomic DNA methylation was not affected by folate status in any of the cell lines (P>.05, Table 2).
Table 2.
Effect of media folic acid concentration on genomic DNA methylation in colon epithelial cell lines
| Cell line | Media folic acid (nM) | ANOVA | |||
|---|---|---|---|---|---|
| 25 | 50 | 75 | 150 | P | |
| HCEC | 3.14±0.29 | 2.53±0.19 | 2.95±0.67 | 1.94±0.06 | .20 |
| NCM460 | 3.81±0.12 | 3.82±0.19 | 3.76±0.32 | 4.19±0.29 | .58 |
| NCM356 | 3.04±0.15 | 3.01±0.28 | 2.27±0.23 | 2.81±0.13 | .09 |
DNA methylation is reported as nanograms of 5-methylcytosine per microgram of DNA. Data are expressed as mean±S.E.M.
Media folate concentration did not affect p53 exon 7–8 strand breakage in HCEC and NCM356 but caused a stepwise increase in strand breaks in NCM460 cells (Fig. 2). A 1.96 cycle difference in ΔCt was observed between the 25- and the 150-nM folate groups. Based on our experiments with restriction-digested DNA [43], we estimate this to represent an approximately 23.1% reduction in intact p53 exon 7–8 DNA template.
Fig. 2.

Effect of media folic acid concentration on p53 exon 7–8 integrity in colon epithelial cell lines. Increased ΔCt indicates increased strand breakage within p53 exons 7–8. Data are expressed as mean±S.E.M. aP<.05 vs. 150 nM. NCM460 P trend (regression)=.004.
4. Discussion
We studied the impact of folate depletion on the expression of select genes involved in folate uptake, cell adhesion and migration and cell-cycle checkpoint regulation, all of which are relevant to carcinogenesis.
A strong up-regulation of FOLR1, but not RFC, due to folate depletion was an anticipated observation due to the high affinity of FOLR1 but not RFC for folic acid [39]. This adaptive mechanism has previously been shown to be mediated by an increase in transcription rate and transcript half-life [33,36]. It is noteworthy that in NCM460 cells, FOLR1 expression and cell proliferation are similar for the upper three folate concentrations and then significantly increase and decrease at 25 nM folate, respectively. Although comparisons of relative expression cannot be made between the cell lines, it is likely that NCM460 cells have a much higher basal FOLR1 expression than HCEC and NCM356 cells, which is sufficient to support maximal cell growth down to folate concentrations of 50 nM.
Folate depletion induced the up-regulation of p16, p21 and p53 tumor suppressor genes, which are involved in DNA damage signaling, inhibition of cell-cycle progression through checkpoints and apoptosis. It is likely that folate depletion caused uracil incorporation and DNA breaks [26], thereby activating the DNA repair protein ATM. ATM is known to activate p53, via Chk2, which, in turn, stimulates p21 transcription. p21 arrests the cell cycle by inhibiting CDK2/cyclin E while p16 inhibits CDK4,6/cyclin D. Because folate-depletion-induced damage is expected to occur during S-phase when uracil is incorporated into newly synthesized DNA, activation of the CDK inhibitors p16 and p21 would block the transition of cells out of this phase of the cell cycle. In support of this, the accumulation of cells in S-phase has been observed in folate-depleted lymphocytes [50]. It should be noted that HCECs were immortalized using the SV40-T antigen, which is known to bind and inhibit p53 protein function [51]. Nevertheless, the folate-depletion-induced increase in p53 transcript in HCECs was of a similar magnitude to the non-SV40 immortalized NCM cells. One might infer that since p21 transcript is also similarly elevated across the three cell lines, the SV40-T antigen does not significantly impair the ability of p53 to induce downstream genes such as p21 transcription in HCECs in this system.
We have previously observed a folate-depletion-induced up-regulation of p53 transcript and protein in primary human lymphocytes despite 26% and 40% reductions in amplifiable template from exons 7–8 and 5–8 of the gene, respectively [43]. This demonstrates that the induction of strand breaks is not invariably accompanied by a reduction in the corresponding transcript. In the current study, NCM460 showed a similar pattern: the degree of p53 exon 7–8 template loss in depleted compared to replete cells (23%) was accompanied by a significant increase in steady-state p53 mRNA. Interestingly, no loss of amplifiable template was observed in either HCEC or NCM356 cells (Fig. 2). As this assay is a measure of intact gene template, the copy number of the target gene is a central determinant of the result and, as such, might only be reliable in cells with a diploid karyotype. We have previously determined the HCECs to be almost tetraploid (72.13±1.03 chromosomes) and NCM460 to be almost diploid (56.47±0.63 chromosomes) but have not determined the karyotype of NCM356.
Folate depletion has been shown to induce p53 stand breaks with a concomitant reduction in steady-state p53 mRNA in the rat colon [40]. In this tissue, unlike our cell culture models, the severity of p53 gene loss or damage may have overwhelmed cellular mechanisms aimed at maintaining elevated p53 during genotoxic stress, thereby increasing the chance that cells with deleterious genetic or epigenetic aberrations continue dividing, a procarcinogenic state.
In agreement with our rat studies [44], folate depletion induced the up-regulation of urokinase in HCEC and NCM460 cells. Others observed an up-regulation in HCT116 but not in Caco-2 cells [42]. Urokinase is involved in ECM remodeling and cell migration and, when elevated in primary tumors, is a predictor of metastasis and poor prognosis for breast cancer patients [52]. The observations that cadherins, urokinase and various other attachment factors and ECM proteases are affected by folate status [31,53] suggest that cell adhesion, migration and ECM remodeling, important processes in progression of established lesions, may be a novel mechanism, in addition to genetic/epigenetic instability, by which folate status modulates our risk for cancer.
It is not clear how or why folate status affects urokinase transcript levels; however, a complex interplay between multiple genes seems to be involved. Activation of urokinase has been shown to be dependent on E-cadherin, at least at the protein level [54]. In our system, the two cell lines that have a folate-depletion-induced elevation of E-cadherin (HCEC and NCM460) are also the ones that display an increase in urokinase. Regulation of urokinase has also been attributed to SMAD-4 [55] and β-catenin [56]. Furthermore, SMAD-4 is an inducer of E-cadherin transcription [57], and in our system, the changes in these two genes are paralleled, with a trend for increase in HCEC and NCM460 and decrease in NCM356 during folate depletion. A detailed survey of protein levels is clearly required to further decipher this scheme.
Our data, along with those of others, indicate that the Wnt pathway is clearly affected by cellular folate status. In addition to the abovementioned changes in β-catenin and E-cadherin, which has an additional role in sequestering β-catenin, APC is also affected by folate status, at least in NCM356 cells. Similarly, we have also observed a reduction in APC transcript in the mouse colon due to multiple B-vitamin deficiency (Liu et al., submitted for publication). Wnt signaling pathway members APC and β-catenin are mutated or lost in the majority of the sporadic colorectal cancer cases, resulting in activation of the pathway, which has a diverse array of transcriptional targets.
We did not observe any alteration in genomic DNA methylation due to folate depletion in our system (Table 2). This may be due to the fact that DMEM, which is the base for A52 media, contains a high concentration of methionine (30 mg/L or 200 µM), thereby negating the cellular need for folate to recycle homocysteine to methionine. In future experiments, titrating the amount of methionine in the media down to the minimal level that supports normal proliferation may precipitate an effect of folate depletion on global DNA methylation and thereby amplify the gene expression response.
Emerging data seem to indicate that folate status affects several cancer-related pathways including the p53 pathway (p53 and p21), the Rb pathway (p16) and the APC/Wnt pathway (CTNN1 and CHD1), as well as pathways involved in cell adhesion (CDH1) and cell migration and invasion (PLAU1). Folate-induced changes in these pathways may underlie the observed associations, whether positive or negative, between folate intake and cancer incidence.
Acknowledgments
This work was supported by grants from the Cancer Research and Prevention Foundation (J.W.C.), NCI (J.B.M.: KO5 CA100048-01) and NIDDK (J.B.M.: T32 DK007651-16). This material is based on work supported by the U.S. Department of Agriculture, under Agreement No. 58-1950-4-401. Any opinions, findings, conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the view of the U.S. Department of Agriculture.
References
- 1.Giovannucci E, Rimm EB, Ascherio A, Stampfer MJ, Colditz GA, Willett WC. Alcohol, low-methionine-low-folate diets, and risk of colon cancer in men [see comments] J Natl Cancer Inst. 1995;87:265–273. doi: 10.1093/jnci/87.4.265. [DOI] [PubMed] [Google Scholar]
- 2.Larsson SC, Giovannucci E, Wolk A. A prospective study of dietary folate intake and risk of colorectal cancer: modification by caffeine intake and cigarette smoking. Cancer Epidemiol Biomarkers Prev. 2005;14:740–743. doi: 10.1158/1055-9965.EPI-04-0581. [DOI] [PubMed] [Google Scholar]
- 3.Ma J, Stampfer MJ, Giovannucci E, Artigas C, Hunter DJ, Fuchs C, et al. Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res. 1997;57:1098–1102. [PubMed] [Google Scholar]
- 4.Shrubsole MJ, Gao YT, Cai Q, Shu XO, Dai Q, Hebert JR, et al. MTHFR polymorphisms, dietary folate intake, and breast cancer risk: results from the Shanghai Breast Cancer Study. Cancer Epidemiol Biomarkers Prev. 2004;13:190–196. doi: 10.1158/1055-9965.epi-03-0273. [DOI] [PubMed] [Google Scholar]
- 5.Beilby J, Ingram D, Hahnel R, Rossi E. Reduced breast cancer risk with increasing serum folate in a case–control study of the C677T genotype of the methylenetetrahydrofolate reductase gene. Eur J Cancer. 2004;40:1250–1254. doi: 10.1016/j.ejca.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 6.Hussien MM, McNulty H, Armstrong N, Johnston PG, Spence RA, Barnett Y. Investigation of systemic folate status, impact of alcohol intake and levels of DNA damage in mononuclear cells of breast cancer patients. Br J Cancer. 2005;92:1524–1530. doi: 10.1038/sj.bjc.6602530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez BY, McDuffie K, Wilkens LR, Kamemoto L, Goodman MT. Diet and premalignant lesions of the cervix: evidence of a protective role for folate, riboflavin, thiamin, and vitamin B12. Cancer Causes Control. 2003;14:859–870. doi: 10.1023/b:caco.0000003841.54413.98. [DOI] [PubMed] [Google Scholar]
- 8.Kwasniewska A, Tukendorf A, Semczuk M. Folate deficiency and cervical intraepithelial neoplasia. Eur J Gynaecol Oncol. 1997;18:526–530. [PubMed] [Google Scholar]
- 9.Butterworth CE, Jr, Hatch KD, Macaluso M, Cole P, Sauberlich HE, Soong SJ, et al. Folate deficiency and cervical dysplasia. JAMA. 1992;267:528–533. [PubMed] [Google Scholar]
- 10.Voorrips LE, Goldbohm RA, Brants HA, van Poppel GA, Sturmans F, Hermus RJ, et al. A prospective cohort study on antioxidant and folate intake and male lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:357–365. [PubMed] [Google Scholar]
- 11.Shen H, Wei Q, Pillow PC, Amos CI, Hong WK, Spitz MR. Dietary folate intake and lung cancer risk in former smokers: a case–control analysis. Cancer Epidemiol Biomarkers Prev. 2003;12:980–986. [PubMed] [Google Scholar]
- 12.Larsson SC, Giovannucci E, Wolk A. Folate intake, MTHFR polymorphisms, and risk of esophageal, gastric, and pancreatic cancer: a meta-analysis. Gastroenterology. 2006;131:1271–1283. doi: 10.1053/j.gastro.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Jang H, Mason JB, Choi SW. Genetic and epigenetic interactions between folate and aging in carcinogenesis. J Nutr. 2005;135:2967S–2971S. doi: 10.1093/jn/135.12.2967S. [DOI] [PubMed] [Google Scholar]
- 14.James SJ, Basnakian AG, Miller BJ. In vitro folate deficiency induces deoxynucleotide pool imbalance, apoptosis, and mutagenesis in Chinese hamster ovary cells. Cancer Res. 1994;54:5075–5080. [PubMed] [Google Scholar]
- 15.Melnyk S, Pogribna M, Miller BJ, Basnakian AG, Pogribny IP, James SJ. Uracil misincorporation, DNA strand breaks, and gene amplification are associated with tumorigenic cell transformation in folate deficient/repleted Chinese hamster ovary cells. Cancer Lett. 1999;146:35–44. doi: 10.1016/s0304-3835(99)00213-x. [DOI] [PubMed] [Google Scholar]
- 16.Crott JW, Mashiyama ST, Ames BN, Fenech MF. Methylenetetrahydrofolate reductase C677T polymorphism does not alter folic acid deficiency-induced uracil incorporation into primary human lymphocyte DNA in vitro. Carcinogenesis. 2001;22:1019–1025. doi: 10.1093/carcin/22.7.1019. [DOI] [PubMed] [Google Scholar]
- 17.Duthie SJ, Hawdon A. DNA instability (strand breakage, uracil misincorporation, and defective repair) is increased by folic acid depletion in human lymphocytes in vitro. FASEB J. 1998;12:1491–1497. [PubMed] [Google Scholar]
- 18.Koury MJ, Horne DW, Brown ZA, Pietenpol JA, Blount BC, Ames BN, et al. Apoptosis of late-stage erythroblasts in megaloblastic anemia: association with DNA damage and macrocyte production. Blood. 1997;89:4617–4623. [PubMed] [Google Scholar]
- 19.Jacob RA, Gretz DM, Taylor PC, James SJ, Pogribny IP, Miller BJ, et al. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J Nutr. 1998;128:1204–1212. doi: 10.1093/jn/128.7.1204. [DOI] [PubMed] [Google Scholar]
- 20.Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pogribny IP, Miller BJ, James SJ. Alterations in hepatic p53 gene methylation patterns during tumor progression with folate/methyl deficiency in the rat. Cancer Lett. 1997;115:31–38. doi: 10.1016/s0304-3835(97)04708-3. [DOI] [PubMed] [Google Scholar]
- 22.Dianov GL, Timchenko TV, Sinitsina OI, Kuzminov AV, Medvedev OA, Salganik RI. Repair of uracil residues closely spaced on the opposite strands of plasmid DNA results in double-strand break and deletion formation. Mol Gen Genet. 1991;225:448–452. doi: 10.1007/BF00261686. [DOI] [PubMed] [Google Scholar]
- 23.Kim YI, Pogribny IP, Basnakian AG, Miller JW, Selhub J, James SJ, et al. Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumor suppressor gene. Am J Clin Nutr. 1997;65:46–52. doi: 10.1093/ajcn/65.1.46. [DOI] [PubMed] [Google Scholar]
- 24.Pogribny IP, Basnakian AG, Miller BJ, Lopatina NG, Poirier LA, James SJ. Breaks in genomic DNA and within the p53 gene are associated with hypomethylation in livers of folate/methyl-deficient rats. Cancer Res. 1995;55:1894–1901. [PubMed] [Google Scholar]
- 25.Halsted CH, Villanueva JA, Devlin AM, Niemela O, Parkkila S, Garrow TA, et al. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc Natl Acad Sci U S A. 2002;99:10072–10077. doi: 10.1073/pnas.112336399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crott JW, Mashiyama ST, Ames BN, Fenech M. The effect of folic acid deficiency and MTHFR C677T polymorphism on chromosome damage in human lymphocytes in vitro. Cancer Epidemiol Biomarkers Prev. 2001;10:1089–1096. [PubMed] [Google Scholar]
- 27.Branda RF, McCormack JJ, Perlmutter CA, Mathews LA, Robison SH. Effects of folate deficiency on the metastatic potential of murine melanoma cells. Cancer Res. 1988;48:4529–4534. [PubMed] [Google Scholar]
- 28.Bonassi S, Hagmar L, Stromberg U, Montagud AH, Tinnerberg H, Forni A, et al. Chromosomal aberrations in lymphocytes predict human cancer independently of exposure to carcinogens. European Study Group on Cytogenetic Biomarkers and Health. Cancer Res. 2000;60:1619–1625. [PubMed] [Google Scholar]
- 29.Duthie SJ, Narayanan S, Blum S, Pirie L, Brand GM. Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr Cancer. 2000;37:245–251. doi: 10.1207/S15327914NC372_18. [DOI] [PubMed] [Google Scholar]
- 30.James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr. 2002;132:2361S–2366S. doi: 10.1093/jn/132.8.2361S. [DOI] [PubMed] [Google Scholar]
- 31.Jhaveri MS, Wagner C, Trepel JB. Impact of extracellular folate levels on global gene expression. Mol Pharmacol. 2001;60:1288–1295. doi: 10.1124/mol.60.6.1288. [DOI] [PubMed] [Google Scholar]
- 32.Laird PW, Jaenisch R. DNA methylation and cancer. Hum Mol Genet. 1994;3:1487–1495. doi: 10.1093/hmg/3.suppl_1.1487. [DOI] [PubMed] [Google Scholar]
- 33.Zhu WY, Alliegro MA, Melera PW. The rate of folate receptor alpha (FR alpha) synthesis in folate depleted CHL cells is regulated by a translational mechanism sensitive to media folate levels, while stable overexpression of its mRNA is mediated by gene amplification and an increase in transcript half-life. J Cell Biochem. 2001;81:205–219. doi: 10.1002/1097-4644(20010501)81:2<205::aid-jcb1036>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 34.Jansen G, Kathmann I, Rademaker BC, Braakhuis BJ, Westerhof GR, Rijksen G, et al. Expression of a folate binding protein in L1210 cells grown in low folate medium. Cancer Res. 1989;49:1959–1963. [PubMed] [Google Scholar]
- 35.Hsueh CT, Dolnick BJ. Altered folate-binding protein mRNA stability in KB cells grown in folate-deficient medium. Biochem Pharmacol. 1993;45:2537–2545. doi: 10.1016/0006-2952(93)90235-o. [DOI] [PubMed] [Google Scholar]
- 36.Sadasivan E, Regec A, Rothenberg SP. The half-life of the transcript encoding the folate receptor alpha in KB cells is reduced by cytosolic proteins expressed in folate-replete and not in folate-depleted cells. Gene. 2002;291:149–158. doi: 10.1016/s0378-1119(02)00591-7. [DOI] [PubMed] [Google Scholar]
- 37.Baggott JE, Morgan SL, Ha T, Vaughn WH, Hine RJ. Inhibition of folate-dependent enzymes by non-steroidal anti-inflammatory drugs. Biochem J. 1992;282:197–202. doi: 10.1042/bj2820197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Said HM, Chatterjee N, Haq RU, Subramanian VS, Ortiz A, Matherly LH, et al. Adaptive regulation of intestinal folate uptake: effect of dietary folate deficiency. Am J Physiol Cell Physiol. 2000;279:C1889–C1895. doi: 10.1152/ajpcell.2000.279.6.C1889. [DOI] [PubMed] [Google Scholar]
- 39.Shane B. Folate chemistry and metabolism. In: Bailey LB, editor. Folate in health and disease. New York (NY): Marcel Dekker, Inc; 1995. pp. 1–22. [Google Scholar]
- 40.Kim YI, Shirwadkar S, Choi SW, Puchyr M, Wang Y, Mason JB. Effects of dietary folate on DNA strand breaks within mutation-prone exons of the p53 gene in rat colon. Gastroenterology. 2000;119:151–161. doi: 10.1053/gast.2000.8518. [DOI] [PubMed] [Google Scholar]
- 41.Sohn KJ, Stempak JM, Reid S, Shirwadkar S, Mason JB, Kim YI. The effect of dietary folate on genomic and p53-specific DNA methylation in rat colon. Carcinogenesis. 2003;24:81–90. doi: 10.1093/carcin/24.1.81. [DOI] [PubMed] [Google Scholar]
- 42.Novakovic P, Stempak JM, Sohn KJ, Kim YI. Effects of folate deficiency on gene expression in the apoptosis and cancer pathways in colon cancer cells. Carcinogenesis. 2006;27:916–924. doi: 10.1093/carcin/bgi312. [DOI] [PubMed] [Google Scholar]
- 43.Crott JW, Liu Z, Choi SW, Mason JB. Folate depletion in human lymphocytes up-regulates p53 expression despite marked induction of strand breaks in exons 5–8 of the gene. Mutat Res. 2007;626:171–179. doi: 10.1016/j.mrgentox.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Crott JW, Choi SW, Ordovas JM, Ditelberg JS, Mason JB. Effects of dietary folate and aging on gene expression in the colonic mucosa of rats: implications for carcinogenesis. Carcinogenesis. 2004;25:69–76. doi: 10.1093/carcin/bgg150. [DOI] [PubMed] [Google Scholar]
- 45.Zhu WY, Melera PW. Metallothionein is overexpressed by hamster fibroblasts selected for growth in 15 pm folinic acid and provides a growth advantage in low folate. Cancer Res. 1999;59:4194–4199. [PubMed] [Google Scholar]
- 46.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 47.Song J, Medline A, Mason JB, Gallinger S, Kim YI. Effects of dietary folate on intestinal tumorigenesis in the apcMin mouse. Cancer Res. 2000;60:5434–5440. [PubMed] [Google Scholar]
- 48.Song J, Sohn KJ, Medline A, Ash C, Gallinger S, Kim YI. Chemopreventive effects of dietary folate on intestinal polyps in Apc +/− Msh2−/− mice. Cancer Res. 2000;60:3191–3199. [PubMed] [Google Scholar]
- 49.Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002;74:4526–4531. doi: 10.1021/ac020050h. [DOI] [PubMed] [Google Scholar]
- 50.Courtemanche C, Huang AC, Elson-Schwab I, Kerry N, Ng BY, Ames BN. Folate deficiency and ionizing radiation cause DNA breaks in primary human lymphocytes: a comparison. FASEB J. 2004;18:209–211. doi: 10.1096/fj.03-0382fje. [DOI] [PubMed] [Google Scholar]
- 51.Ozer HL, Banga SS, Dasgupta T, Houghton J, Hubbard K, Jha KK, et al. SV40-mediated immortalization of human fibroblasts. Exp Gerontol. 1996;31:303–310. doi: 10.1016/0531-5565(95)00024-0. [DOI] [PubMed] [Google Scholar]
- 52.Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer. 1997;72:1–22. doi: 10.1002/(sici)1097-0215(19970703)72:1<1::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 53.Crott JW, Choi SW, Ditelberg JS, Mason J. Washington (DC): American Association for Cancer Research; 2003. Effects of folic acid and aging on gene expression in the colonic mucosa of rats: implications for carcinogenesis; p. 894. [Google Scholar]
- 54.Sasaki CY, Lin H, Passaniti A. Regulation of urokinase plasminogen activator (uPA) activity by E-cadherin and hormones in mammary epithelial cells. J Cell Physiol. 1999;181:1–13. doi: 10.1002/(SICI)1097-4652(199910)181:1<1::AID-JCP1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 55.Schwarte-Waldhoff I, Klein S, Blass-Kampmann S, Hintelmann A, Eilert C, Dreschers S, et al. DPC4/SMAD4 mediated tumor suppression of colon carcinoma cells is associated with reduced urokinase expression. Oncogene. 1999;18:3152–3158. doi: 10.1038/sj.onc.1202641. [DOI] [PubMed] [Google Scholar]
- 56.Hiendlmeyer E, Regus S, Wassermann S, Hlubek F, Haynl A, Dimmler A, et al. Beta-catenin up-regulates the expression of the urokinase plasminogen activator in human colorectal tumors. Cancer Res. 2004;64:1209–1214. doi: 10.1158/0008-5472.can-3627-2. [DOI] [PubMed] [Google Scholar]
- 57.Reinacher-Schick A, Baldus SE, Romdhana B, Landsberg S, Zapatka M, Monig SP, et al. Loss of Smad4 correlates with loss of the invasion suppressor E-cadherin in advanced colorectal carcinomas. J Pathol. 2004;202:412–420. doi: 10.1002/path.1516. [DOI] [PubMed] [Google Scholar]