

Abstract
MassMatrix is a program that matches tandem mass spectra with theoretical peptide sequences derived from a protein database. The program uses a mass accuracy sensitive probabilistic score model to rank peptide matches. The tandem mass spectrometry search software was evaluated by use of a high mass accuracy data set and its results compared with those from Mascot, SEQUEST, X!Tandem, and OMSSA. For the high mass accuracy data, MassMatrix provided better sensitivity than Mascot, SEQUEST, X!Tandem, and OMSSA for a given specificity and the percentage of false positives was 2%. More importantly all manually validated true positives corresponded to a unique peptide/spectrum match. The presence of decoy sequence and additional variable post-translational modifications did not significantly affect the results from the high mass accuracy search. MassMatrix performs well when compared with Mascot, SEQUEST, X!Tandem, and OMSSA with regard to search time. MassMatrix was also run on a distributed memory clusters and achieved search speeds of ~100,000 spectra per hour when searching against a complete human database with 8 variable modifications. The algorithm is available for public searches at http://www.massmatrix.net.
Keywords: Tandem mass spectra, Database search, High mass accuracy, Proteomics, Post-translational modification
1 INTRODUCTION
Database search in combination with shotgun proteomics is the major tool used to identify peptides and proteins in complex protein mixtures. Database search programs match experimental spectra with theoretical spectra created from the database. They are classified into four categories according to their score algorithms: descriptive, interpretative, stochastic and statistical/probabilistic [1]. SEQUEST [2] is an example of a descriptive model and one of the most commonly used database search programs. Other programs of this type include Sonar [3] and SALSA [4]. PeptideSearch [5] is based on an interpretative model for database search. SCOPE [6] and OLAV [7] use stochastic models for database search. A group of programs that are based on statistical and probabilistic models have also been developed [8–15]. Among them, Mascot [9] is the most commonly used. The probability based score is a direct measure of the probability that the match is significant. Probability based scores from different search algorithms can be directly compared, whereas descriptive score must be converted to probabilities for comparisons [16].
These programs score candidate matches for the experiment spectra with theoretically generated counterparts from a protein database. Low mass accuracy, noise and low signal to noise ratio can compromise search results from database search programs [8, 9]. There are inconsistencies between search results from different search programs due to their different score algorithms. Kapp et al [17] performed a systematic comparison of various database search algorithms including Mascot [9], SEQUEST [2], Sonar [3], Spectrum Mill (www.chem.agilent.com) and X!Tandem [18]. A data set containing 3952 tandem MS spectra created from human B3-Cit-plasma (Asian-American) on an LCQ Deca XP ion trap mass spectrometer was searched by all five algorithms. The trypsin-constrained search results from the five algorithms achieved consensus for 345 spectral matches. Considering all search results the algorithms matched 662 spectra [17].
It has been reported that improving mass accuracy of precursor and product ions produced during data-dependent LC-MS/MS can significantly improve the confidence of identification of peptides and lower the rates of false and ambiguous identifications [19–22]. More instruments capable of performing LC-MS/MS with high mass resolution and high mass accuracy are becoming available to the research community [23–28].
Meng et al developed a score algorithm based on a Poisson distribution for protein identification in top-down proteomics that incorporates mass accuracy during scoring [29]. The algorithm is implemented in a web based application named ProSight PTM [30]. Some algorithms take advantage of mass accuracy in bottom-up proteomics, however, the full potential of mass accuracy has not been fully exploited. Here we describe a new search program that uses a mass accuracy sensitive probabilistic score algorithm for peptide identification in shotgun proteomics based on a binomial distribution and central limit theorem [31]. This approach is separate and distinct from algorithms that filter matches based on mass accuracy. In the latter high mass accuracy can be used to filter spectra by only searching tandem mass spectra whose precursor ion falls within the stated mass tolerance, and filtering product ions by high mass accuracy can further reduce the likelihood of a random match [19, 22, 32]. However, a mass accuracy sensitive probabilistic score model implicitly takes mass accuracy into account during scoring and high mass accuracy will not only reduce false positives, but also improves the scores of true positive matches. By incorporating mass accuracy in the peptide scores, MassMatrix achieved better sensitivity for high accuracy data sets than Mascot, SEQUEST, X!Tandem, and OMSSA [14]. Furthermore, in MassMatrix high mass accuracy lowers false positive rate and improves confidence in peptide assignment and protein identification. The presence of decoy sequences and additional variable PTMs has limited impact on the database search results of high mass accuracy, which allows for high-throughput unsupervised searches. Comparisons are made between the search results from MassMatrix, Mascot, SEQUEST, X!Tandem, and OMSSA for a data set obtained with an LTQ-Orbitrap mass spectrometer. Furthermore the algorithm was evaluated against the five algorithms mentioned above by the publicly available data set describe by Kapp et al [17].
2 METHODS
2.1 Search Engine
MassMatrix is the name of the software package that implements the new score algorithm described previously [31]. The algorithm was developed with ANSI C++. The software is portable and has been compiled successfully on personal computers and high-performance clusters running Microsoft Windows or Linux operating systems. A parallel version, MPI MassMatrix, based on the message passing interface (MPI) has been developed for use on distributed memory clusters. A web interface to a public form of the algorithm is also available at http://www.massmatrix.net. MassMatrix supports mzDATA format, mzXML format and Mascot generic format (.MGF) as input for tandem MS data. Databases must be formatted as FASTA or converted to the MassMatrix database format (.BAS) for protein database. This manuscript describes the implementation of the algorithm and its performance on real data sets.
MassMatrix contains two independent score models, including a mass accuracy sensitive statistical model and a descriptive model. These models are used to calculate three distinct scores for a peptide match. The two statistical scores, pp and pp2, represent the negative logarithm of the probability that a peptide match is a random occurrence. For example, a pp or pp2 score of 6 indicates that the peptide match is random with a one-out-of-a-million chance. The pp score evaluates a peptide match based on the number of matched product ions in the experimental spectrum and the pp2 score is based on the total abundance of matched product ions in the experimental spectrum. Because each score is distinct, the combination of scores is useful for validating each peptide match. Among these three scores, pp value returned from the statistical model is the primary standard used to assess the quality of peptide matches [31].
MassMatrix searches PTMs and chemical modifications of the peptide sequence. There are two types of modifications included: 1) fixed modifications, and 2) variable modifications. Fixed modifications are those that modify all occurrences of certain amino acid residues in the protein sequences. Fixed modifications do not add complexity to the database because the search space does not increase. Variable modifications are those that may or may not modify the occurrence of certain amino acid residues in the protein sequences. Variable modifications add complexity as there are a great number of permutations of variably modified peptides for each sequence. MassMatrix searches all possible permutations of modified peptides for each peptide sequence. For example, a peptide sequence with three lysine residues and two serine residues will create (2+1)3×(1+1)2 = 108 permutations of unmodified and modified peptides when two variable modifications for lysine and one variable modification for serine are allowed. This paradigm results in high computational expense due to the increased number of theoretically possible modified peptides. The need for such a comprehensive search was born from our extensive analysis of histone proteins which possess this level of complexity in their patterns of PTMs. MassMatrix was specifically designed to be able to solve such large problems.
A flow diagram for the database search is shown in Figure 1. MassMatrix initially digests the protein sequences according to the enzyme or cleavage sites specified by the user. The resulting peptide sequences (and any permutations due to PTMs) are fragmented and then matched against the experimental data. To improve efficiency, it skips redundant peptide sequences. Three scores (score, pp, and pp2 values) are calculated for each potential match by use of the algorithm described previously [31]. After the scores for all potential matches are calculated, matches below the critical thresholds are discarded. Peptide hits are then matched with its corresponding protein sequences. MassMatrix then outputs the results as html files for portability.
Figure 1.

The overall scheme for the MassMatrix search software.
Searches of big data sets against large databases with many variable modifications are computationally expensive. The number of peptides searched can exceed 1010 and the job could require days to complete. MPI MassMatrix was designed for use on distributed memory clusters. The search algorithm lends itself to being an embarrassingly parallel application. Since the search processes for each peptide sequence are fully independent, they can be easily split into sub-jobs that are then distributed to many processors on the cluster. The master node initializes the peptide list to be searched and distributes peptide search as sub-jobs to the slave processors. The master maintains a balanced load among all slaves throughout the job.
2.2 Sample Preparation and Mass Spectrometry
Human histones were isolated from Kasumi-1 cells as described previously [33]. The mixture of core histones was digested by the protease trypsin in 100 mM ammonium bicarbonate buffer. The digested peptides were identified by nano liquid chromatography tandem mass spectrometry (nano-LC-MS/MS). The high mass accuracy data set was obtained on an LTQ-Orbitrap mass spectrometer (ThermoElectron Finnigan, San Jose, CA, USA) by use of data-dependent LC-MS/MS. Ions were fragmented by use of collision induced dissociation in the linear ion trap and mass analyzed by the Orbitrap mass analyzer. The high accuracy data set contains precursor and product ions obtained with the Orbitrap.
2.3 Database Search and Search Parameters
The .RAW data files obtained from the mass spectrometer were converted to .DTA files by use of extract_msn.exe, a windows console application provided by Thermo Electron (ThermoElectron Finnigan, San Jose, CA, USA). Tandem MS spectra with more than five product ions were extracted to .DTA files and then merged into .MGF files by use of a Perl script. Spectra were not grouped based on precursor mass. The data set in .MGF format is available at http://www.massmatrix.net/download/. The data set was then searched by use of MassMatrix against the NCBInr human databases with the following options: i) Modifications: variable acetylation of lysine, variable acetylation of N-terminus; ii) Enzyme: trypsin; iii) Missed Cleavages: 3; iv) Peptide Length: 4 to 30 amino acid residues; v) Precursor Ion Charge: 1+, 2+, 3+; and VI) A mass tolerance of 0.02 Da and 0.01 Da for the precursor and product ions respectively. The same data set was also evaluated by Mascot, SEQUEST (SEQUST v.28 on BioWorks 3.3), X!Tandem, and OMSSA. The search parameters in Mascot, SEQUEST, X!Tandem, and OMSSA were identical to those in MassMatrix where appropriate. Critical values for scores in the three programs were set as follows: pp or pp2 value > 6 in MassMatrix; score > 30 in Mascot; XCorr > 1.5 for +1 peptides, 2.0 for 2+ peptides and 2.5 for 3+ peptides in SEQUEST; expectation value < 0.1 in X!Tandem; and e-value < 0.1 in OMSSA. If multiple peptide matches were found for a given spectrum only the match with the highest score was considered. In order to improve the performance of SEQUEST, the protein database was indexed prior to database searches.
The true positives and false positives for the three algorithms were determined by searches against a human database containing reversed decoy sequence as described by Elias [34]. The total number of false positive peptide matches was calculated by multiplying the number of peptide matches to reversed sequences by two. The number of true positives was then calculated by subtracting total number of false positives from the total number of peptide matches in the forward and reversed databases [34].
2.4 Comparisons with Other Algorithms
In order to test the consensus between MassMatrix and five publicly available search algorithms (Mascot [9], SEQUEST [2], Sonar [3], Spectrum Mill, and X!Tandem [18]), a data set provided by Kapp et al [17] was searched by MassMatrix against the Human International Protein Index database (IPI, version 3.19 July 2003, 60397 entries, European Bioinformatics Institute) [17, 35] with the following options: i) Precursor ion tolerance: 3.0 Da; ii) Product ion tolerance: 0.8 Da iii) No fixed or variable modifications; iv) Enzyme: trypsin; v) Missed Cleavages: 2; vi) Peptide Length: 4 to 40 amino acid residues; vii) Precursor Ion Charge: 1+; 2+, 3+; and viii) Maximum Product Ion Charge: 2+. The result was compared with those reported by Kapp et al [17]. The search parameters in MassMatrix were set according to the counterpart parameters that Kapp et al. used for the other five search programs. Mascot, SEQUEST, X!Tandem and Sonar Searches were performed against the Human International Protein Index database with the following parameters: trypsin-constrained (two missed cleavages); 3.0 Da precursor ion tolerance and 0.5 Da fragment ion tolerance, and ESIIT selected as instrument setting. Searches in Spectrum Mill used the same parameters except that the precursor mass tolerance is 2.5 Da and the fragmentation ion tolerance is 0.7 Da. The search parameters in the five algorithms reported by Kapp et al were set to maximize and optimize the performances of the search algorithms [17].
3 RESULTS AND DISCUSSION
3.1 Consensus with Other Search Algorithms
The reliability and consensus of MassMatrix results compared to those of other algorithms was determined by searching the publicly available data set evaluated by Kapp et al [17]. This data set contains 3952 tandem MS spectra obtained from an LCQ Deca XP ion trap mass spectrometer (Thermo-Finnigan, San Jose, CA, USA) [17]. The search parameters were set according to those described previously. Spectra that were not determined to be singly charged were extracted as both doubly and triply charged resulting in 5806 spectra searched [17]. The MassMatrix search was performed at the critical values of pp value = 6 and pp2 value = 8. 826 spectra were identified by MassMatrix with significant peptide matches. The Venn Diagram for peptide matches of the LCQ data set from MassMatrix, Mascot and SEQUEST are shown in Figure 2.
Figure 2.
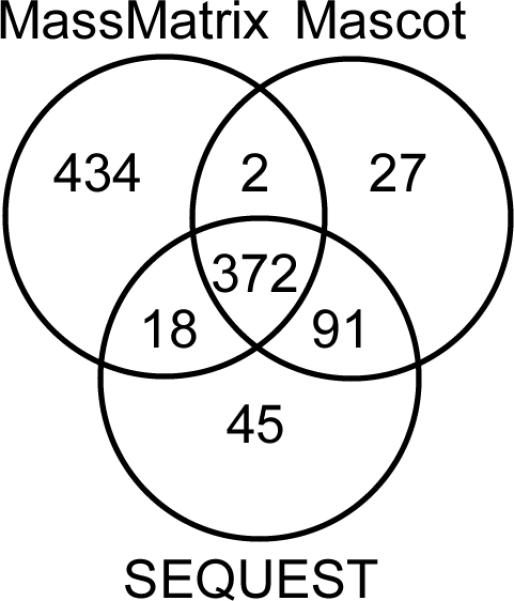
A Venn Diagram showing consensus of peptide matches from MassMatrix, Mascot, and SEQEUST for the LCQ data set.
The complete lists of all “first pass” peptide matches from the five searches in Mascot, SEQUEST, Sonar, Spectrum Mill, and X!Tandem by Kapp et al [17] were downloaded from http://www.ludwig.edu.au/archive/. In Kapp's report, 662 out of 3952 spectra were identified with “first pass” matches. Out of these 662 “first pass” matches, the five algorithms achieved consensus on 349 spectral matches. When factoring in the results from MassMatrix, the consensus peptide lists from all six algorithms drops to 345. The 4 spectra that were different had low signal to noise ratio, and did not appear to be good matches based on the protocol of manual validation described by Tabb et al [36].
The consensus results between MassMatrix and each search algorithm were individually compared. MassMatrix achieved high levels of consensus with each algorithm as follows: a) 498 spectra out of all 660 Mascot “first pass” matches; b) 519 out of 662 SEQUEST matches; c) 453 out of 646 Sonar matches; d) 428 out of 544 Spectrum Mill matches; and e) 434 out of 557 X!Tandem matches. For the 660 “first pass” peptide matches from Mascot, 492 were verified by Kapp and et al. as correct identifications [17]. Most of these correct ID (488) were captured by MassMatrix. The other 4 were manually checked and they all have very low signal to noise ratio and low scores. MassMatrix also returned many peptide matches there were not caught by any of the five other algorithms. This might be due to the fact that the later version of IPI human database was used in the MassMatrix search and the majority of the additional peptides found by MassMatrix were of low scores and less than 6 amino acid residues in length, which were too small to be returned by other algorithms.
3.2 Comparison of High Accuracy Search Results
The mass accuracy sensitive score model in MassMatrix was evaluated against a high mass accuracy data set and the results were compared with those obtained by the commercial search engines Mascot, SEQUEST, X!Tandem, and OMSSA [2, 9, 14, 18]. Experimental MS/MS data for a tryptic digest of a protein mixture containing histones were obtained on a ThermoElectron Corp LTQ-Orbitrap mass spectrometer. Histones were chosen as they represent a unique challenge to search engines due to the large number of PTMs [33]. A primary goal in developing MassMatrix was to be able to obtain meaningful results from data that contain a large number of PTMs. Precursor and product ions were mass analyzed in the Orbitrap to achieve high mass accuracy (< 5ppm).
Figure 3 displays the Venn Diagrams for peptide matches from MassMatrix, Mascot, SEQUEST, X!Tandem, and OMSSA. 197 of 1837 spectra were scored as potential peptide matches in MassMatrix. Mascot returned 104 peptide matches where 98 were found by MassMatrix; SEQUEST returned 86 peptide matches of which 77 were found by MassMatrix; X!Tandem returned 125 peptide matches where 110 were found by MassMatrix; OMSSA returned 144 peptide matches of which 106 were found by MassMatrix. The complete lists of peptide matches from the five algorithms for the high mass accuracy data set are listed in Supplementary Tables 1–5.
Figure 3.

A Venn Diagram showing consensus of peptide matches from MassMatrix, Mascot, SEQEUST, X!Tandem, and OMSSA for the Orbitrap data set.
Further comparison between MassMatrix, Mascot, SEQUEST, X!Tandem, and OMSSA was made by use of receiver operating characteristic (ROC) analysis [14, 37, 38]. The number of true positives and false positives in the ROC curves was determined by searches with the presence of the reversed human database in addition to the human database. The ROC analysis of the data set was performed with and without allowing the modifications of acetylation of K and acetylation of N-terminus. Because the majority of correctly identified peptide matches are unmodified, ROC curves of the data set for the five programs without allowed modifications were very similar to those with allowed modifications. The result from ROC analysis for the five programs with allowing modifications of acetylation of K and acetylation of N-terminus is discussed in details below (Figure 4).
Figure 4.
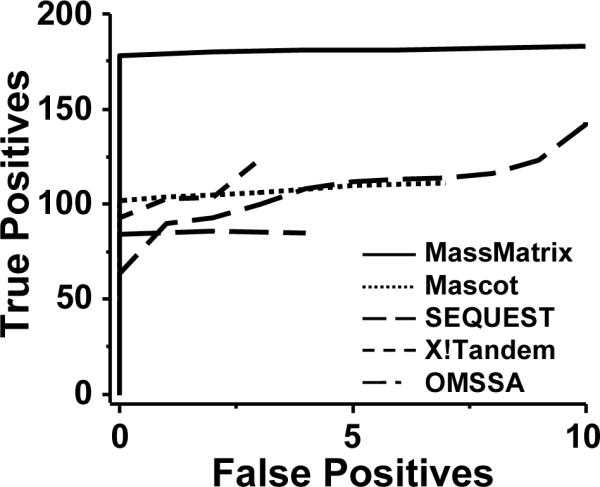
ROC curves of MassMatrix, Mascot, SEQEUST, X!Tandem, and OMSSA for the Orbitrap data set.
Among the peptides with significant scores returned by MassMatrix, the relative number of false positives was as low as 2%. Furthermore all true positives corresponded to a unique peptide/spectrum match in MassMatrix. MassMatrix achieved better sensitivity than Mascot, SEQUEST, X!Tandem, and OMSSA for a given specificity when analyzing high mass accuracy data obtained on the Orbitrap. This result is due to the mass accuracy sensitive score model that achieves better separation between the distributions of pp values for the true positives and false positives [31]. A more detailed discussion of this effect is present in the next section.
3.3 Effect of High Mass Accuracy in MassMatrix
The statistical model in MassMatrix does not provide any benefit or penalty for mass error. Rather the model relies on the occurrence of the mass within the give mass tolerance window. Therefore we were able to simulate the effect of mass spectrometer accuracy on the pp values by altering the mass tolerance used to search the high mass accuracy data set. This approach eliminates any possible factors that may arise due to the use of data sets obtained on different instruments. The distributions of pp values for true and false positives at simulated mass accuracy of 1.0 Da, 0.1 Da and 0.01 Da are shown in Figure 5. Because the pp values implicitly include mass accuracy in the statistical model, their distribution for true matches improves with the mass accuracy and search tolerance. For the high mass accuracy data set, we observed an increase of 10 in the pp values of true positives for every 10 fold increase in mass accuracy. Thus MassMatrix improves the scores for true matches as the mass accuracy increases. Because the pp value is the negative logarithm of the probability that a match is random, the overall statistical confidence in peptide assignment was significantly improved. Additionally the number of reported false positives substantially decreased as mass accuracy increased. In contrast to true matches, the pp scores for false positives decreased at higher accuracy and thus fewer false positives exist above the critical threshold and were reported by MassMatrix. Thus raising mass accuracy has an immediate effect of reducing false positives and improving peptide scores for true positives.
Figure 5.

Distributions of pp values for (a) true positives and (b) false positives at different mass accuracies: 1.0 Da, 0.1 Da and 0.01 Da.
3.4 Distraction from Additional Modifications
Including additional modifications also increases the number of peptides searched and result in a greater likelihood for peptide false positives. The high mass accuracy data set was also searched against the human database with additional modifications of phosphorylation of serine, threonine and tyrosine. The resulting number of peptides searched increased from 6.1 million to 665.7 million. The additional modification imposed no negative effect on the search results of high accuracy data in MassMatrix.
3.5 Search Speed
The search time in MassMatrix is proportional to the number of theoretical peptides searched. Proteins containing redundant sequences in the databases do not increase the search time because redundant peptide sequences are skipped. The search time also increases with the number of experimental tandem MS spectra. However, this increase is not linear (nearly logarithmic) as all tandem MS spectra are stored in a sorted table and recalled by use of a binary search algorithm. For the search of Orbitrap data set, MassMatrix and SEQUEST took 61 and 255 sec respectively on a single AMD 3200+ (2.2GHz) PC. Mascot spent 240 sec on a dual-Intel Xeon server (2.8 GHz × 2). X!Tandem and OMSSA took 73 and 80 sec respectively on an Intel quad core PC (2.4 GHz). Despite the difference in architecture the search results show that MassMatrix performs as well as or better than Mascot, SEQUEST, X!Tandem, and OMSSA with regard to search time. On a modern PC, MassMatrix was able to search >100,000 tandem MS spectra per hour against the human protein database with 2 variable modifications or >100,000 peptides per second against a data set containing 1837 tandem MS spectra.
The scalability of MPI MassMatrix was determined by searching the high mass accuracy data set against a human database with 8 variable modifications. In this example the number of peptides searched exceeded 4 × 108. The search was performed on a Linux Itnium2 cluster (900 MHz processors). As shown in Figure 6, the search speed of MPI MassMatrix was nearly proportional to the number of processors used. The speedup showed obvious nonlinearity when more than 40 processors were used. This nonlinear scaling was due to the high communication overhead between the master and slave nodes. The non-ideal scaling was exacerbated by searching a narrow mass tolerance that in turn reduced the number of spectral comparisons on each slave and increased the rate of request between slave and master nodes. Load balancing algorithms are currently being examined to improve the scaling on clusters that exceed 20 processors. The parallel version of MassMatrix achieved a maximum speed of 94,474 tandem MS spectra per hour against the human protein database with 8 PTMs or 5,959,229 peptides per second against a data set containing 1837 tandem MS spectra while running on 80 processors with a peak performance of 72 Gflops.
Figure 6.
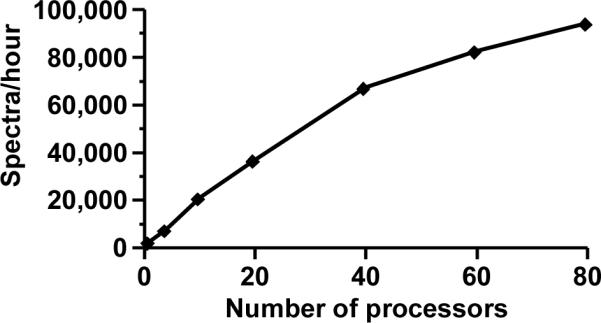
Parallel job performance of the MPI version of MassMatrix when searching a data set containing 1837 tandem MS spectra against a complete human database with 8 variable modifications. The jobs were performed on a 900 MHz Itanium cluster.
4 CONCLUDING REMARKS
A new tandem MS database search program, MassMatrix, employs a mass accuracy sensitive score algorithm. The mass accuracy sensitive score algorithm gives rise to higher sensitivity and specificity for searches at high mass accuracy. The high mass accuracy also improves the pp values for true matches, reduces the pp value for random matches and results in improved confidence in peptide identification. The program was first tested and compared with other five search algorithms by use of a data set that was validated and published by Kapp and et al. It was shown that MassMatrix has consensus with other publicly available programs for searches at low mass accuracy and it returned most of peptide matches from other programs that were validated as correct identification by Kapp et al [17]. The peptide matches that MassMatrix missed were then manually checked and found to be of low scores and quality. The program was also tested and compared with Mascot, SEQUEST, X!Tandem, and OMSSA by use of a high mass accuracy data set. The ROC analysis shows that MassMatrix has a higher sensitivity than Mascot, SEQUEST, X!Tandem, and OMSSA for the high mass accuracy data. The relative number of false positives for our high accuracy data set was 2% and each spectrum corresponds to a validated unique peptide match. The presence of decoy sequences and additional modifications did not significantly alter the search results. The high confidence achieved when searching high mass accuracy data reduces the requirement for manual validation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Chen Ren (OSU, Department of Chemistry), Josh Ellis (OSU, Campus Chemical Instrument Center), Kari B. Green-Church (OSU, Campus Chemical Instrument Center), Vlad Zabrouskov (ThermoElectron Finnigan) and Eugene A. Kapp & Frédéric Schütz (Joint Proteomics Laboratory, Ludwig Institute for Cancer Research - Parkville, Victoria, Australia) for providing data sets and access to resources used in this study. The study was funded by the Ohio State University, the National Institutes of Health CA107106, and the V Foundation/American Association for Cancer Research Translational Cancer Research Grant and the Leukemia & Lymphoma Society.
Abbreviations
- MPI
message passing interface
- ECD
electron capture dissociation
- ROC
receiver operating characteristic
5 REFERENCES
- [1].Sadygov RG, Cociorva DC, Yates JR. Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nature Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- [2].Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- [3].Field HI, Fenyö D, Beavis RC. RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimizes protein identification, and archives data in a relational database. Proteomics. 2002;2:36–47. [PubMed] [Google Scholar]
- [4].Hansen BT, Jones JA, Mason DE, et al. SALSA: a pattern recognition algorithm to detect electrophile-adducted peptides by automated evaluation of CID spectra in LC-MS-MS analyses. Anal. Chem. 2001;73:1676–1683. doi: 10.1021/ac001172h. [DOI] [PubMed] [Google Scholar]
- [5].Mann M, Wilm M. Error tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- [6].Bafna V, Edwards N. SCOPE: a probabilistic model for scoring tandem mass spectra against a peptide database. Bioinformatics. 2001;17:S13–S21. doi: 10.1093/bioinformatics/17.suppl_1.s13. [DOI] [PubMed] [Google Scholar]
- [7].Colinge J, Masselot A, Giron M, et al. OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics. 2003;3:1454–1463. doi: 10.1002/pmic.200300485. [DOI] [PubMed] [Google Scholar]
- [8].Zhang N, Aebersold R, Schwikowshi B. ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics. 2002;2:1406–1412. doi: 10.1002/1615-9861(200210)2:10<1406::AID-PROT1406>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- [9].Perkins DN, Pappin DJC, Creasy DM, et al. Probability-based protein identification by searching sequence database using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [10].MacCoss MJ, Wu CC, Yates JR. Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal. Chem. 2002;74:5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- [11].Havilio M, Haddad Y, Smilansky Z. Intensity-based statistical scorer for tandem mass spectrometry. Anal. Chem. 2003;75:435–444. doi: 10.1021/ac0258913. [DOI] [PubMed] [Google Scholar]
- [12].Sadygov RG, Yates JR. A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal. Chem. 2003;75:3792–3798. doi: 10.1021/ac034157w. [DOI] [PubMed] [Google Scholar]
- [13].Sadygov RG, Liu H, Yates JR. Statistical models for protein validation using tandem mass spectral data and protein amino acid sequence databases. Anal. Chem. 2004;76:1664–1671. doi: 10.1021/ac035112y. [DOI] [PubMed] [Google Scholar]
- [14].Geer LY, Markey SP, Kowalak JA, et al. Open mass spectrometry search algorithm. J. Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- [15].Sadygov R, Wohlschlegel J, Park SK, et al. Central limit theorem as an approximation for intensity-based scoring function. Anal. Chem. 2006;78:89–95. doi: 10.1021/ac051206r. [DOI] [PubMed] [Google Scholar]
- [16].Keller A, Nesvizhskii A, Kolker E, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- [17].Kapp EA, Schütz F, Connolly LM, et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5:3475–3490. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- [18].Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J. Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- [19].Olsen JV, de Godoy LMF, Li G, et al. Parts per million mass accuracy on an Or-bitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- [20].Steen H, Mann M. The abs's (and xyz's) of peptide sequencing. Mol. Cell. Proteomics. 2004;5:699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- [21].Fernández FM, Smith LL, Kuppannan K, et al. Peptide sequencing using a patchwork approach and surface-induced dissociation in sector-TOF and dual quadrupole mass spectrometers. J. Am. Soc. MassSpectrom. 2003;14:1387–1401. doi: 10.1016/j.jasms.2003.09.003. [DOI] [PubMed] [Google Scholar]
- [22].Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (±10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal. Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- [23].Makarov A, Denisov E, Kholomeev A, et al. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006;78:2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- [24].Makarov A, Denisov E, Lange O, et al. Dynamic range of mass accuracy in LTQ Or-bitrap hybrid mass spectrometer. J. Am. Soc. Mass Spectrom. 2006;17:977–982. doi: 10.1016/j.jasms.2006.03.006. [DOI] [PubMed] [Google Scholar]
- [25].Yates JR, Cociorva D, Liao L, et al. Performance of a linear ion trap-Orbitrap hybrid for peptide analysis. Anal. Chem. 2006;78:493–500. doi: 10.1021/ac0514624. [DOI] [PubMed] [Google Scholar]
- [26].Everley PA, Bakalarski CE, Elias JE, et al. Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. J. Proteome Res. 2006;5:1224–1231. doi: 10.1021/pr0504891. [DOI] [PubMed] [Google Scholar]
- [27].Haas W, Faherty BK, Gerber SA, et al. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell. Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- [28].Nielsen ML, Bennett KL, Larsen B, et al. Peptide end sequencing by orthogonal MALDI tandem mass spectrometry. J. Proteome Res. 2002;1:63–77. doi: 10.1021/pr0155174. [DOI] [PubMed] [Google Scholar]
- [29].Meng F, Cargile BJ, Miller LM, et al. Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat. Biotechnol. 2001;19:952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- [30].LeDuc RD, Taylor GK, Kim YB, et al. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004;32:W340–W345. doi: 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu H, Freitas AF. A high mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jensen ON, Podtelejnikov A, Mann M. Delayed extraction improves specificity in database searches by matrix-assisted laser desorption/ionization peptide maps. Rapid Commun. Mass Spectrom. 1996;10:1371–1378. doi: 10.1002/(SICI)1097-0231(199608)10:11<1371::AID-RCM682>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- [33].Ren C, Zhang L, Freitas MA, et al. Peptide mass mapping of acetylated Isoforms of histone H4 from mouse lymphosarcoma cells treated with histone deacetylase (HDACs) inhibitors. J. Am. Soc. Mass Spectrom. 2005;16:1641–1653. doi: 10.1016/j.jasms.2005.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- [35].Kersey PJ, Duarte J, Williams A, et al. The international protein index: an integrated database for proteomics experiments. Proteomics. 2004;4 doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- [36].Tabb DL, Friedman DB, Ham AL. Verification of automated peptide identifications from proteomic tandem mass spectra. Nat. Protocols. 2006;1:2213–2222. doi: 10.1038/nprot.2006.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Baker SG. The central role of receiver operating characteristic (ROC) curves in evaluating tests for the early detection of cancer. J. Natl. Cancer Inst. 2003;95:511–515. doi: 10.1093/jnci/95.7.511. [DOI] [PubMed] [Google Scholar]
- [38].Tabb DL, Saraf A, Yates JR. GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal. Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.