

Abstract
Purpose
Elevated phospholipase D (PLD) activity provides a survival signal in several human cancer cell lines and suppresses apoptosis when cells are subjected to the stress of serum withdrawal. Thus, targeting PLD survival signals has potential to suppress survival in cancer cells that depend on PLD for survival. Honokiol is a compound that suppresses tumor growth in mouse models. The purpose of this study was to investigate the effect of honokiol on PLD survival signals and the Ras dependence of these signals.
Experimental Design
The effect of honokiol upon PLD activity was examined in human cancer cell lines where PLD activity provides a survival signal. The dependence of PLD survival signals on Ras was investigated, as was the effect of honokiol on Ras activation.
Results
We report here that honokiol suppresses PLD activity in human cancer cells where PLD has been shown to suppress apoptosis. PLD activity is commonly elevated in response to the stress of serum withdrawal, and, importantly, the stress-induced increase in PLD activity is selectively suppressed by honokiol. The stress-induced increase in PLD activity was accompanied by increased Ras activation, and the stress-induced increase in PLD activity in MDA-MB-231 breast cancer cells was dependent on a Ras. The PLD activity was also dependent on the GTPases RalA and ADP ribosylation factor. Importantly, honokiol suppressed Ras activation.
Conclusion
The data provided here indicate that honokiol may be a valuable therapeutic reagent for targeting a large number of human cancers that depend on Ras and PLD for their survival.
Phospholipase D (PLD), which catalyzes the hydrolysis of phosphatidylcholine to phosphatidic acid and choline, has been implicated in cellular signals that suppress apoptosis and contribute to the survival of cancer cells (1, 2). Elevated PLD activity leads to the elevated expression of Myc (3) and stimulates the activation of mammalian target of rapamycin (mTOR; refs. 4, 5), which has been implicated in survival signals in a wide variety of human cancers (6). Elevated PLD activity also suppresses the tumor suppressors p53 (7) and protein phosphatase 2A (8). Thus, there is a growing body of evidence implicating PLD in survival signaling—in that many of the downstream targets of PLD have been implicated in the suppression of apoptosis and the survival of cancer cells.
Consistent with the ability of PLD to stimulate signals implicated in cancer cell survival, elevated PLD activity has been observed in several human cancers, including breast, kidney, colon, and gastric cancer (2). In addition, elevated PLD activity has been observed in several human cancer cell lines, including those from breast, lung, pancreatic, prostate, and kidney cancers (9–13). More importantly, suppression of PLD activity in cancer cells with elevated PLD activity resulted in apoptosis (9, 10, 13–15). Thus, a substantial body of evidence indicates that elevated PLD activity stimulates survival signals in many human cancers.
Activated Ras proteins stimulate increases in PLD activity (16). The PLD1 isoform is constitutively associated with RalA (16, 17), a downstream target of Ras signaling (1). Many of the cancer cell lines with elevated PLD activity have activating mutations in Ras (12).5 Thus, a critical target of Ras signaling in human cancers with activating mutations could be PLD. Consistent with this hypothesis, Counter and colleagues (18) reported that the most critical target of Ras in the transformation of human cells was guanine nucleotide exchange factor for RalA, further implicating PLD in Ras-induced tumorigenesis. Thus, it is possible that the survival of cancer cells with activating mutations to Ras genes may depend on PLD and the downstream targets of PLD signaling that includes mTOR (5). These studies suggest that suppression of the Ras signals that target RalA could suppress the survival signals mediated by PLD.
The therapeutic targeting of survival signals in cancer cells has obvious appeal because, in principle, suppression of survival signals will result in apoptosis. Honokiol is a natural product isolated from an extract of seed cones from Magnolia grandiflora with antimicrobial activity (19). Honokiol has more recently been found to have antiangiogenic properties and blocked tumor growth in mouse xenografts (20). Honokiol was reported to induce caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells (21), and to inhibit the bone metastatic growth of human prostate cancer cells (22). In this report, we have evaluated the effect of honokiol on PLD activity in human cancer cells with elevated PLD activity. We report here that honokiol suppresses Ras activation and potently suppresses Ras-dependent PLD activity induced by the stress of serum withdrawal in cancer cells and induces apoptosis.
Materials and Methods
Cells and cell culture conditions
All human cancer cell lines used in this study were obtained from the American Type Culture Collection and were maintained in DMEM with 10% bovine calf serum (Sigma). Cos1 cells were maintained in DMEM and 5% fetal bovine serum as described previously (23).
Materials
[3H]Myristic acid was obtained from New England Nuclear. Precoated silica 60A TLC plates were from Whatman. Honokiol was purified according to the method of Amblard et al. (24). Antibodies against HIF2α and hemagglutinin were obtained from Santa Cruz Biotechnology. Antibodies raised against poly-(ADP-ribose) polymerase (PARP), actin, ribosomal subunit S6 kinase (S6K), phosphorylated-S6K, eukaryotic initiation factor 4E binding protein 1 (4E-BP1), and phosphorylated-4EBP1 were purchased from Cell Signaling Technologies. Phosphatidylethanolamine, phosphatidylcholine, and phosphatidylinositol-4,5-bis-phosphate were purchased from Avanti Polar Lipids. [Methyl-3H]phosphatidylcholine was purchased from Perkin-Elmer Life Sciences.
Plasmid vectors
Vectors used were pcDNA3.1(-) (Invitrogen) and pcDNA3.1(-)-S17NRas, which was constructed by inserting the S17N Ras gene from pCMV-S17NRas (Clonetech) using flanking EcoR1 and BamH1 sites. They were constructed by PCR amplification of the corresponding cDNAs and cloned into the EcoRI site of pcDNA3.1 (-) (Invitrogen). The ADP ribosylation factor (ARF) vectors pcDNA3.1-ARF1T31N and pcDNA3.1-ARF6T27N were described previously (25). The generation of the pCGN vectors expressing hemagglutinin-tagged Sos and Ras were described previously (26). The S28N RalA mutant cloned into pcDNA3.1(-) was described previously (27).
Western blot analysis
Extraction of proteins from cultured cells and Western blot analysis of extracted proteins was done using the enhanced chemiluminescence system (Amersham) as described previously (9).
Ras activation assays
Cells were harvested and lysates were prepared and treated with the Ras-binding domain of Raf1 (Pierce) according to the vendor's instructions. Ras-GTP bound to the Raf1-binding domain was recovered and subjected to Western blot analysis using a Pan-Ras antibody supplied with the Ras activation assay kit. For the Cos1 cell assays, a hemagglutinin antibody was used to bind the ectopically expressed Ras and Sos proteins.
Cell viability and apoptosis
Cell viability was determined by trypan blue exclusion. After various treatments, cells were harvested, washed, and treated with trypan blue at a concentration of 0.4% w/v. After 10 min, trypan blue uptake (dead cells) was determined by counting on a hemocytometer. Apoptosis was evaluated by examination of cleavage of the caspase-3 substrate PARP as described previously (9).
Phospholipase D assays
For in vivo PLD activity, cells were plated in 60-mm culture dishes in DMEM with 10% serum at a density that would allow them to grow to ∼80% confluence after 2 d; then, cells were shifted to DMEM containing 0.5% or 10% serum, as indicated in the figure legends, overnight. Cells were then prelabeled for 4 h with [3H]myristate (3 μCi, 40 Ci/mmol) in 3 mL of medium. PLD catalyzed transphosphatidylation in the presence of 0.8% 1-BtOH, and the extraction and characterization of lipids by TLC were done as described previously (28). For in vitro PLD activity, assays were done with exogenous substrate as described previously (29). Briefly, PLD activity was measured by the release of [methyl-3H]choline from [choline-methyl-3H]dipalmitoyl-phosphatidylcholine. PLD (1-10 nmol/L) was reconstituted with phospholipid vesicle substrates composed of 10 μmol/L dipalmitoyl-phosphatidylcholine, 100 μmol/L phosphati-dylethanolamine, 6.2 μmol/L phosphatidylinositol-4,5-bisphosphate, and 1.4 μmol/L cholesterol. Assays were conducted for 30 min at 37°C in 50 mmol/L HEPES (pH 7.5), 80 mmol/L KCl, 3 mmol/L EGTA, 0.1 mmol/L DTT, 3.6 mmol/L MgCl2, 3.6 mmol/L CaCl2, and 10 μmol/L GTPγS. Reactions were stopped by the addition of trichloroacetic acid and bovine serum albumin. Free [methyl-3H]choline was separated from precipitated lipids and proteins by centrifugation and was analyzed by liquid scintillation counting. The counts present in the absence of added enzyme (i.e., background) were subtracted from other samples. ADP-ribosylation factor 1 (Arf1), PLD1, and PLD2 used in these assays were prepared as described previously (29).
Results
Stress-induced PLD activity in MDA-MB-231 cells is dependent on Ras
We previously reported that serum withdrawal led to increased PLD activity in MDA-MB-231 cells (10). As shown in Fig. 1A, the increased PLD activity was not due to any changes in the levels of the PLD isoforms PLD1 and PLD2. This suggested that the increased PLD activity observed in response to the stress of serum withdrawal was due to increased activity, not increased protein levels. The regulation of PLD activity has been reported previously to involve a GTPase cascade of Ras and RalA (1). RalA, which constitutively associates with PLD1 (17), recruits ARF GTPases into PLD complexes (27, 30), where ARF actually stimulates the PLD activity of PLD1 (31). We therefore examined the effect of dominant negative mutants for H-Ras, RalA, and ARF1 and ARF6, on the PLD activity in serum-deprived MDA-MB-231 cells. As shown in Fig. 1B, dominant negative mutants for H-Ras, RalA, and ARF all suppressed the stress-induced PLD activity in the MDA-MB-231 cells, indicating that Ras-RalA-PLD1 pathway (1) is activated. We next examined the effect of serum withdrawal on Ras activation in the MDA-MB-231 cells. As shown in Fig. 1C, serum withdrawal led to an increase in GTP-bound Ras, while having no effect on total Ras levels. This effect could be detected within 1 hour (Fig. 1C). Because serum growth factors can also induce Ras activation, the surprising increase in Ras GTP binding observed on serum withdrawal may actually be more significant than what is observed. The data in Fig. 1 reveal that the increased PLD activity observed in response to the stress of serum withdrawal is dependent on both Ras and RalA, and that there is a rapid increase in Ras activation in MDA-MB-231 cells subjected to the stress of serum withdrawal.
Fig. 1.
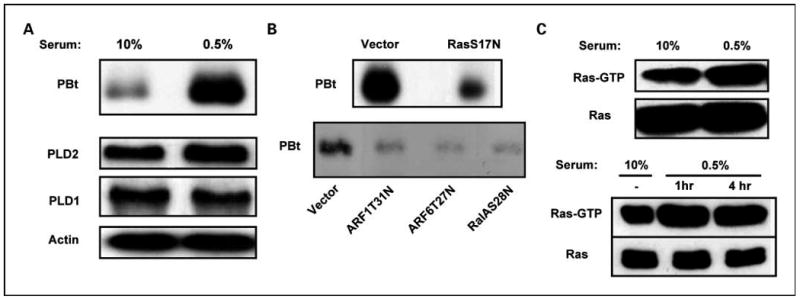
Stress-induced PLD activity in MDA-MB-231 cells is dependent on Ras and RalA. A, MDA-MB-231 cells were plated; 24 h later, the cells were placed in fresh medium containing either 10% or 0.5% serum. Eighteen hours later, [3H]myristate was added; 4 h later, BtOH (0.8%) was added for 20 min, at which time the cells were harvested and the extracted membrane lipids were separated by TLC to determine the levels of the transphosphatidylation product phosphatidyl-BtOH (PBt). The levels of PLD1, PLD2, and actin were determined using Western blot analysis using the corresponding antibodies. B, MDA-MB-231 cells were plated and then transiently transfected with an empty vector control or vectors that express either an S17N Ras, aT31N ARF1, aT27N ARF6, or an S28N RalA mutant 24 h later. The cells were placed in medium containing 0.5% serum 24 h later, and the PLD activity was determined as in A after an additional 24 h. C, MDA-MB-231 cells were plated as in A and placed in either 10% or 0.5% serum 24 h later. At this point, the cells were harvested and lysates were prepared and treated with the Ras-binding domain of Raf1 (Pierce) according to the vendor's instruction. Ras-GTP bound to the Raf1-binding domain was recovered and subjected to Western blot analysis using a Pan-Ras antibody supplied with the Ras activation assay kit. One milligram of protein from whole-cell lysates was used for the Ras pull-down assay, and 20 μg of whole cell lysates were loaded onto the gel used for the Western blot. Each experiment was repeated at least twice with equivalent results.
Honokiol suppresses stress-induced PLD activity in MDA-MB-231 cells
Honokiol (Fig. 2A) was recently reported to suppress the growth of MDA-MB-231 human breast cancer cells in a mouse xenograft tumorigenesis assay (32). We reported previously that these cells depend on PLD activity for their survival and their ability to migrate in culture (9, 10, 14). We therefore examined whether honokiol had any effect on the highly elevated PLD activity observed in MDA-MB-231 cells subjected to serum withdrawal. MDA-MB-231 cells were placed in medium containing either 10% or 0.5% serum for 18 hours, at which time the PLD activity was evaluated in the presence and absence of honokiol for 4 hours. As shown in Fig. 2B, honokiol completely suppressed PLD activity in the cells in 0.5% serum while having little effect on the basal PLD activity in the cells maintained in 10% serum. These data indicate that honokiol specifically suppresses the PLD activity elevated in response to the stress of serum withdrawal while having minimal effect on the basal PLD activity in the MDA-MB-231 cells.
Fig. 2.
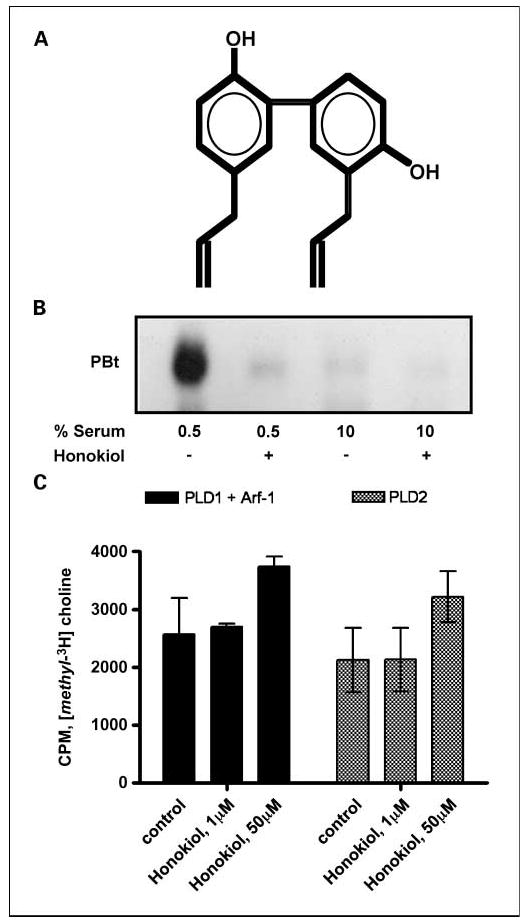
Honokiol suppresses stress-induced PLD activity in MDA-MB-231 human cancer cells. A, the structure of honokiol. B, MDA-MB-231 cells were plated as in Fig. 1 and then shifted to either 10% serum or 0.5% serum overnight as indicated. Honokiol (20 μmol/L) or control ethanol was then added for 4 h as indicated, at which time the cells were harvested and the levels of phosphatidyl-BtOH were determined as in Fig. 1. The experiment shown is representative of at least three independent experiments. C, the effect of honokiol on the activity of PLD isoforms was measured using exogenous substrate assay as described previously (29). Partially purified proteins were incubated in the presence or absence of the indicated concentrations of honokiol for 30 min at 37°C. ARF-1 was included in the PLD1 assay because there is no activity without an Arf protein. The amount of background was determined and subtracted using a bovine serum albumin standard. Error bars, SD for triplicate values.
Because honokiol suppressed the PLD activity elevated in response to the stress of serum withdrawal, we examined the effect of honokiol on in vitro PLD activity using purified recombinant PLD1 and PLD2 protein. As shown in Fig. 2C, honokiol had no significant effect on the activity of either PLD1 or PLD2. ARF-1 is required for the in vitro activity of PLD1 and was included in the reaction with PLD1. Thus, the effect of honokiol is likely upstream of either PLD1 or PLD2 in the MDA-MB-231 cells and targets a regulatory mechanism for activating PLD in response to the stress of serum withdrawal.
Honokiol suppresses Ras activation
As indicated in Fig. 1, the PLD activity in MDA-MB-231 cells is dependent on Ras and RalA. Because honokiol suppressed the PLD activity in the MDA-MB-231 cells and this PLD activity was dependent on Ras, we examined the effect of honokiol on Ras activation in the MDA-MB-231 cells. We used a pull-down assay that uses the Ras-binding domain of Raf1, which recognizes GTP-bound Ras. As shown in Fig. 3A, honokiol suppressed the level of GTP-bound Ras. The effect honokiol on Ras activation was small but reproducible. This is likely due to the activated K-Ras present in these cells (33, 34), which would give a high background of Ras GTP binding. MDA-MB-231 cells express high levels of the epidermal growth factor (EGF) receptor (35) and EGF stimulates both Ras activation and increased PLD activity (28, 36). We therefore examined the effect of honokiol on Ras activation in response to EGF in the MDA-MB-231 cells. As shown in Fig. 3B, honokiol also suppressed the Ras activation induced by EGF in the MDA-MB-231 cells.
Fig. 3.

Honokiol suppresses Ras activation. A, MDA-MB-231 cells were plated and then placed in medium containing 0.5% medium 24 h later. Twenty-four hours later, the cells were treated with either DMSO or honokiol (Hon; 20 μmol/L) as indicated for 2 h. At this point, the cells were harvested and lysates were prepared and treated with the immobilized Ras-binding domain of Raf1 as in Fig. 1. Ras-GTP bound to the Raf1-binding domain was recovered and subjected to Western blot analysis using the Pan-Ras antibody. Total Ras in the cell lysates and actin levels were also examined by Western blot analysis. B, MDA-MB-231 cells were plated and 24 h later were shifted to medium containing 0.5% serum. The cells were then treated with EGF (200 ng/mL) for 10 min. The cells were also treated with honokiol (20 μmol/L) before the addition of EGF for the times indicated. The levels of GTP-bound Ras, total Ras, and actin were then determined as in A. C, Cos1 cells were transfected with plasmids expressing hemagglutinin-tagged Ras (50 ng) and Sos (500 ng) for 24 h. The cells were placed in serum-free medium overnight in the presence of increasing concentration of honokiol. Cells were then collected and Ras activation was determined using the pull-down assay as above. Levels of Ras-GTP, Sos, and Ras were determined by Western blot analysis using an antibody that recognized hemagglutinin. D, Cos1 cells were transfected with the plasmid expressing hemagglutinin-tagged Ras (50 ng) for 24 h. The cells were placed in serum-free medium overnight in the presence and absence of honokiol (50 μmol/L) as indicated. Cells were then stimulated with 100 ng/mL EGF for the indicated times (min) and Ras activation was determined as in A. E, Cos1 cells were transfected with plasmids expressing hemagglutinin-tagged Ras (10 ng) and Sos (100 ng) for 24 h. The Sos plasmid was not included in the lane at the far right. The cells were placed in serum-free medium overnight in the presence and absence of honokiol (50 μmol/L) as indicated. Cells were then stimulated with 100 ng/mL EGF for the indicated times (min) and Ras activation was determined as in A. Experiments shown are representative of at least two independent experiments.
To further establish that honokiol was suppressing Ras activation, we examined the effect in Cos1 cells where background Ras activation was lower. Hemagglutinin-tagged Ras and Sos were transiently transfected into the Cos1 cells with excess Sos to stimulate Ras activation as described previously (37). Twenty-four hours later, the cells were then shifted to serum-free conditions in the presence of increasing concentrations of honokiol and the levels of Ras, Sos, and GTP-bound Ras was evaluated. As shown in Fig. 3C, honokiol strongly suppressed Ras-GTP levels at concentrations of 20 μmol/L and higher. We also examined the effect of honokiol on the ability of EGF to induce Ras activation in Cos1 cells. As shown in Fig. 3D, honokiol suppressed the ability of EGF to increase the level of GTP-bound Ras. This experiment used endogenous Sos and ectopically expressed Ras. As shown in Fig. 3E, the effect of honokiol was more pronounced when ectopically expressed Sos was introduced, indicating that Ras activation by Sos was affected. These data indicate that honokiol suppresses Ras activation.
Honokiol suppresses downstream targets of PLD survival signals
Elevated PLD activity in MDA-MB-231 cells has been reported to activate mTOR (9, 38), which has been correlated with survival signals in human cancer cells (6). Elevated expression of PLD was also shown to lead to increased phosphorylation of the mTOR substrates S6K and 4E-BP1 (7, 38). We therefore examined the effect of honokiol on the phosphorylation of S6K and 4E-BP1. As shown in Fig. 4A, honokiol suppressed phosphorylation of both S6K and 4E-BP1. These data further indicate that honokiol suppresses PLD activity in MDA-MB-231 cells and moreover suppresses the phosphorylation of two mTOR substrates induced by PLD activity that correlate with survival signals in cancer cells.
Fig. 4.
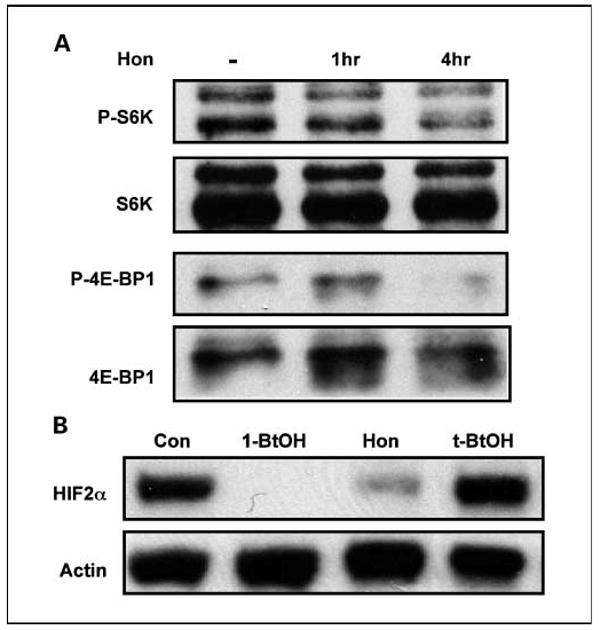
Honokiol suppresses downstream targets of PLD survival signals. A, MDA-MB-231 cells were plated in DMEM with 10% serum. Forty-eight hours later, the cells were shifted to 0.5% for 16 h. Honokiol (20 μmol/L) or control ethanol was then added for the indicated times. The cells were then harvested and analyzed for levels of S6K, phosphorylated S6K (P-S6K), 4E-BP1, and P-4E-BP1 by Western blot analysis as described previously (10). B, 786-O cells were plated and then shifted to medium containing 0.5% serum 24 h later. Eighteen hours later, the cells were treated with 1-BtOH (0.8%), honokiol (20 μmol/L), and t-BtOH (0.8%) as indicated. Cell lysates were prepared 4 h later and examined for HIF2α and actin expression by Western lot analysis. The experiment shown is representative of two independent experiments.
We recently reported that PLD activity was required for the expression of HIF2α in the renal cell carcinoma cell line 786-O cells (13). We therefore examined the effect of honokiol on HIF2α expression in these cells. As reported previously, 1-butanol, which prevents the production of the PLD metabolite phosphatidic acid, suppressed HIF2α expression in these cells (Fig. 4B), confirming the dependence of HIF2α expression on PLD. Also shown in Fig. 4B is that honokiol suppresses HIF2α expression. The data in Fig. 4 show that honokiol suppresses two downstream effects of PLD—the activation of mTOR and HIF2α expression.
Honokiol induces apoptosis in MDA-MB-231 cells deprived of serum
We previously reported that suppression of PLD activity under conditions of serum withdrawal resulted in apoptosis (9, 13, 14). We therefore examined the effect of honokiol on cell viability and cleavage of the caspase-3 substrate PARP—an indicator of apoptotic cell death—in the MDA-MB-231 and 786-O cells. We examined the effect of honokiol on cell viability and PARP cleavage in 0.5% serum after 4- and 24-hour honokiol treatment and in 10% serum after 24-hour honokiol treatment in the MDA-MB-231 cells. As shown in Fig. 5, after 24 hours of honokiol treatment, cell viability was reduced and PARP cleavage increased in 0.5% serum but not in 10% serum. Importantly, the cells were still viable after 4 hours of honokiol treatment—the time point used in Figs. 1 and 2 where we evaluated the effect of honokiol upon PLD activity and on S6K and 4E-BP1 phosphorylation. Honokiol also induced apoptosis in the 786-O cells (data not shown), where it has been shown that PLD and HIF2α are critical for survival (13, 39, 40). These data indicate that honokiol can be used to suppress survival in these two cancer cell lines that rely on PLD activity for their survival under reduced serum conditions.
Fig. 5.

Honokiol induces apoptosis in MDA-MB-231 deprived of serum. MDA-MB-231 cells were plated in DMEM with 10% serum for 48 h then changed to DMEM with either 10% or 0.5% serum overnight as indicated. Honokiol (20 μmol/L) or control ethanol was then added either at the time of changing medium (24-h time point), or 4 h before harvesting (4-h time point) as indicated. The cells were then examined for cell viability and PARP cleavage as described previously (9). The Western blot is representative of at least three independent experiments. The error bars for cell viability represent the SD for triplicate samples from a representative experiment repeated thrice.
Honokiol suppresses stress-induced PLD activity and induces apoptosis in T24 bladder and Calu1 lung cancer cells
PLD activity is elevated in T24 bladder and Calu-1 lung cancer cells, and the PLD activity in these cells is elevated in response to serum withdrawal (10, 12). T24 cells have an activated H-Ras mutant (41, 42) and Calu-1 cells an activated K-Ras mutant (43). The PLD activity in both of these cell lines is dependent on Ras (12). We therefore examined the effect of honokiol on the PLD activity and survival of these human cancer cell lines. As shown in Fig. 6A, honokiol suppressed the stress-induced PLD activity while having little effect on the PLD activity observed in the presence of serum in both T24 and Calu-1 cells. We reported previously that the PLD activity in these cells was required for the survival of these cells in the absence of serum (12). We therefore examined whether honokiol would induce apoptosis in T24 and Calu-1 cells subjected to serum withdrawal. As shown in Fig. 6B, honokiol induced cell death and PARP cleavage in these cells when these cells were placed in 0.5% serum. These data indicate that the effects of honokiol observed in MDA-MB-231 and 786-O cells could also be observed in other cancer cells where PLD has been implicated in survival signals.
Fig. 6.

Honokiol suppresses stress-induced PLD activity in T24 bladder and Calu1 lung cancer cells. A, T24 and Calu1 cells (obtained from the American Type Culture Collection) were plated in DMEM containing 10% serum for 48 h and then shifted to either 10% serum or 0.5% serum overnight as indicated. Honokiol (20 μmol/L) or control ethanol was then added for 4 h as indicated. [3H]myristate was added with the honokiol. After 4 h, BtOH (0.7%) was added for 20 min, at which time the cells were harvested and the level of phosphatidyl-BtOH was determined as in Fig. 1. The experiments shown are representative of at least two independent experiments. B, T24 and Calu-1 cells were plated in DMEM with 10% serum for 48 h then changed to DMEM containing either 10% or 0.5% serum overnight as indicated. Honokiol (20 μmol/L) or control ethanol was then added either at the time of changing medium (24 h) or 4 h before harvesting as in Fig. 3, at which time the cells were examined for cell viability or PARP cleavage. The Western blot is representative of at least two independent experiments. The error bars for cell viability represent the SD for triplicate samples from a representative experiment repeated twice.
Discussion
In this report, we have provided evidence that honokiol, a natural product isolated from Magnolia grandiflora, suppresses PLD survival signals in human cancer cells. The PLD activity in MDA-MB-231 cells examined was dependent on Ras, and honokiol also suppressed Ras activation in these cells. Honokiol was especially effective in cells where Sos was ectopically expressed, indicating that honokiol is suppressing Ras activation by Sos. Importantly, honokiol suppressed the PLD activity that is elevated in response to stress in MDA-MB-231, Calu1, and T24 cancer cells. This is the PLD activity that is critical for the survival of the cancer cells under the poorly vascularized conditions of an emerging tumor. These data indicate that honokiol could be a valuable reagent to target an apparent large number of human cancers that depend on PLD activity and Ras for their survival.
The PLD activity elevated in response to stress in the MDA-MB-231 cells was dependent on Ras in the MDA-MB-231 cells and also in the Calu1 and T24 cancer cells (12). As shown in Fig. 1, there was an increase in Ras activation in the MDA-MB-231 cells. This is interesting in that there is an activated K-Ras gene in the MDA-MB-231 cells. Similarly, there are activating mutations to Ras genes in both the Calu1 and T24 cells. We recently reported that the PLD activity in the T24 cells, where H-Ras is activated, is dependent on the expression of H-Ras; the PLD activity in the Calu-1 cells, where K-Ras is activated, is dependent on the expression of K-Ras (12). This suggests that there is increased GTP binding on the mutated Ras genes upon serum withdrawal. However, it is also possible that the increased GTP-bound Ras represents activation of the non-mutated Ras—either the Ras protein encoded by the non-mutated wild-type allele or other Ras isoforms. In the case of the MDA-MD-231, that would mean that H-Ras was being activated or the wild-type K-Ras encoded by the nonmutant allele. We were unable to distinguish which Ras isoforms were being activated using isoform-specific antibodies to identify the Ras in the pull-down assays due to the lack of specificity of the antibodies. Honokiol blocks PLD activity in the 786-O cells where there is no activated Ras,6 indicating that honokiol can suppress PLD activity in cells where there is no activated Ras. Thus, although it is not yet clear which Ras isoforms are being activated in response to the stress of serum withdrawal, it is possible that the activation of wild type Ras isoforms could play an important role in the survival of human cancer cells, at least in part by activating PLD.
Previous studies have suggested that honokiol can stimulate apoptosis by modulation of nuclear factor-κB (NF-κB) activation pathway (44–46). Interestingly, NF-κB has been shown to be downstream of both Ral and RalA (47, 48). Thus, it is possible that both PLD and NF-κB are critical targets of honokiol action. We do not know whether there is any dependence of NF-κB action on PLD activity; however, the data reported here are consistent with the previous studies identifying NF-κB as a target of honokiol in that there are common upstream elements that are targeted by honokiol—that being Ras activation.
Honokiol has been shown previously to suppress tumor growth in mouse xenograft studies (20, 32, 44). Recently, honokiol was shown to suppress the growth of MDA-MB-231 cells in a mouse xenograft study (32). Importantly, the levels of honokiol used in the mouse xenograft studies were much higher than previously reported to get levels of plasma honokiol sufficient to suppress PLD activity in this study (49). The observation that tumor growth is arrested, but not regressed, suggests that honokiol is not killing all of the cancer cells. As reported here, honokiol was more effective at killing cells when they were in low serum. Emerging tumors are poorly vascularized (50) and, therefore, with less exposure to serum growth factors and perhaps other factors in serum, cancer cells in a solid tumor mass could be more susceptible to the apoptotic effects honokiol. Because honokiol is apparently more effective in stressed cells, it might be possible to make honokiol more effective in vivo with combination therapies that target vascularization. The findings here indicate that honokiol has strong potential as an anticancer agent because it targets survival signal in cancer cells and has the potential to resurrect default apoptotic programs. Importantly, early studies with mouse models indicate that honokiol is well tolerated at high concentrations (20, 22, 44, 49).
Acknowledgments
We thank Lee Henage for assistance with the in vitro PLD assays; Dr. Michael Ohh (University of Toronto, Toronto, Ontario, Canada) for the 786-O cells; Dr. Larry Feig (Tufts University, Boston, MA) for the dominant negative Ras plasmid; and Crislyn D'Souza-Schorey (Notre Dame University, Notre Dame, IN) for the dominant negative ARF plasmids.
Grant support: National Cancer Institute, NIH, CA46677 (D.A. Foster); a Support of Continuous Research Excellence grant, NIH, GM60654 (D.A. Foster); NIH grants GM58516 and P50CA098131 (H.A. Brown); NIH grants AR42687 and AR02030 (J.L. Arbiser); and a Research Centers in Minority Institutions grant, NIH, RR-03037, which supports infrastructure and instrumentation at Hunter College. A. Garcia was supported by a Gene Center fellowship from the Research Centers in Minority Institutions.
Footnotes
Note: A. Garcia and Y. Zheng contributed equally to this work.
Disclosure of Potential Conflicts of Interest: J. Arbiser, patent filed for international rights. The other authors disclosed no potential conflicts of interest.
Our unpublished observations.
Our unpublished results.
References
- 1.Foster DA, Xu L. Phospholipase D in cell proliferation and cancer. Mol Cancer Res. 2003;1:789–800. [PubMed] [Google Scholar]
- 2.Foster DA. Phospholipase D survival signals as a therapeutic target in cancer. Current Signal Trans Ther. 2006;1:295–303. [Google Scholar]
- 3.Rodrik V, Gomes E, Hui L, Rockwell P, Foster DA. Myc stabilization in response to estrogen and phospholipase D in MCF-7 breast cancer cells. FEBS Lett. 2006;580:5647–52. doi: 10.1016/j.febslet.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–5. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 5.Foster DA. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007;67:1–4. doi: 10.1158/0008-5472.CAN-06-3016. [DOI] [PubMed] [Google Scholar]
- 6.Sawyers CL. Will mTOR inhibitors make it as cancer drugs? Cancer Cell. 2003;4:343–8. doi: 10.1016/s1535-6108(03)00275-7. [DOI] [PubMed] [Google Scholar]
- 7.Hui L, Abbas T, Pielak R, Joseph T, Bargonetti J, Foster DA. Phospholipase D elevates the level of MDM2 and suppresses DNA damage-induced increases in p53. Mol Cell Biol. 2004;24:5677–88. doi: 10.1128/MCB.24.13.5677-5686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hui L, Rodrik V, Pielak RM, Zheng Y, Foster DA. mTOR-dependent suppression of protein phosphatase 2A is critical for phospholipase D survival signals in human breast cancer cells. J Biol Chem. 2005;280:35829–35. doi: 10.1074/jbc.M504192200. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene. 2005;24:672–9. doi: 10.1038/sj.onc.1208099. [DOI] [PubMed] [Google Scholar]
- 10.Zheng Y, Rodrik V, Toschi A, et al. Phospholipase D couples survival and migration signals in response to stress in human breast cancer cells. J Biol Chem. 2006;281:15862–8. doi: 10.1074/jbc.M600660200. [DOI] [PubMed] [Google Scholar]
- 11.Gadir N, Lee E, Garcia A, Toschi A, Foster DA. Suppression of TGF-β signaling by phospholipase D. Cell Cycle. 2007;6:2840–5. doi: 10.4161/cc.6.22.4921. [DOI] [PubMed] [Google Scholar]
- 12.Shi M, Zheng Y, Garcia A, Foster DA. Elevated phospholipase D activity in human cancer cells with activating Ras mutations provides survival signal. Cancer Lett. 2007;258:268–75. doi: 10.1016/j.canlet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA. Elevated HIF2α expression in VHL deficient renal cell carcinoma cells is dependent on phospholipase D. Oncogene. 2008;27:2746–53. doi: 10.1038/sj.onc.1210927. [DOI] [PubMed] [Google Scholar]
- 14.Zhong M, Shen Y, Zheng Y, et al. Phospholipase D prevents apoptosis in v-Src-transformed rat fibro-blasts and MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun. 2003;302:615–9. doi: 10.1016/s0006-291x(03)00229-8. [DOI] [PubMed] [Google Scholar]
- 15.Gadir N, Jackson D, Lee E, Foster DA. Defective TGF-β signaling sensitizes human cancer cells to rapamycin. Oncogene. 2007;27:1055–62. doi: 10.1038/sj.onc.1210721. [DOI] [PubMed] [Google Scholar]
- 16.Jiang H, Luo JQ, Urano T, et al. Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature. 1995;378:409–12. doi: 10.1038/378409a0. [DOI] [PubMed] [Google Scholar]
- 17.Luo JQ, Liu X, Hammond SM, et al. Ral interacts directly with the Arf-responsive PIP2-dependent phospholipase D1. Biochem Biophys Res Commun. 1997;235:854–9. doi: 10.1006/bbrc.1997.6793. [DOI] [PubMed] [Google Scholar]
- 18.Lim KH, Baines AT, Fiordalisi JJ, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–45. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 19.Clark AM, El-Feraly FS, WS Li. Antimicrobial activity of phenolic constituents of Magnolia grandiflora L. J Pharm Sci. 1981;70:951–2. doi: 10.1002/jps.2600700833. [DOI] [PubMed] [Google Scholar]
- 20.Bai X, Cerimele F, Ushio-Fukai M, et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–7. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 21.Battle TE, Arbiser J, Frank DA. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood. 2005;106:690–7. doi: 10.1182/blood-2004-11-4273. [DOI] [PubMed] [Google Scholar]
- 22.Shigemura K, Arbiser JL, Sun SY, et al. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer. 2007;109:1279–89. doi: 10.1002/cncr.22551. [DOI] [PubMed] [Google Scholar]
- 23.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol. 2007;9:706–12. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 24.Amblard F, Delinsky D, Arbiser JL, Schinazi RF. Facile purification of honokiol and its antiviral and cytotoxic properties. J Med Chem. 2006;49:3426–7. doi: 10.1021/jm060268m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for ARF6 in receptor-mediated endocytosis. Science. 1995;267:1175–8. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- 26.Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D. Regulation of Sos activity by intramolecular interactions. Mol Cell Biol. 1998;18:880–6. doi: 10.1128/mcb.18.2.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo JQ, Liu X, Frankel P, et al. Functional association between RalA and Arf in active phospholipase D complexes. Proc Natl Acad Sci U S A. 1998;95:3632–7. doi: 10.1073/pnas.95.7.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Y, Xu L, Foster DA. Phospholipase D requirement for receptor-mediated endocytosis. Mol Cell Biol. 2001;21:595–602. doi: 10.1128/MCB.21.2.595-602.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henage LG, Exton JH, Brown HA. Kinetic analysis of a mammalian phospholipase D: allosteric modulation by monomeric GTPases, protein kinase C, polyphosphoinositides. J Biol Chem. 2006;281:3408–17. doi: 10.1074/jbc.M508800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu L, Frankel P, Jackson D, et al. Elevated phospholipase D activity in H-Ras-, but not K-Ras-transformed cells by the synergistic action of RalA and Arf6. Mol Cell Biol. 2003;23:645–64. doi: 10.1128/MCB.23.2.645-654.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweis PC. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell. 1993;75:1137–44. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- 32.Wolf I, O'Kelly J, Wakimoto N, et al. Honokiol, a natural biphenyl, inhibits in vitro and in vivo growth of breast cancer through induction of apoptosis and cell cycle arrest. Int J Oncol. 2007;30:1529–37. [PubMed] [Google Scholar]
- 33.Kozma SC, Bogaard ME, Buser K, et al. The human c-Kirsten ras gene is activated by a novel mutation in codon 13 in the breast carcinoma cell line MDA-MB-231. Nucleic Acids Res. 1987;15:5963–71. doi: 10.1093/nar/15.15.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogata H, Sato H, Takatsuka J, De Luca LM. Human breast cancer MDA-MB-231 cells fail to express the neurofibromin protein, lack its type I mRNA isoform and show accumulation of P-MAPK and activated Ras. Cancer Lett. 2001;172:159–64. doi: 10.1016/s0304-3835(01)00648-6. [DOI] [PubMed] [Google Scholar]
- 35.Lev DC, Kim LS, Melnikova V, Ruiz M, Ananthaswamy HN, Price JE. Dual blockade of EGFR and ERK1/2 phosphorylation potentiates growth inhibition of breast cancer cells. Br J Cancer. 2004;91:795–802. doi: 10.1038/sj.bjc.6602051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Z, Hornia A, Joseph T, et al. Phospholipase D and RalA cooperate with the EGF receptor to transform 3Y1 rat fibroblasts. Mol Cell Biol. 2000;20:462–7. doi: 10.1128/mcb.20.2.462-467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boykevisch S, Zhao C, Sondermann H, et al. Regulation of ras signaling dynamics by Sos-mediated positive feedback. Curr Biol. 2006;16:2173–9. doi: 10.1016/j.cub.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–42. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 39.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:439–44. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 41.Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature. 1982;298:343–7. doi: 10.1038/298343a0. [DOI] [PubMed] [Google Scholar]
- 42.Taparowsky E, Suard Y, Fasano O, Shimizu K, Goldfarb M, Wigler M. Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature. 1982;300:762–5. doi: 10.1038/300762a0. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu K, Birnbaum D, Ruley MA, et al. Structure of the Ki-ras gene of the human lung carcinoma cell line Calu-1. Nature. 1983;304:497–500. doi: 10.1038/304497a0. [DOI] [PubMed] [Google Scholar]
- 44.Ahn KS, Sethi G, Shishodia S, Sung B, Arbiser JL, Aggarwal BB. Honokiol potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through modulation of nuclear factor-κB activation pathway. Mol Cancer Res. 2006;4:621–33. doi: 10.1158/1541-7786.MCR-06-0076. [DOI] [PubMed] [Google Scholar]
- 45.Lee SY, Yuk DY, Song HS, et al. Growth inhibitory effects of obovatol through induction of apoptotic cell death in prostate and colon cancer by blocking of NF-κB. Eur J Pharmacol. 2008;582:17–25. doi: 10.1016/j.ejphar.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 46.Tse AK, Wan CK, Shen XL, Yang M, Fong WF. Honokiol inhibits TNF-α-stimulated NF-κB activation and NF-κB-regulated gene expression through suppression of IKK activation. Biochem Pharmacol. 2005;70:1443–57. doi: 10.1016/j.bcp.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Henry DO, Moskalenko SA, Kaur KJ, et al. Ral GTPases contribute to regulation of cyclin D1 through activation of NF-κB. Mol Cell Biol. 2000;20:8084–92. doi: 10.1128/mcb.20.21.8084-8092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan S, Meng Q, Laterra JJ, Rosen EM. Ras effector pathways modulate scatter factor-stimulated NF-κB signaling and protection against DNA damage. Oncogene. 2007;26:4774–96. doi: 10.1038/sj.onc.1210271. [DOI] [PubMed] [Google Scholar]
- 49.Tsai TH, Chou CJ, Cheng FC, Chen CF. Pharmacokinetics of honokiol after intravenous administration in rats assessed using high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994;655:41–5. doi: 10.1016/0378-4347(94)00031-x. [DOI] [PubMed] [Google Scholar]
- 50.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]