

Summary
Activation of Toll-Like Receptors (TLRs) leads to de-repression and subsequent activation of inflammatory response genes that play essential roles in innate and acquired immunity. De-repression requires signal-dependent turnover of the nuclear receptor corepressor NCoR from target promoters, but the mechanisms remain poorly understood. Here, we report that TLR4 utilizes NFκB to deliver IKKε to target promoters that contain ‘integrated circuits’ of κB and AP-1 sites, resulting in local phosphorylation of c-Jun and subsequent NCoR clearance. In contrast, TLR2-signaling leads to rapid activation of CaMKII and phosphorylation of the TBLR1 component of NCoR complexes, bypassing the requirement for c-Jun-phosphorylation and enabling NCoR clearance from promoters lacking integrated κB elements. Intriguingly, the IKKε-dependent clearance pathway is sensitive to transrepression by Liver X Receptors, while the CaMKII-dependent pathway is not. These findings reveal mechanisms for integration of TLR, calcium and nuclear receptor signaling pathways that underlie pathogen-specific responses and disease-specific programs of inflammation.
Keywords: Inflammatory genes, NCoR, IKK, CaMKII, TBLR1, c-Jun, de-repression
Introduction
TLRs comprise a highly conserved family of receptors for pathogen-associated molecular patterns that play essential roles in regulating innate and acquired immune responses (reviewed in (Medzhitov, 2001; Trinchieri and Sher, 2007). For example, TLR4 recognizes the lipopolysaccharide (LPS) component of gram-negative bacteria, while TLR2 is involved sensing peptidoglycan, bacterial lipoproteins and yeast cell walls. Although activation of a particular TLR could provide the innate immune system with important clues as to the nature of an infectious pathogen in advance of the development of specific immunity, effective utilization of this information would require that the initial recognition step be coupled to receptor-specific cellular responses. In addition to roles in pathogen recognition, TLRs have been found to contribute significantly to a number of inflammatory diseases in model systems, including atherosclerosis, arthritis, diabetes, Alzheimer's disease, and cancer (Cook et al., 2004; Frantz et al., 2007; Gibson et al., 2004; Shiraki et al., 2006). Overall, the emerging picture suggests that TLR-responsive genes must not only be capable of being rapidly and highly induced in response to infection to confer effective immunity, but must also be tightly regulated under basal conditions to prevent the consequences of chronic inflammation.
TLR ligation regulates gene expression through the utilization of common and receptor-specific signalling pathways. All TLRs, except TLR3, utilize MyD88 as an adaptor protein to activate signalling cascades that control the activities of numerous downstream transcription factors, including NF-κB, AP-1 and IRF family members (reviewed in (O'Neill and Bowie, 2007)). TLR1, 2, and 6 also contain a phosphatidyl inositol 3 kinase (PI3K) binding motif (YXXM) not found in other TLRs (Arbibe et al., 2000). Activation of PI3K and consequent calcium mobilization have been shown to be particularly important for TLR2 signalling in several cell types (Chun and Prince, 2006; Kim et al., 2008).
Under basal conditions, the promoters of many highly inducible TLR target genes in macrophages are occupied by the Nuclear receptor CoRepressor (NCoR) or the related Silencing Mediator of Retinoic acid and Thyroid hormone receptors (SMRT) (Ghisletti et al., 2009). NCoR and SMRT are core components of corepressor complexes that contain histone deacetylase 3 (HDAC3) and the transducin beta-like proteins, TBL1 and TBLR1 (Li et al., 2000; Yoon et al., 2003), and mediate active repression functions of unliganded nuclear receptors and other sequence-specific repressors (Guenther et al., 2001; Ogawa et al., 2005; Ogawa et al., 2004; Pascual et al., 2005; Perissi et al., 2004). Recent studies indicate that NCoR is recruited to broad sets of inflammatory response genes by c-Jun, while SMRT is recruited to both distinct and overlapping genes by the Ets repressor, TEL (Ghisletti et al., 2009; Ogawa et al., 2004). TLR4-dependent activation of these genes requires active removal of NCoR/SMRT in addition to the recruitment of activators and coactivators. Studies of several nuclear receptors and signal-dependent transcription factors suggest a final common de-repression step involving activation of TBL1 and TBLR1, F-box/WD-40 adaptor proteins that are important for recruitment of the ubiquitin-conjugating enzymes, including an ubiquitin E2 ligase, UbcH5, resulting in ubiquitylation of NCoR/SMRT complexes and their subsequent removal from gene promoters by the 19S proteosome complex (Frasor et al., 2005; Ogawa et al., 2004; Perissi et al., 2004). Clearance of NCoR/SMRT complexes in response to TLR4 signaling can be prevented by the peroxisome proliferator-activated receptor γ (PPARγ) or liver X receptors (LXRs) in a ligand-dependent manner, resulting in attenuated transcriptional responses and inhibition of inflammation (Blaschke et al., 2006; Ghisletti et al., 2007; Pascual et al., 2005).
While the requirement for NCoR/SMRT clearance from inflammatory response genes has clearly been established as a prerequisite to gene activation, the mechanisms utilized by TLRs to initiate this step are unknown. Here, we define parallel but distinct phosphorylation-dependent signaling pathways that are differentially utilized by TLR2 and TLR4 to direct removal of NCoR complexes from target genes. These signaling pathways are interpreted at the level of individual promoters to enable derepression of overlapping but distinct sets of genes and impose different sensitivities to anti-inflammatory actions of LXRs.
Results
Different signaling inputs engage different NCoR turnover mechanisms
The inducible nitric oxide synthase gene (nos2, aka inos) was chosen as a starting point for evaluating mechanisms mediating signal-dependent NCoR clearance because it is occupied by the NCoR complex in the basal state (Ghisletti et al., 2007; Pascual et al., 2005) and is induced by Pam3CSK4 (Pam3, a specific agonist for TLR2/1), LPS (a specific agonist for TLR4), and tetradecanoyl phorbol acetate (TPA, an activator of PKC signaling) in primary macrophages (Figure 1A). These substances induce different levels of inos expression in part because of the relative extents to which they stimulate the activities of NFκB, AP-1, IRF, and other signal-dependent factors that act in a cooperative manner to induce inos transcription. Unexpectedly, chromatin immunoprecipitation (ChIP) experiments revealed that the three inducers promoted NCoR clearance with different kinetics; loss of NCoR was most rapid in response to Pam3, followed by TPA, and then LPS (Figure 1B). This pattern was observed using three different antibodies, supporting a mechanism of NCoR removal rather than signal-dependent alterations in the accessibility of epitopes required for immunoprecipitation (IP) (Figure S1A). Furthermore, clearance of NCoR was temporally preceded by the transient recruitment of the S1 subunit of the 19S proteasome (Figure S1B). Clearance of NCoR in response to LPS required the F-box domains of both TBL1 and TBLR1, while clearance in response to Pam3 or TPA required the F-box domain of TBLR1 exclusively, indicating distinct requirements for TBL1- and TBLR1- mediated recruitment of ubiquitin-conjugating machinery (Figure S2A). Cell fractionation assays demonstrated that total NCoR protein levels in the nucleus were not altered following treatments (Figure S2B), suggesting that the signal-dependent clearance of NCoR occurs in a promoter-specific manner. Intriguingly, LPS-induced dismissal of NCoR from the inos promoter was blocked by the proteasome activity inhibitor, MG132, but this was not the case for TPA or Pam3 (Figure 1C).
Figure 1. c-Jun phosphorylation-dependent and independent mechanisms for NCoR dismissal.

(A) inos induction by TPA, LPS, and Pam3. BMDM were treated with each stimulus for 6hrs. inos expression was evaluated by QPCR. (B) Differential NCoR turnover kinetics in response to different signal inputs. ChIP timecourse assays were performed in BMDM challenged with TPA, LPS or Pam3 for 2, 6, 10, and 20 minutes using antibodies against NCoR. Immunoprecipitated DNA was analyzed by QPCR using primers specific for the inos promoter. (C) Effect of MG132 on NCoR turnover. ChIP assays for NCoR were performed in BMDM pretreated with or without 6 μM MG132 for 30min and then challenged with TPA, LPS, or Pam3 for 1hr. (D) Effect of c-Jun S63/73A mutations on transcriptional activation of an inos-luc reporter gene. RAW264.7 cells were transfected with indicated expression plasmids and treated with indicated ligands 48h later. Luciferase activities were assayed after 6hrs of treatment. (E) Effect of c-Jun S63/73A mutations on NCoR clearance from the inos promoter. RAW264.7 cells were transfected as in D. NCoR clearance was assayed by ChIP after 1h of treatment with the indicated ligands. *p<0.05 versus non-treated controls. Errors bars represent standard deviation (SD).
Basal recruitment of NCoR to the inos promoter is dependent on c-Jun (Ghisletti et al., 2009) and the AP-1 site located 270bp upstream of the transcriptional start site (Figure S2C). Previous studies have demonstrated that phosphorylations of c-Jun at S63 and S73 are required for TPA-induced NCoR clearance from AP-1 target genes (Ogawa et al., 2004). To examine the possibility that c-Jun phosphorylation may also be involved in NCoR clearance in response to TLR signaling, inos promoter activity was evaluated in RAW264.7 macrophages that were co-transfected with wild type (WT) or S63/73A mutant c-Jun expression plasmids and then treated with vehicle, TPA, LPS, or Pam3. Induction of the inos promoter by TPA and LPS required the S63/73 phospho-acceptor sites, while activation by Pam3 did not (Figure 1D). ChIP assays demonstrated that NCoR dissociated from the inos promoter after TPA and LPS treatment in RAW264.7 macrophages transfected with WT c-Jun, but not with the mutant (Figure 1E), suggesting an essential role of c-Jun phosphorylation in promoting the TPA- and LPS-mediated NCoR dismissal.
Although the de-repression steps of TPA and LPS converged on c-Jun, the differences in their corepressor clearance kinetics and sensitivity to MG132 (shown in Figure 1B-C) suggested that the mechanisms upstream of the c-Jun phosphorylation step are likely to be different. c-Jun-N-terminal kinase 1 (JNK1) can phosphorylate Jun proteins in many cell types (Derijard et al., 1994; Pulverer et al., 1991). Knockdown of JNK1 (Figure S9A, S10A) dramatically reduced TPA induced inos expression (Figure S2D) but did not affect induction of inos by LPS (Figure S2E). ChIP studies showed that TPA, but not LPS or Pam3 signaling, recruited JNK onto the inos promoter (Figure 2A). Furthermore, knockdown of JNK1 expression inhibited TPA-, but not LPS- or Pam3-induced, NCoR clearance (Figure 2B). Together, these results suggested that TPA, TLR4 and TLR2 utilize distinct mechanisms for corepressor clearance: the TPA-induced clearance requires c-Jun phosphorylation and JNK1, the TLR4-induce clearance requires c-Jun phosphorylation, but not JNK1, and the TLR2-mediated NCoR turnover is independent of both c-Jun phosphorylation and JNK1.
Figure 2. TLR4-mediated NCoR clearance is p65-dependent.

(A) Signal-specific recruitment of JNK onto the inos promoter. ChIP assays for JNK were performed in BMDM treated with TPA, LPS, or Pam3 for the indicated times using antibodies against JNK. (B) ChIP assay for NCoR in BMDM transfected with siCTL or siJNK1 siRNAs. BMDM were then treated with TPA, LPS, or Pam3 for 1h prior to ChIP assays. **p<0.05 versus siCTL treated with TPA. (C) ChIP assay for p65 recruitment following LPS treatment for the indicated times. (D) ChIP assay for NCoR occupancy on the inos promoter in WT or p65-/- MEFs treated with LPS or Pam3 for 1hr. (E) ChIP assay for NCoR in BMDM following transfection with siCTL or sip65 siRNAs. ChIP assay was performed 1h after stimulation with LPS. (F) Effect of mutation of the κB element in the inos promoter on TLR4-mediated NCoR clearance detected by ChIP assay. RAW cells were transfected with WT or mutant inos luciferase reporter for 24h and then challenged with LPS for 1h. **p<0.05 versus WT p65 treated with LPS. *p<0.05 versus non-treated controls. Errors bars represent SD.
LPS induced NCoR clearance is dependent on p65 recruitment of IKKε
The identification of TLR4-induced NCoR clearance as c-Jun- and proteasome-dependent, but JNK-independent, raised the question of what molecular messenger(s) might be involved in TLR4-specific NCoR turnover. NFκBs play major roles in driving pro-inflammatory gene expression and their activation requires a combination of IKKs and proteasome activities (reviewed in (Li and Verma, 2002). ChIP time course experiments showed an early peak of p65 recruited onto the inos promoter prior to NCoR clearance (Figure 2C). As TLR4-mediated NCoR clearance was dependent on proteasome activities important in IκB degradation and p65 activation (reviewed in (Karin and Ben-Neriah, 2000), we speculated that the recruitment of p65 to target promoters may participate in events leading to corepressor dismissal. Consistent with this possibility, LPS activated inos and promoted NCoR clearance in wildtype (WT) MEFs, but not in p65-/- MEFs (Figure 2D and S3A). Similarly, knockdown of p65 in bone marrow derived macrophages (BMDM) (Figure S9B, S10B) confirmed that p65 was required for LPS induced NCoR dismissal from the inos promoter (Figure 2E) and mRNA induction (Figure S3B). A κB-binding element at −110bp from the transcriptional start site is in close proximity to the c-Jun/AP-1 anchor site for the NCoR complex (Figure S2C), suggesting that it may be important for p65-dependent NCoR turnover. Consistent with this possibility, mutation of this κB-site in the context of a functional inos promoter effectively diminished NCoR dismissal (Figure 2F) and promoter activities upon TLR4 signaling, but this mutation did not affect NCoR clearance in response to TLR2 signaling (data not shown).
Since p65 itself lacks enzymatic activities that may be necessary for c-Jun-phosphorylation, we hypothesize that it might recruit such activities to the promoter. NFκB family members interact with several kinases, including the inhibitor of κB kinase epsilon, IKKε (Nomura et al., 2000). IKKε is structurally similar to IKKα and IKKβ, (Nomura et al., 2000) and can phosphorylate and/or alter cellular localizations of IκBα, cREL, IRF3, and IRF7, resulting in transcriptional activation of downstream target genes (Harris et al., 2006; Kravchenko et al., 2003; Sweeney et al., 2005). Consistent with this, knockdown of IKKε dramatically reduced LPS-induced inos expression (Figure S2E). In addition, IKKε has been reported to phosphorylate c-Jun in fibroblast-like synoviocytes (Harris et al., 2006; Kravchenko et al., 2003). We found that IKKε can phosphorylate c-Jun in vitro and that LPS-mediated c-Jun phosphorylation was IKKε-dependent (Figure 3A-B). Although the majority of IKKε resides in the cytoplasm, 5-10% of total IKKε was found in the nucleus of macrophages even under basal conditions (Figure S3C), consistent with a previous report of nuclear IKKε in cancer cells (Peant et al., 2009). In addition to inducing nuclear translocation of p65, TLR4 signaling stimulated the interaction of p65 with IKKε (Figure S3D-E). The LPS-induced interaction of IKKε appears to be dependent on phosphorylation of p65 at serine 534 (equivalent to S536 of human p65) because it was abolished by a serine to alanine point mutation (Figure S3F). These observations raised the possibility that IKKε could be recruited to gene promoters containing κB sites by activated p65 to facilitate local phosphorylation of c-Jun and NCoR clearance.
Figure 3. p65 delivers IKKε to the inos promoter to mediate TLR4-dependent NCoR clearance.
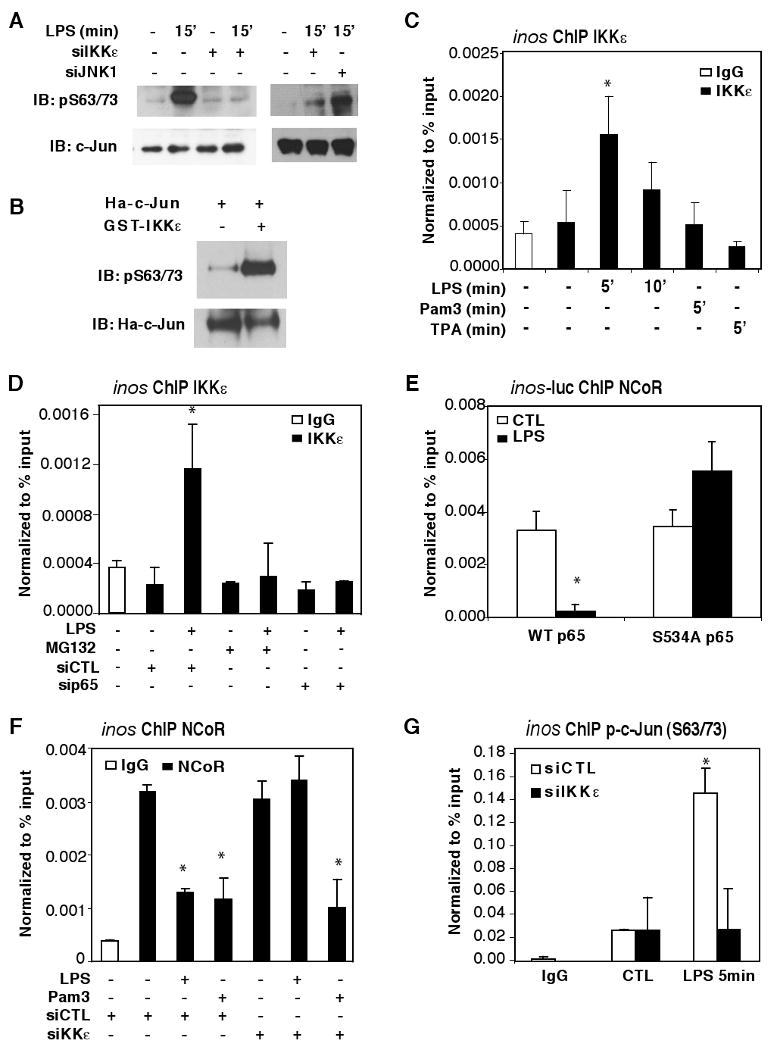
(A) Detection of phosphorylated c-Jun in whole cell lysate by IB using phospho-c-Jun (S63/73)-specific antibodies. BMDM were transfected with siCTL, siJNK1, or siIKKε siRNAs, and challenged with LPS for the indicated times prior to analysis. (B) Assay of immunoprecipitated c-Jun for phosphorylation in vitro by GST-IKKε. Ha-c-Jun was overexpressed in HeLa cells, pulled down using Ha-beads, and incubated with 50ng of GST-IKKε. p-c-Jun (S63/73) antibody was used for IB. (C) ChIP assay for IKKε on the inos promoter in BMDM treated with LPS, TPA, or Pam3 for the indicated times using antibodies against IKKε or control IgG. (D) ChIP assay for IKKε on the inos promoter in BMDM transfected with siCTL or sip65 siRNAs. ChIP assays were performed 5 min after LPS treatment. (E) ChIP assay for NCoR on the inos promoter in the context of a transfected inos–luciferase reporter in p65-/- MEFs co-transfected with WT or the S534A mutant of p65. (F) ChIP assay for NCoR on the inos promoter in BMDM transfected with siCTL or siIKKε siRNAs. ChIP assays were performed after treatment with LPS or Pam3 for 1hr. (G) ChIP assay using p-c-Jun (S63/73)-specific antibodies was performed after treatment with LPS for 5 min. *p<0.05 versus non-treated controls. Errors bars represent SD.
Consistent with this possibility, ChIP studies showed that TLR4 signaling leads to recruitment of IKKε onto the inos promoter (Figure 3C), but not to the mmp12 promoter lacking a κB-element (data not shown). Pretreating macrophages with the proteasome inhibitor, MG132, which inhibits IκB degradation and prevents the translocation of NF-κB into the nucleus (Alkalay et al., 1995; Traenckner et al., 1994), abolished the recruitment of IKKε onto the inos promoter. A similar effect was observed upon knockdown of p65 expression (Figure 3D). ChIP time course and re-ChIP studies demonstrated that p65 and IKKε were recruited onto the inos promoter in a common complex (Figure S1B, S4A). Furthermore, the phospho-null mutant of p65 (S534A) that failed to interact with IKKε upon TLR4 signaling (Figure S3F) was unable to initiate NCoR clearance upon TLR4 signaling (Figure 3E). These results suggest that p65-recruitment of IKKε to promoters likely plays an important role in initiation of corepressor turnover. Indeed, knockdown of IKKε impaired LPS-induced NCoR clearance on the inos promoter (Figure 3F), but left p65 recruitment intact (Figure S4B).
As c-Jun phosphorylation was implicated in TLR4-mediated NCoR clearance, we used phospho-c-Jun S63/73 specific antibodies to examine the phosphorylation status of c-Jun on the inos promoter. These assays confirmed that promoter-bound c-Jun was phosphorylated in response to TLR4 signaling in an IKKε-dependent manner (Figure 3G). To examine whether c-Jun is the sole target of p65-IKKε responsible for NCoR turnover on the inos promoter, we transfected RAW264.7 macrophages with p65-specific siRNA along with plasmids directing expression of either WT or a phospho-mimic c-Jun mutant (S63/73E) and then treated with LPS for 1hr. The loss of NCoR clearance in p65 knockdown macrophages was restored upon overexpression of c-Jun phospho-mimic mutant (Figure S4C). Thus, c-Jun is likely to be the primary target of p65-dependent NCoR clearance in response to TLR4 activation. However, knockdown of NCoR simultaneously with knockdown of IKKε was insufficient to rescue reduction in inos mRNA transcription (Figure S2E), consistent with roles of IKKε in activation of other transcription factors (e.g., IRF3) required for maximum transcriptional output. The IKK-related kinase, TBK1, has also been shown to be important in TLR3 and TLR4-signaling pathways based on its ability to activate transcription factors such as IRF3 (Chau et al., 2008). Consistent with these previous findings, knockdown of TBK1 expression reduced transcriptional activation of inos in response to LPS (Figure S5A, C, and E). In contrast, global- and local-promoter-c-Jun phosphorylation, and NCoR clearance were not affected by TBK1 knockdown (Figure S5H-I), suggesting that TLR4-mediated phosphorylation of c-Jun was primarily IKKε-dependent. Although cytoplasmic functions of IKKε require interactions with scaffold proteins, including TANK/I-TRAF (Chau et al., 2008), knockdown of TANK/I-TRAF did not alter IKKε-mediated c-Jun phosphorylation or NCoR clearance on the inos promoter (Figure S5B, D, H, and J), suggesting that nuclear IKKε involved in NCoR clearance may exist in a distinct complex.
A parallel calcium-CaMKII pathway mediates TLR2-dependent NCoR clearance
In contrast to the TPA- and TLR4-induced NCoR clearance, TLR2-mediated NCoR turnover occurred more rapidly and bypassed the requirement for c-Jun phosphorylation (Figure 1B, C and data not shown). Because TLR2 can engage calcium-dependent signaling pathways (Chun and Prince, 2006; Kim et al., 2008), we evaluated the effect of an intracellular calcium chelator, BAPTA-AM, on TLR2-induced NCoR turnover. BAPTA-AM treatment significantly delayed the kinetics of TLR2-induced corepressor clearance (Figure 4A), suggesting that calcium played a role in the rapid TLR2 derepression of inflammatory genes. As CaMKII has been implicated in regulating NCoR-TBLR1-dependent retinoic acid receptor target genes (Perissi et al., 2004; Si et al., 2007), and similarly, Pam3 activation of inos expression was solely dependent on the F-box domain of TBLR1 (Figure 4B, S2A), we hypothesized that TBLR1 as well as CaMKII may participate in the TLR2 derepression pathway. Immunoblot (IB) results indicated that CaMKII was highly expressed in macrophages and was activated selectively by TLR2, and not by TLR4, ligands (Figure 4C) and was transiently recruited onto the inos promoter in a signal-dependent manner (Figure 4D). Re-ChIP experiments showed that upon TLR2 signaling, CaMKII was recruited in conjunction with the NCoR complex on the inos promoter (Figure 4E), suggesting that component(s) of the NCoR complex may act as a scaffold for CaMKII recruitment. Knockdown of TBLR1, but not TBL1 (Figure S9D, S10D), blocked the recruitment of CaMKII (Figure 4F), consistent with the specific functional requirement for TBLR1 during Pam3 activation of the inos promoter (Figure 4B, S2A). Similar to BAPTA-AM treatment, knockdown of either CaMKIIγ (Figure S9E, S10E) or TBLR1 was sufficient to switch the fast TLR2-mediated NCoR turnover to the slower, TLR4-like, c-Jun-dependent mechanism (Figure 4G, S6A-B). Together, these results suggest that CaMKIIγ and TBLR1 are important determinants of the fast kinetics of NCoR clearance observed upon TLR2 activation.
Figure 4. TLR2 induced NCoR corepressor clearance was calcium dependent.
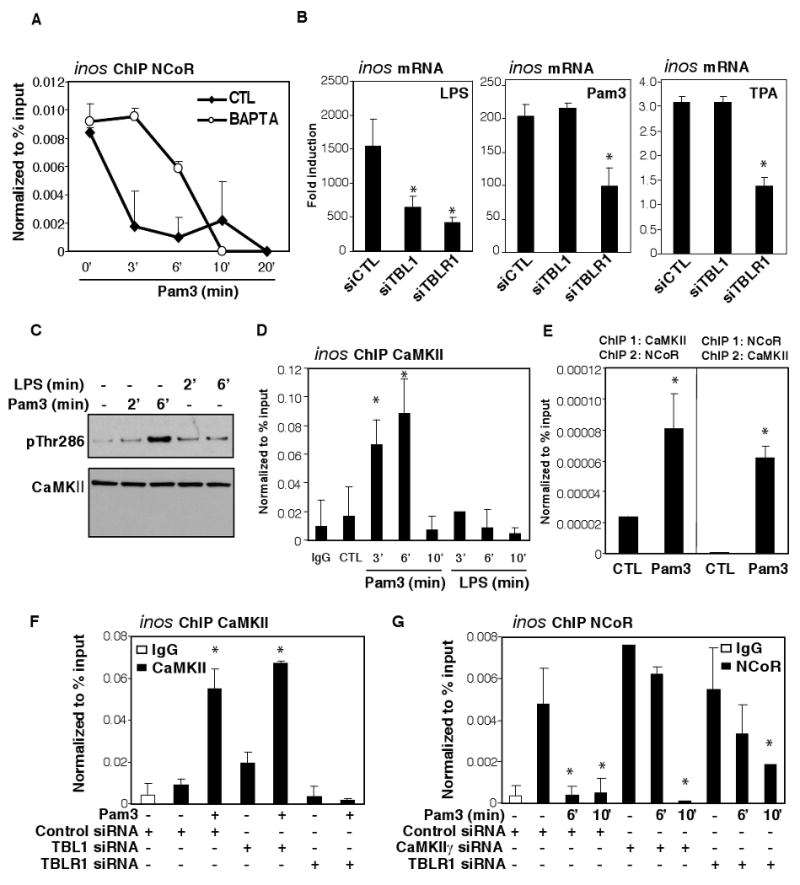
(A) ChIP assay for NCoR on the inos promoter following treatment of BMDM with Pam3 for 3, 6, 10, and 20 minutes in the presence or absence of 6μM BAPTA. (B) Activation of inos in response to 6hr TPA, LPS and Pam3 stimulation in macrophages transfected with siCTL, siTBL1 or siTBLR1 siRNAs. (C) BMDM were treated with LPS or Pam3 for 2 and 6 min. Nuclear extracts were immunoblotted for CaMKII and p-Thr186 CaMKII. (D) ChIP assay for CaMKII on the inos promoter in BMDM following treatment with LPS or Pam3 for 3, 6, and 10 min. (E) ChIP/re-ChIP assays for CaMKII and NCoR on the inos promoter in BMDM challenged with Pam3 for 6 min. (F) ChIP assay for CaMKII in BMDM transfected with siCTL, siTBLR1, or siTBL1 and stimulated with Pam3 for 3 min. (G) ChIP assay for NCoR in BMDM transfected with siCTL, siCamKIIγ or siTBLR1 siRNAs and stimulated with Pam3 for 6 and 10 min. *p<0.05 versus non-treated controls. Errors bars represent SD.
Phosphorylation of TBLR1 has recently been shown to be important for corepressor clearance from nuclear receptors (Perissi et al., 2008). Consistent with these studies, overexpression of a phosphorylation-null mutant of TBLR1 blocked NCoR clearance upon TLR2 signaling. In contrast, overexpression of the phospho-mimic mutant of TBLR1 partially reduced basal occupancy of NCoR on the inos promoter (Figure S6C-D respectively). As both CaMKII and TBLR1 were linked to TLR2-mediated NCoR clearance on the inos promoter, we hypothesized that CaMKII may directly interact with and phosphorylate TBLR1 upon calcium signaling. Indeed, Co-IP studies found that TBLR1 interacted with CaMKII in a TLR2-dependent manner in RAW264.7 macrophages (Figure 5A). In addition, CaMKIIγ can directly phosphorylate full-length GST-TBLR1 in vitro (Figure 5B). Using phospho-specific antibodies directed against either S123 (a PKCδ site) or S199 (kinase unknown) (Perissi et al., 2008) (diagramed in Figure 5C), we found that TBLR1 was phosphorylated at the S199 site in the nucleus (Figure 5D) and that pS199-TBLR1 can be readily detected on the inos promoter in ChIP assays upon TLR2-signaling (Figure 5E). Furthermore, knockdown of CaMKIIγ resulted in the loss of phosphorylation of TBLR1 on the inos promoter (Figure 5F). In concert, these experiments suggest that TLR2 signaling activates CaMKIIγ, which then directly phosphorylates TBLR1 to initiate rapid NCoR clearance.
Figure 5. TLR2 activation of CaMKII leads to phosphorylation of TBLR1.
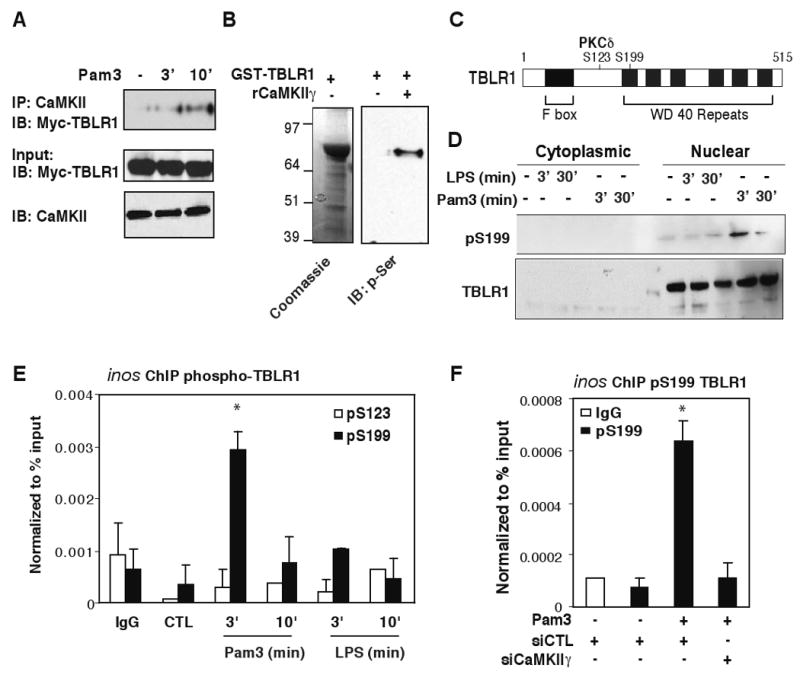
(A) RAW264.7 cells were transfected with Myc-TBLR1 expression construct and treated with Pam3 for 3 and 10 min. IP was carried out with CaMKII-specific antibodies and TBLR1 was detected by anti-Myc antibodies by IB. Bottom panel illustrates inputs for Myc-TBLR1 and CaMKII in each sample. (B) Recombinant GST-TBLR1 proteins were used as substrates for CaMKII in vitro. Bacterial GST-TBLR1 was captured on glutathione agarose and incubated with or without 0.5 μg of activated rCaMKIIγ. Anti-phospho-serine antibody was used to detect phosphorylated TBLR1 proteins by IB. (C) Diagram of TBLR-specific phosphorylation sites. (D) BMDM were challenged with LPS or Pam3 for 3 and 30 min. Cytoplasmic and nuclear extracts were immunoblotted for total and pS199 TBLR1. (E) ChIP assays for phospho-TBLR1 in BMDM treated with Pam3 or LPS for the indicated times using antibodies against p-S199-TBLR1 or p-S123-TBLR1. (F) ChIP assays for p-S199-TBLR1 on the inos promoter in BMDM transfected with siCTL or siCaMKIIγ siRNAs and challenged with Pam3 for 3 minutes. *p<0.05 versus non-treated controls. Errors bars represent SD.
TLR4/IKK and TLR2/CaMKII pathways are utilized to regulate common and distinct sets of inflammatory response genes
We next evaluated potential roles of the p65/IKKε and CaMKII/TBLR1 pathways in clearance of NCoR from other TLR2 and TLR4 target genes. ChIP studies coupled to siRNA knockdowns confirmed the requirement of IKKε for TLR4- and CaMKIIγ for fast TLR2-mediated NCoR clearance on the cxcl2, cxcl9, cxcl10, and ccl4 promoters (Figure 6A-D). ChIP assays using phospho-c-Jun S63/73 specific antibodies also confirmed that IKKε was necessary for c-Jun phosphorylation on each of these promoters (Figure S7).
Figure 6. TLR4/IKKε and TLR2/CaMKII pathways regulate distinct subsets of proinflammatory target genes.

(A-D) ChIP assay for NCoR on the indicated promoters in BMDM transfected with siCTL, siIKKε or siCaMKII siRNAs and stimulated with LPS for 20min or Pam3 for 5 min. (E, F) Effect of LPS and Pam3 treatment of BMDM on NCoR occupancy and mRNA induction of dtx2 (E) and sphk1 (F). (G,H) Induction of sphk1 (G) and dtx2 (H) mRNA in BMDM transfected with siCTL or siCaMKIIγ siRNAs and stimulated with Pam3 for 2hrs. *p<0.05 versus treated siCTL sample. Errors bars represent SD.
The ability of TLR2 to bypass the c-Jun phosphorylation-dependent mechanism involving the recruitment of IKKε through κB elements suggests that this may allow TLR2 to activate a subset of genes that are not inducible by TLR4. A search in publicly available microarray databases identified a small subset of genes that can be activated by TLR2, but not TLR4, using a six-fold induction cut-off (Table S1) (Kim et al., 2007) and (Lipid Maps Consortium, lipidmaps.org). Interestingly, the inability of TLR4 to activate two of these genes, a pro-inflammatory target gene, Spingosine kinase 1 (Sphk1) and a Notch pathway ubiquitin ligase gene, Deltex2 (dtx2), correlates with the absence of any κB elements in the vicinity of AP-1 sites within their respective proximal promoters. We confirmed that these genes were selectively activated by TLR2, but not TLR4, and their transcriptional activation and corepressor dismissal were dependent on CaMKIIγ (Figure 6E-H). These findings illustrate that parallel, yet distinct, NCoR clearance pathways are activated by TLR2 and TLR4 and may contribute to the TLR-specific de-repression and activation of different subsets of pro-inflammatory genes.
NCoR clearance pathways and transrepression by LXRs
PPARγ and LXRs have been shown to exert anti-inflammatory effects in macrophages and hepatocytes by preventing the signal-dependent turnover of NCoR complexes (Blaschke et al., 2006; Ghisletti et al., 2007; Pascual et al., 2005). Recent studies have demonstrated that these anti-inflammatory activities can be modulated in a signal-specific manner. For example, LXR agonists were shown to be effective at inhibiting transcriptional activation of inos in response to LPS, but were inactive when the stimulus was Pam3 (Ghisletti et al., 2007). We confirmed this finding (Figure S8A) and investigated whether the differential sensitivity of the TLR4 and TLR2 pathways to LXR repression maybe related to activation of CaMKII by the TLR2 pathway. Interestingly, conversion of the TLR2-CaMKII pathway to a TLR4-like pathway by removing calcium-CaMK signaling with CaMKIIγ specific siRNA (Figure 7A-B) or BAPTA (Figure S8B) restored the ability of the LXR ligand, GW3965, to mediate transrepression of inos. In contrast, converting the TLR4-p65 pathway to the TLR2-CaMKII-like pathway by simultaneous treatment with calcium ionophore (A23187) (Figure S8C) and TLR4 ligand desensitized the corepressor to the anti-inflammatory input of LXRs. This observation predicted that cell surface receptors that exert pro-inflammatory effects through calcium-dependent pathways, such as the P2Y receptors, may also prevent the ability of LXR agonists to antagonize inflammatory responses. Consistent with this prediction, activation of purinergic receptor signaling with ATP also blocked the ability of the LXR agonist to prevent NCoR clearance and inhibit inos induction (Figure 7C-D).
Figure 7. TLR signaling and anti-inflammatory crosstalk.

(A) Induction of inos mRNA in BMDM transfected with siCTL or siCaMKIIγ siRNAs and treated with Pam3 in the presence or absence of the LXR agonist GW3965 (GW) as indicated. (B) ChIP assay for NCoR on the inos promoter in BMDM transfected with siCTL or siCaMKIIγ siRNAs and treated as indicated. (C) Induction of inos mRNA in BMDM treated with LPS, ATP and/or GW as indicated. (D) ChIP assay for NCoR on the inos promoter in BMDM treated with LPS, ATP and/or GW as indicated. *p<0.05 versus non-treated controls. Errors bars represent standard deviation. (E) Model for utilization of p65/IKKε and CaMKII pathways in TLR4 and TLR2-mediated corepressor turnover. See discussion for details.
Discussion
TLR4 signaling induces a p65/IKKε-dependent NCoR clearance mechanism
Although many lines of evidence suggest that NCoR and SMRT function as key checkpoints for regulating inflammatory responses, the clearance mechanisms remain poorly defined (Hoberg et al., 2006; Ogawa et al., 2004). Here, we described two distinct NCoR clearance pathways involving phosphorylation-dependent mechanisms that can be exploited to enable gene-specific transcriptional responses. In the case of TLR4 signaling, p65 functions as a messenger to bring IKKε-enzymatic activity to κB-containing inflammatory gene promoters. This in turn enables IKKε to phosphorylate adjacent c-Jun/NCoR complexes and initiate corepressor clearance (Figure 7E). In addition to the inos, cxcl2, cxcl9, cxcl10, and ccl4 promoters, the promoters of many LPS inducible genes exhibit AP-1 and κB-sites in close proximity (Table S2), suggesting that coupled AP-1/κB elements may function as ‘integrated circuits’ for toggling promoters from a repressed to an activated state in response to an inflammatory stimulus. Although phosphorylation of S536 of p65 has been linked to activation of target genes, mechanisms accounting for this activity have remained enigmatic. The present findings suggest that phosphorylation of S536 creates a docking site for recruitment of IKKε to specific target gene promoters, enabling it to initiate NCoR clearance. It will be of interest to determine whether analogous kinase delivery mechanisms are utilized by other signal-dependent transcription factors, such as those involved in interferon signaling, to achieve corepressor clearance.
A Ca++/CaMKIIγ-dependent NCoR Clearance Mechanism
The present studies demonstrate that upon TLR2 signaling, CaMKIIγ is rapidly recruited to NCoR complexes on promoters of pro-inflammatory target genes, where it phosphorylates TBLR1 at S199. This in turn is proposed to activate TBLR1 to recruit ubiquitylation machinery, resulting in corepressor clearance from the target promoter. The observation that MG132 blocked TLR4-dependent, but not TLR2-mediated, NCoR clearance, delineates the distinct roles of proteosome complexes in these two derepression pathways. In the case of TLR4-dependent clearance, proteosome-dependent degradation of IκB is a prerequisite for nuclear entry of p65, and hence the localization of IKKε to target promoters. Since TLR2 signaling utilizes CaMKII to directly activate TBLR1, proteolysis of IκB is not required. Evidence that the signal-dependent removal of NCoR is nonetheless ubiquitin-dependent in response to TLR2 or TLR4 signaling is supported by the requirement for the F box domains of TBL1 and TBLR1 that are critical in recruiting ubiquitin-conjugating enzymes (Perissi et al., 2004), and ChIP data demonstrating the transient recruitment of the S1 component of the 19S proteasome to the inos promoter prior to NCoR clearance. The 19S proteasome complex is capable of recognizing and unfolding ubiquitinated proteins and has been observed to be recruited to promoters in the absence of the 20S complex that mediates degradation (Perissi et al., 2004; Perissi et al., 2008). Our results are thus most consistent with a model in which the 19S subunit mediates the active removal, but not degradation, of ubiquitinated components of the NCoR complex.
We found that the CaMKII-dependent de-repression mechanism utilized by TLR2 operates on the inos, cxcl2, cxcl9, cxcl10, ccl4 and Il1β genes (Figure 6 and data not shown), suggesting that it is a widely used mechanism. Furthermore, the ability of TLR2-dependent calcium signaling to bypass the κB-AP-1 composite requirement allows TLR2 to activate genes that are not activated by TLR4 signaling, including Sphk1 and the Notch pathway gene Dtx2. Sphks generate the lysosphingolipid sphingosine-1-phosphate (S1P), which acts as an extracellular ligand for S1P receptors that play important roles in lymphocyte trafficking and may participate in the pathophysiology of inflammatory diseases (Spiegel and Milstien, 2003). Notch signaling directly regulates immune responses by regulating TLR signaling and modulating inflammatory mediator production, including IFNγ and IL10 (Palaga et al., 2008). These findings thus raise a number of intriguing possibilities regarding other pathways that similarly mobilize intracellular calcium signaling and CaMKII activation, such as ATP/P2Y signaling pathway and the non-canonical CaMKII-dependent Wnt signaling induced by Wnt5A in macrophages (Blumenthal et al., 2006; del Rey et al., 2006; Hanley et al., 2004).
Differential Sensitivity of NCoR Clearance Pathways to LXR Signaling
The ability of PPARγ and LXRs to repress inflammatory gene expression by preventing NCoR turnover supports a physiologic role of NCoR corepressor complexes as regulatory checkpoints for a subset of TLR-inducible genes. Previous studies demonstrated that TLR4-dependent gene activation was sensitive to repressive effects of LXR agonists, while TLR2-dependent gene activation was not. Here we demonstrate that resistance to LXR repression is conferred by the Ca++/CaMKII-dependent derepression pathway. Inhibition of this pathway restores sensitivity of TLR2 signaling to LXR repression, while combining TLR4 signaling with calcium mobilization renders LXRs unable to transrepress. These findings imply that the ability of LXR ligands to inhibit inflammatory gene expression will be context-dependent.
Overall, the present studies reveal promoter-specific mechanisms for integration of TLR, calcium and nuclear receptor signaling pathways that underlie pathogen-specific responses and disease-specific programs of inflammation. TLR-specific NCoR clearance mechanisms may be used to achieve distinct programs of gene regulation that underlie innate immune responses tailored to distinct classes of pathogens. Furthermore, anti-inflammatory drugs that target specific signal-induced corepressor clearance events may be of therapeutic utility in diseases in which inflammation plays a pathogenic role.
Experimental Procedures
Reagents and plasmids
LPS, TPA, calcium ionophore (A23187), and ATP were obtained from Sigma. Pam3CSK4 was from InvivoGen. GW3965 was kindly provided by GlaxoSmithKline. BAPTA/AM was obtained from Calbiochem. Proteasome inhibitor (MG132) was obtained from BioMol. Anti-CaMKII, JNK, p65, TBK, TANK, c-Jun, p-S63/73c-Jun, IKKε, and normal rabbit/goat antibodies were from Santa Cruz Biotech. Anti-NCoR antibodies were from ABR, Abcam and Santa Cruz Biotech. Anti-S1-proteasome complex component antibodies were from Upstate. Anti-p-S123 and p-S199 TBLR1 antibodies were described in (Perissi et al., 2008). The reporter plasmid inos-luc has been previously described (Pascual et al., 2005). Mutations in inos-luc vector were made using the QuickChange site-directed mutagenesis kit (Stratagene). Expression vectors for WT and S63/73A c-Jun mutant were described in (Ogawa et al., 2004). Expression vectors for WT and delta-F-box mutants of TBL1 and TBLR1 were described in (Perissi et al., 2004; Perissi et al., 2008).
Cell culture, treatment, and transfection
BMDM were generated from 5 wk old C57BL/6 (Harlan) mice as described in (Sawka-Verhelle et al., 2004). WT and p65-/- MEFs were cultured as described in (Werner et al., 2005). RAW 264.7 cells were cultured in DMEM containing 10% FBS as described in (Ghisletti et al., 2007). LPS, TPA, Pam3CSK4, and GW3965 were used at a concentration of 100ng/ml, 100nM, 300ng/ml, and 1μM respectively.
For RNAi experiments, scrambled control or smart-pool siRNAs (Dharmacon) against JNK1, RelA/p65, IKKε, CaMKIIγ, TBL1, TBLR1, TANK, and TBK were transfected into primary macrophages using lipofectamine 2000 (Invitrogen) as described (Ghisletti et al., 2009). Transient transfections in RAW 264.7 cells and MEFs were performed as described in (Ghisletti et al., 2007) using Superfect reagent (Qiagen). For luciferase assays, β-galactosidase expression vector was also cotransfected as internal control. For siRNA experiments, RAW264.7 cells were transfected with siRNAs (100nM) using Superfect reagent for 48 h before activation with TLR ligands. Luciferase activity was normalized to β-galactosidase activity. Transfection data are represented as mean +/- SD of three independent experiments in triplicates.
ChIP assays
ChIP assays were performed as described in detail in (Ghisletti et al., 2009). For ReChIP assay, after the first IP, beads were eluted in 10mM DTT and diluted with ReChIP dilution buffer (50nM Tris/HCl pH 7.45, 150mM NaCl, 1% Triton, 2mM EDTA) and subjected to a second IP. Primer sequences are listed in Table S3.
RNA isolation and real time PCR
Total RNA (isolated by RNeasy kit, Qiagen) was prepared from MEFs or macrophages treated as indicated in each legend. One μg of total RNA was used for cDNA synthesis, and 1 μl of cDNA was used for real time PCR analysis was performed on an Applied Biosystems 7300 Real-time PCR system. Values are normalized with GAPDH content. Data are represented as mean +/- SD of three independent experiments in duplicates. Primer sequences are listed in Table S3.
Co-IP assay
RAW cells were transfected with Myc-TBLR1 or Myc-p65 (WT or S534A mutant) and lysed in 10 mM Tris-HCl ph 7.5, 450 mM NaCl, 0.5% NP40, 1mM EDTA with protease inhibitors. IPs were carried out with anti-CaMKII or IKKε antibody and TBLR1 or p65 was detected using anti-Myc antibody (Abcam).
Kinase Assays
For in vitro CaMK kinase assays, 0.5ug recombinant CaMKIIg (Upstate) was incubated with bacterially purified GST-TBLR1 (full-length) fusion proteins attached to glutathione beads. The beads were washed, boiled, and then subjected to SDS-PAGE. Serine phosphorylations were detected by anti-p-Serine antibody (Chemicon). For in vitro IKKε kinase assays, 50ng recombinant GST-IKKε (Cell Signalling) was incubated with immunoprecipitated Ha-c-Jun proteins from HeLa whole cell extracts. The Ha-beads were washed, boiled, and then subjected to SDS-PAGE. p-S63/73-c-Jun was detected by p-S63/73-specific antibody.
Supplementary Material
Acknowledgments
We thank Alexander Hoffmann for WT and p65-/- MEFs and critical reading of the manuscript. We thank Lynn Bautista for assistance with preparation of the manuscript. These studies were supported by a Fondation Leducq Transatlantic Network Grant and by NIH grants CA52599, DK074868, DK063491, HL088093, HL065445 and GM069338. W.H. was supported by Gates Millennium Scholars Program and NIDDK predoctoral training grant 5T32DK007541-22. S.G. was supported by a fellowship from the American Heart Association. M.G.R. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nature immunology. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Takata Y, Caglayan E, Collins A, Tontonoz P, Hsueh WA, Tangirala RK. A nuclear receptor corepressor-dependent pathway mediates suppression of cytokine-induced C-reactive protein gene expression by liver X receptor. Circulation research. 2006;99:e88–99. doi: 10.1161/01.RES.0000252878.34269.06. [DOI] [PubMed] [Google Scholar]
- Blumenthal A, Ehlers S, Lauber J, Buer J, Lange C, Goldmann T, Heine H, Brandt E, Reiling N. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood. 2006;108:965–973. doi: 10.1182/blood-2005-12-5046. [DOI] [PubMed] [Google Scholar]
- Chau TL, Gioia R, Gatot JS, Patrascu F, Carpentier I, Chapelle JP, O'Neill L, Beyaert R, Piette J, Chariot A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008;33:171–180. doi: 10.1016/j.tibs.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Chun J, Prince A. Activation of Ca2+-dependent signaling by TLR2. J Immunol. 2006;177:1330–1337. doi: 10.4049/jimmunol.177.2.1330. [DOI] [PubMed] [Google Scholar]
- Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nature immunology. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- del Rey A, Renigunta V, Dalpke AH, Leipziger J, Matos JE, Robaye B, Zuzarte M, Kavelaars A, Hanley PJ. Knock-out mice reveal the contributions of P2Y and P2X receptors to nucleotide-induced Ca2+ signaling in macrophages. The Journal of biological chemistry. 2006;281:35147–35155. doi: 10.1074/jbc.M607713200. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nature clinical practice. 2007;4:444–454. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- Frasor J, Danes JM, Funk CC, Katzenellenbogen BS. Estrogen down-regulation of the corepressor N-CoR: mechanism and implications for estrogen derepression of N-CoR-regulated genes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13153–13157. doi: 10.1073/pnas.0502782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes & development. 2009;23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Molecular cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, Wong J, Genco CA. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Molecular and cellular biology. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Musset B, Renigunta V, Limberg SH, Dalpke AH, Sus R, Heeg KM, Preisig-Muller R, Daut J. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9479–9484. doi: 10.1073/pnas.0400733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, Oliere S, Sharma S, Sun Q, Lin R, Hiscott J, Grandvaux N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J Immunol. 2006;177:2527–2535. doi: 10.4049/jimmunol.177.4.2527. [DOI] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Molecular and cellular biology. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kim HG, Kim JY, Gim MG, Lee JM, Chung DK. Mechanical stress induces tumor necrosis factor-alpha production through Ca2+ release-dependent TLR2 signaling. Am J Physiol Cell Physiol. 2008 doi: 10.1152/ajpcell.00085.2008. [DOI] [PubMed] [Google Scholar]
- Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, Jang E, Lee HA, Youn J, Akira S, Lee MS. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity. 2007;27:321–333. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. The Journal of biological chemistry. 2003;278:26612–26619. doi: 10.1074/jbc.M303001200. [DOI] [PubMed] [Google Scholar]
- Li J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. The EMBO journal. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Nomura F, Kawai T, Nakanishi K, Akira S. NF-kappaB activation through IKK-i-dependent I-TRAF/TANK phosphorylation. Genes Cells. 2000;5:191–202. doi: 10.1046/j.1365-2443.2000.00315.x. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, Glass CK. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14461–14466. doi: 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SH, Osborne BA. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol. 2008;38:174–183. doi: 10.1002/eji.200636999. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peant B, Diallo JS, Dufour F, Le Page C, Delvoye N, Saad F, Mes-Masson AM. Over-expression of IkappaB-kinase-epsilon (IKKepsilon/IKKi) induces secretion of inflammatory cytokines in prostate cancer cell lines. Prostate. 2009 doi: 10.1002/pros.20912. [DOI] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG. TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Molecular cell. 2008;29:755–766. doi: 10.1016/j.molcel.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- Sawka-Verhelle D, Escoubet-Lozach L, Fong AL, Hester KD, Herzig S, Lebrun P, Glass CK. PE-1/METS, an antiproliferative Ets repressor factor, is induced by CREB-1/CREM-1 during macrophage differentiation. The Journal of biological chemistry. 2004;279:17772–17784. doi: 10.1074/jbc.M311991200. [DOI] [PubMed] [Google Scholar]
- Shiraki R, Inoue N, Kobayashi S, Ejiri J, Otsui K, Honjo T, Takahashi M, Hirata K, Yokoyama M, Kawashima S. Toll-like receptor 4 expressions on peripheral blood monocytes were enhanced in coronary artery disease even in patients with low C-reactive protein. Life sciences. 2006;80:59–66. doi: 10.1016/j.lfs.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Si J, Mueller L, Collins SJ. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J Clin Invest. 2007;117:1412–1421. doi: 10.1172/JCI30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Sweeney SE, Hammaker D, Boyle DL, Firestein GS. Regulation of c-Jun phosphorylation by the I kappa B kinase-epsilon complex in fibroblast-like synoviocytes. J Immunol. 2005;174:6424–6430. doi: 10.4049/jimmunol.174.10.6424. [DOI] [PubMed] [Google Scholar]
- Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. The EMBO journal. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defense. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. The EMBO journal. 2003;22:1336–1346. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.