

Abstract
Synaptic vesicle endocytosis is critical to maintain synaptic communication during intense stimulation. Here we describe Tweek, a conserved protein that is required for synaptic vesicle recycling. tweek mutants show reduced FM 1–43 uptake, cannot maintain release during intense stimulation and harbor larger than normal synaptic vesicles, implicating it in vesicle recycling at the synapse. Interestingly, the levels of a fluorescent PI(4,5)P2 reporter are reduced at tweek mutant synapses and the probe is aberrantly localized during stimulation. In addition, various endocytic adaptors known to bind PI(4,5)P2 are mislocalized and the defects in FM 1–43 dye uptake and adaptor localization are partially suppressed by removing one copy of the phosphoinositide-phosphatase synaptojanin, suggesting a role for Tweek in maintaining proper phosphoinositide levels at synapses. Our data implicate Tweek in regulating synaptic vesicle recycling via an action mediated at least in part by the regulation of PI(4,5)P2 levels or availability at the synapse.
Introduction
Synaptic vesicle recycling relies heavily on clathrin dependent endocytosis, ensuring a continuous supply of new synaptic vesicles during stimulation (Haucke, 2003; Kasprowicz et al., 2008). Numerous proteins involved in the process have been identified through biochemical screening approaches and forward genetic screens in model organisms (Babcock et al., 2003; Jung and Haucke, 2007). Several protein adaptors and lipids coordinately bind and recruit downstream effectors that mediate membrane bending to shape new vesicles. The coordinated action of dynamin and its effectors results in separation of new vesicles that are subsequently prepared for a new round of fusion (van der Bliek and Meyerowitz, 1991). While numerous proteins and lipids have been assigned a specific function during this process, others play a supporting role to scaffold synaptic zones, ensuring a high fidelity of recycling (Koh et al., 2004; Majumdar et al., 2006).
Phosphoinositide lipids (PIs) play important regulatory roles in various cellular processes, including vesicle recycling (Martin, 1998; Vicinanza et al., 2008). PI(4,5)P2 is concentrated at the plasma membrane of most cells, including neurons, and plays multiple roles in the regulation of synaptic transmission (Cremona et al., 1999; Di Paolo et al., 2004; Harris et al., 2000; Micheva et al., 2001; Van Epps et al., 2004; Verstreken et al., 2003). PI(4,5)P2 is a precursor for the phospholipase C-derived metabolites, IP3 and DAG, which are involved in the regulation of Ca2+ release from intracellular stores and vesicle priming and fusion, respectively (Davis and Patrick, 1990; Rhee et al., 2002; Sladeczek, 1987). PI(4,5)P2 also appears to regulate the size of the readily releasable pool of synaptic vesicles and secretory granules (Di Paolo et al., 2004; Gong et al., 2005; Milosevic et al., 2005). In addition, PI(4,5)P2 is implicated in the formation of new synaptic vesicles at the plasma membrane (Cremona et al., 1999; Harris et al., 2000; Mani et al., 2007; Van Epps et al., 2004; Verstreken et al., 2003). Hence, at the synapse, altered levels of PI(4,5)P2 may affect several steps in the synaptic vesicle cycle. However, despite these pleiotropic functions, PI metabolism within the cell is surprisingly spatially restricted and PI-dependent processes appear to be locally regulated (Di Paolo et al., 2002; Golub and Caroni, 2005; Schuske et al., 2003).
Here we report the identification and characterization of a Tweek, a conserved and previously uncharacterized protein. Tweek is required to maintain normal synaptic vesicle recycling and affects PI availability. Our data suggest that Tweek may regulate synaptic vesicle recycling, at least in part, by affecting PI(4,5)P2 pools at the synapse.
Results
Isolation of tweek mutants
To identify proteins that affect synaptic transmission and plasticity, we performed genetic screens using ey-FLP technology (Newsome et al., 2000; Stowers and Schwarz, 1999; Verstreken et al., 2003). These screens allow us to identify flies that carry random chemically induced (EMS) mutations that affect synaptic transmission in photoreceptors, circumventing the organismal lethality associated with these mutations. Flies with mutant eyes were screened by recording electroretinograms (ERG) (Figure 1A). ERGs measure differences in extracellular potential between photoreceptors (PRs) and the body during a short light flash (1 s). In controls, ERG recordings show (1) a de- and repolarization of the PRs, reflecting an intact phototransduction mechanism, and (2) ‘on’ and ‘off’ transients at the onset and conclusion of the light pulse, indicating that the PRs can activate postsynaptic neurons (Figure 1A, grey arrowheads). By isolating mutants with defective on and off transients but normal depolarization, we and others have been able to identify genes that affect synaptic function or development (Babcock et al., 2003; Verstreken et al., 2003). Two mutations in one of the complementation groups on chromosome arm 2L, tweek, show ‘on’ and ‘off’ transient amplitudes that are severely reduced compared to controls (Figure 1A). These data suggest that tweek mutant PRs fail to properly transmit information to post-synaptic neurons.
Figure 1. tweek mutant photoreceptors show synaptic defects.

(A) Electroretinograms of controls (yw eyFLP; P{y+} FRT40A / l(2)cl-2L P{w+} FRT40A), tweek mutants (yw eyFLP; tweek1 or 2 P{y+} FRT40A / l(2)cl-2L P{w+} FRT40A) and rescued tweek animals (yw eyFLP; tweek 2 P{y+} FRT40A; tweek+(HB69) / +). The positions of ‘on’ and ‘off’ transients (or lack thereof) are indicated by grey arrows.
(B–C) Electron microscopy of control (yw eyFLP; P{y+} FRT40A / l(2)cl-2L P{w+} FRT40A) and tweek1 mutant (yw eyFLP; tweek1 P{y+} FRT40A / l(2)cl-2L P{w+} FRT40A) lamina cartridges. PR terminals of one cartridge are artificially labeled in green.
(D–E) Electron microscopy images of single PR terminals of control (C) and tweek1 mutant (D) animals. Capitate projections (arrowhead) and mitochondria (m) are indicated.
To better understand the underlying cause of the defect in neuronal communication in tweek mutant eyes, we performed transmission electron microscopy (TEM), revealing ultrastructural features of the mutant PR synapses, including mitochondrial density and morphology, active zone integrity, glial cell morphology, and synaptic vesicle density. PRs invade the lamina, the first optic neuropil of the fly brain, cluster in groups of six and form characteristic topographic connections on post synaptic cells. One such unit, containing six PRs, labeled green in Figure 1B–C, is a lamina cartridge. Qualitative analyses indicate that most synaptic components are normal in tweek mutant PR terminals (Figure 1B–E), however synaptic vesicle density appears markedly reduced (Figure 1D–E). In addition, although the number of glial invaginations in the PR cells (capitate projections) is not different between controls and mutants (controls: 0.355±0.026 capitates µm−1; tweek1: 0.352±0.041 µm−1; p<0.01), capitate projections are not deeply invaginated or headed in the mutants but often remain shallow (controls: 17.9±4.1% shallow projections and tweek1: 68.3±13.9% shallow projections; p<0.01; Figure 1D, E). Since glial invaginations are thought to be sites of endocytosis in PR terminals (Fabian-Fine et al., 2003), these data, together with the lower vesicle density, suggest that while development of the PR terminals is not much impaired, mutations in tweek may affect synaptic function, possibly endocytosis.
tweek mutants further sparked our interest as rare homozygous flies survive to adulthood. These flies are unable to walk or stand upright for long periods, and exhibit seizures, suggestive of severe neurological defects. Based on the adult behavior of the mutants we named the gene “tweek” as it reminded us of the cartoon character “Tweek” from the TV series “Southpark”. However, even when grown under optimal conditions (see Methods) most homozygous tweek2 or trans-heterozygous tweek1/Df and tweek2/Df mutants die as late pupae and only very few animals eclose (<1/2,000) and die soon after eclosion. Hence, tweek is an essential gene.
tweek encodes a large protein of unknown domain structure
To identify the gene encoding tweek, we mapped the lesions in the mutants using meiotic recombination (Zhai et al., 2003). Rough mapping placed tweek in the 36A–C cytological interval and showed that the mutations fail to complement Df(2L)Exel8036 (Parks et al., 2004). Meiotic fine-mapping (Figure 2A), allowed us to map the lesions in the tweek alleles between EY12630 and KG05250.
Figure 2. Characterization of tweek mutants.
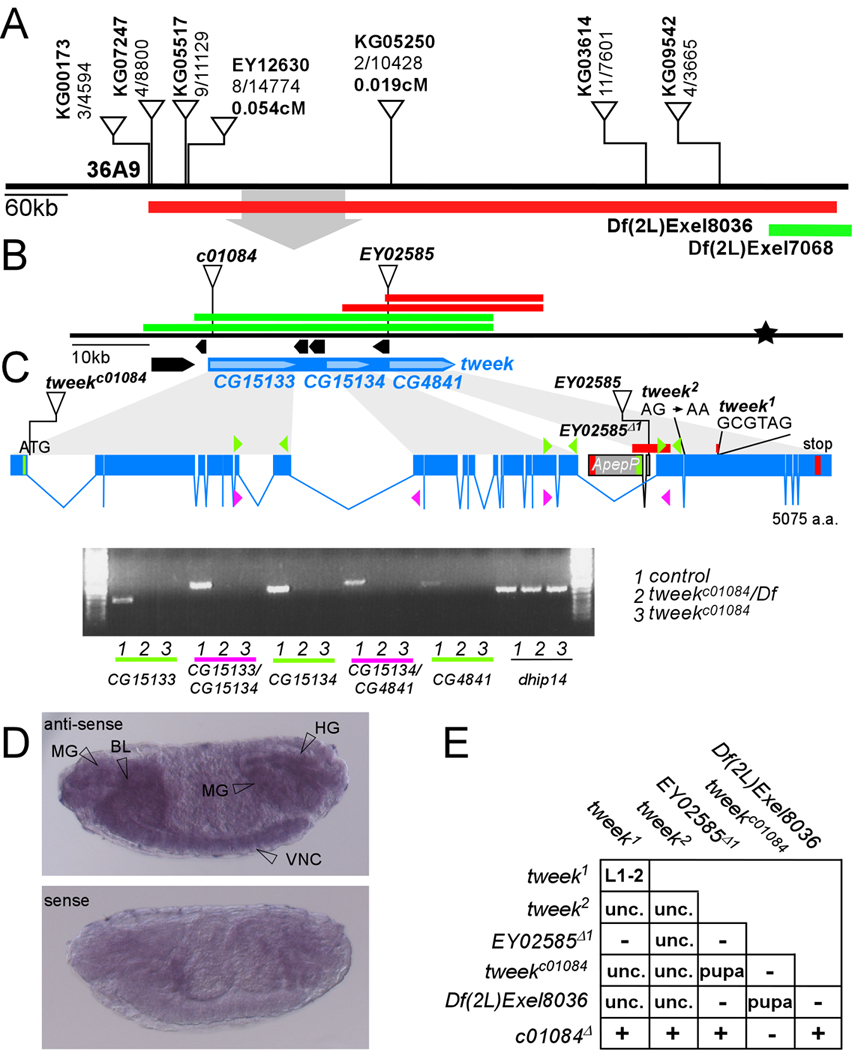
(A) P-element mapping. P-elements used for mapping: numbers separated by a “/” indicate the number of recombinants out of flies scored. Recombination distance in cM for two nearby P-elements is indicated. The cytological interval and the Exelixis deficiencies that complement (Green) or not (Red) are shown. The area magnified in (B) is shown by a grey arrow
(B) The mapping location of tweek (Blue) based on recombination data (Star). CG15133, CG15134 and CG4841 correspond to the tweek gene. EY02528, as well as c01084 fail to complement the tweek1 and 2 alleles. The regions cloned in P[acman] to create rescue constructs are indicated: red constructs do not rescue the tweek alleles while the green constructs do rescue the tweek alleles.
(C) Intron-exon structure of tweek and RT-PCR analysis. Start codons are marked in green and stop codons in red. The P-element excision EY02585Δ1 and the molecular nature of both tweek alleles are indicated. tweek1 harbors a 74 bp deletion (red) and a 6 bp insertion (indicated) and tweek2 harbors a splice acceptor mutation before exon 20. RT-PCR on yw eyFLP; FRT40Aiso control, tweekc01084 and tweekc01084/Df(2L)Exel8036 using the primers shown in Supplemental Table 1. (D) In situ hybridization of dioxygenin labeled RNA to whole stage 15 embryos using a CG4841 probe revealing labeling in the mid gut (MG), hind gut (HG), brain lobe (BL) and ventral nerve cord (VNC). An independent probe against CG15134 shows an identical labeling pattern (see Figure S3) while sense probes do not show specific labeling.
(E) Lethal stage of tweek mutant combinations. L1-2: animals do not survive beyond the first or second instar larval stage. Pupa or Unc: most animals die during the pharate adult (late pupal) stage. However, very few (<1/2,000) manage to eclose but are severely uncoordinated. −: failure to complement, +: complement.
To confirm the mapping position we crossed the tweek EMS alleles to flies that carry lethal transposon insertions in the region. The PiggyBac insertion c01084 (Parks et al., 2004) located downstream of the start codon in CG15133 as well as the P-element insertion EY02585 (Bellen et al., 2004) located between ApepP and CG4841 and an imprecise excision of this P-element (EY02585Δ1), removing the start codons of ApepP and CG4841, all fail to complement the tweek alleles and Df(2L)Exel8036 (Figure 2B,C and E). An excision of the PiggyBac insertion (c01084Δ in Figure 2E) and a precise excision of the P element reverts the lethality and complement the tweek alleles and the deficiency. Although CG15133 and CG4841 are separated by 4 genes encompassing more than 12 kb, c01084 and EY02585Δ1 also fail to complement one another, suggesting that both mutations affect the same gene.
As shown in Figure 2B CG15134 is the only gene located between CG15133 and CG4841 that is transcribed in the same orientation. To determine if CG15133, CG15134 and CG4841 encode a single gene, we performed RT-PCR experiments using cDNAs prepared from control, c01084Δ, c01084/Df and homozygous c01084 flies. Although we were able to amplify and sequence control and c01084Δ cDNA fragments using primers in CG15133 and CG15134, primers in CG15134 and CG4841 and primers in CG15133 and CG4841 (Figure 2C, data not shown and supplemental Table 1), no such fragments could be recovered from homozygous c01084 animals. Hence, our data indicate that CG15133, CG15134 and CG4841, can form one transcript. Furthermore, the PiggyBac (c01084) insertion in CG15133 greatly reduces or abolishes expression of these three separately annotated CGs, suggesting it is a strong hypomorphic or null allele of tweek. Hence, CG15133, CG15134 and CG4841 are jointly transcribed as a large transcript and this transcript corresponds to tweek.
The 5’UTR of tweek is not annotated and we therefore performed Rapid Amplification of 5’ Complementary DNA Ends (5’RACE). We were able to amplify 108 bp 5’ of the ATG start codon in CG15133, identifying the 5’UTR of tweek. These data also place the c01084 insertion inside tweek and we therefore renamed c01084 as tweekc01084. However, the previously annotated second exon of CG15133 does not appear amplifiable in our assays and the gene structure based on our RT-PCR and 5’RACE results is shown in Figure 2C.
We also found a 74 bp deletion and a 6 bp insertion leading to a premature stop codon in tweek1 and an AG to AA splice acceptor mutation before exon 20 in tweek2, predicting a frame shift (Figure 2C). This splice acceptor mutation in tweek2 was also confirmed by RT-PCR (Figure S1 and supplemental Table 1). Since tweekc01084/tweek1, tweekc01084/tweek2, tweek1/Df(2L)Exel8036, tweek2/Df(2L)Exel8036, tweekc01084/ Df(2L)Exel8036 and tweek2/tweek2 show a similar lethal phase, our data suggest that tweekc01084, tweek1 and tweek2 are all severe hypomorphic alleles or null alleles of tweek (Figure 2E). Finally, tweek corresponds to CG15133, CG15134, and CG4841 as all mutations in trans over a Df can be rescued using P(acman) clones (Venken et al., 2006) that contain genomic DNA encompassing CG4841, CG15133 and CG15134 (Figure 1A, green bar in Figure 2B and supplemental Table 2); however, the tweek alleles cannot be rescued with a 21 kb construct encompassing the CG4841 sequence (red bar in Figure 2B). Based on sequence annotation and RT-PCR data, our data indicate that the tweek mRNA transcript is 16,124 bp and the Tweek protein encompasses and ORF of 5,076 amino acids (~565 kDa). Although tweek encodes a very large and evolutionary conserved protein, it contains no known protein domains or motifs, including transmembrane domains or nuclear localization signals. Nonetheless, BLAST searches reveal that Tweek has homologues from nematode to man (Figure S2). In most species including various Drosophilids, mosquitoes, and mouse, the three CGs identified as tweek, correspond to two or three adjacent transcripts annotated as two or three genes. In human, the tweek homologue is annotated as a single 138 kb gene encoding a very large protein named fsa (Cao et al., 2006; Kuo et al., 2006). Although the predicted amino acid sequence of Drosophila Tweek aligns well with its human counterpart over the full length protein (e.g. 30 % identity with mouse Tweek), there are numerous regions that show much higher similarity and identity than the remaining areas (Figure S2). Hence, the sequence of Tweek does not reveal any particular information about its possible function.
To determine the expression pattern of tweek we performed in situ hybridization to embryos. As shown in Figure 2D, tweek is widely expressed but enriched in the brain lobes and in the ventral nerve cord after stage 14 of embryogenesis. We also observe expression in the midgut anlagen. Expression in the CNS becomes stronger in stage 15 embryos. These data are consistent with RT-PCR analyses that reveal expression of human Tweek/Fsa in the brain (McKay et al., 2003). Anti-sense dioxygenin labeled RNA probes complementary to the different 2 CGs show identical RNA expression patterns (Figure 2D and Figure S3). Moreover, the gene located 5’ of CG15133 reveals a very different expression pattern than that of the tweek CGs (data not shown). Hence, tweek encodes a very large transcript that is expressed in the nervous system.
tweek mutants display defects in neurotransmitter release upon repetitive stimulation
Drosophila mutations that affect synaptic vesicle exocytosis show reduced neurotransmitter release during low frequency stimulation, (e.g. SNAP25, syntaxin, CSP, hip14, cacaphony) whereas mutations that affect endocytosis or recycling (e.g. endophilin, AP180, synaptojanin, eps15, dap160, synapsin) do not affect release under such conditions. Hence to determine if tweek affects synaptic transmission we first stimulated motorneurons at low frequency in the presence of 1 or 5 mM Ca2+ and recorded excitatory junctional potentials (EJPs). Neither the resting membrane potential, nor the amplitude of the EJPs recorded during low frequency stimulation (<1 Hz) in various tweek mutants are different from controls (Figure 3A and Figure S4A, B). These data indicate that tweek mutations do not dramatically impair exocytosis under these conditions.
Figure 3. Synaptic vesicle endocytosis is impaired in tweek mutants.
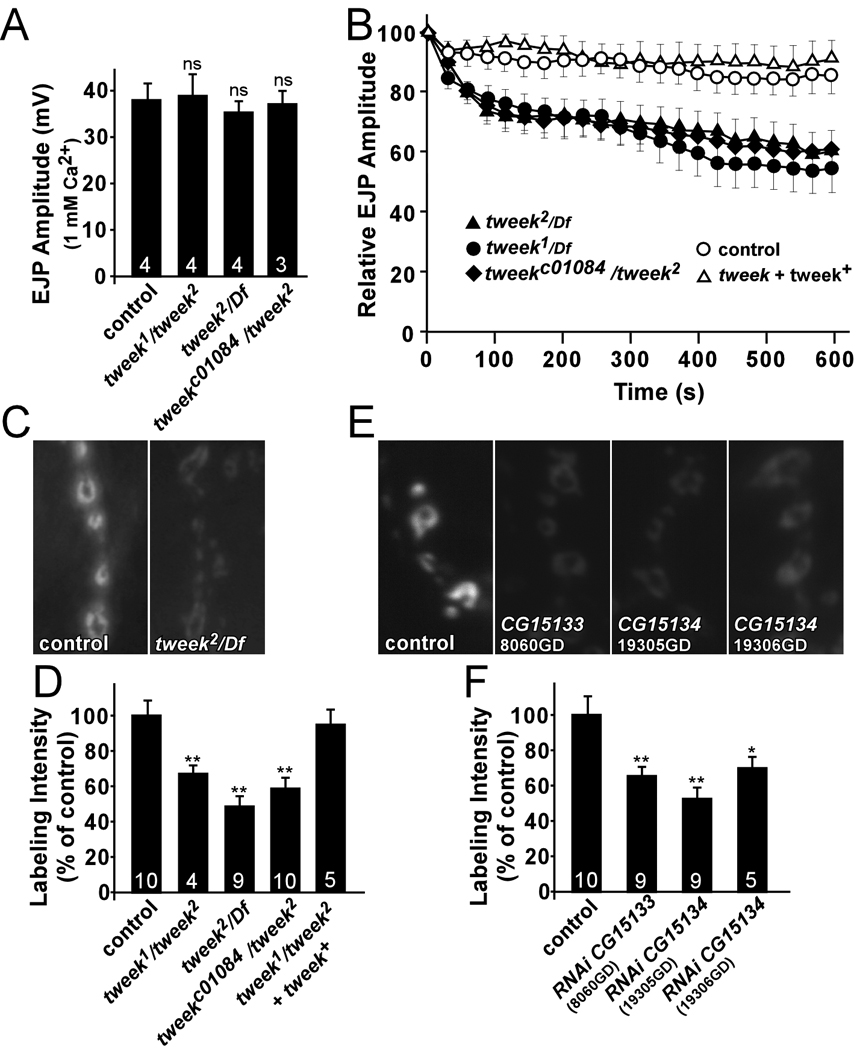
(A) Average EJP amplitude recorded in 1 mM Ca2+ in controls, tweek1/tweek2, tweek2/Df and tweekc01084/tweek2. Recordings were performed for 1 min at 1 Hz and 60 EJP amplitudes were averaged per recording. Under these conditions, there are no exocytic defects in tweek mutants.
(B) Average EJP amplitudes recorded at 10 Hz for 10 min in 5 mM external Ca2+ in controls, tweek1/Df, tweek2/Df, tweekc01084/tweek2 and tweek1/tweek2; tweek+(+HB69) rescued animals. Average EJP amplitudes (binned per 30 s) are normalized to the initial response (an average of the first 5 EJPs).
(C–F) FM 1–43 dye uptake in controls (yw; P{y+} FRT40A) (C,D) or (UAS-DCR2 / w1118; nSyb-Gal4 / +) (E,F), tweek1/tweek2 mutants (yw eyFLP; tweek1 P{y+} FRT40A / tweek2 P{y+} FRT40A), tweek2/Df mutants (eyFLP; tweek2 P{y+} FRT40A / Df(2L)Exel8036), tweekc01084 / tweek2 mutants (eyFLP; tweek2 P{y+} FRT40A / tweekc01084), tweek1/tweek2 mutants with a rescue construct (yw eyFLP; tweek1 P{y+} FRT40A / tweek2 P{y+} FRT40A; tweek+(HB69)/+) (C,D) and flies that express RNAi directed against TWEEK (UAS-DCR2 / w1118; 8060GD / +; nSyb-Gal4 / + or UAS-DCR2 / w1118; nSyb-Gal4 / 19305GD or 19306GD) (E,F). Preparations were incubated in 4 µM dye and were stimulated with 1 min of 90 mM KCl to label the exo-endo cycling pool.
* p<0.05; ** p<0.01 (t-test), ns: not significant, Error bars: SEM, n (the number of animals tested) is indicated in the bars.
Next we tested the ability of tweek mutants to maintain release during intense stimulation. As shown in Figure 3B, EJP amplitudes in tweek mutants declined to about 55–60% of the initial response when stimulated at 10 Hz in 5 mM Ca2+. In contrast, controls maintain release at about 90% of the initial response after 10 min at 10 Hz (Figure 3B). The inability of tweek mutants to maintain normal levels of transmission during intense activity is consistent with a defect in vesicle trafficking or recycling.
Tweek is required in neurons for proper FM1–43 dye uptake
Synaptic vesicle density in tweek mutant PRs is lower than in controls (Figure 1), suggesting a defect to maintain a normal synaptic vesicle pool. To assess if vesicle recycling is affected, we performed live imaging with FM 1–43 dye at third instar NMJs (Ramaswami et al., 1994). In aqueous environments, FM 1–43 is non-fluorescent, but when bound to membranes it increases its fluorescence quantum yield. Hence, newly endocytosed vesicles in the presence of FM 1–43 will be fluorescently labeled, providing a quantitative measure of vesicle uptake (Betz and Bewick, 1992; Verstreken et al., 2007). As shown in Figure 3C and D and Figure S4, when controls, including tweek mutants that carry the genomic rescue construct, are stimulated for 1 min with 90 mM KCl or for 10 min with 10 Hz nerve stimulation in the presence of FM 1–43, synapses are brightly labeled, indicating efficient vesicle retrieval from the membrane during stimulation. In contrast, tweek mutant synapses are labeled less.
To test if this defect is caused by loss of tweek in motor neurons we expressed different RNAi constructs (VDRC) (Dietzl et al., 2007) against tweek. RT-PCR and quantitative RT-PCR shows that expression of three RNAi constructs leads to decreased TWEEK mRNA levels (Figure S5 and data not shown). We stimulated synapses of animals that express these RNAi constructs in neurons using nsyb::Gal4 for 1 min in 90 mM KCl and assessed FM 1–43 dye uptake efficiency. As shown in Figure 3E and F, expression of 8060GD (CG15133), 19305GD (CG15134) or 19306GD (CG15134) results in a significant reduction of FM 1–43 dye uptake, whereas expression of RNAi constructs that do not significantly affect tweek expression show normal uptake. These data indicate that tweek acts in the presynaptic neuron and are consistent with an endocytic defect and/or a reduced synaptic vesicle pool.
tweek affects synaptic vesicle density and size
To further explore presynaptic defects in tweek mutants we performed TEM at third instar NMJ boutons. As shown in Figure 4, synaptic vesicle number is reduced in tweek mutant boutons. While many synaptic features such as mitochondrial number or structure (Figure 4A–C, G), dense body number (T-bars) (Figure 4H–I) dense body morphology (Figure 4 E and F) and the number of docked synaptic vesicles within a 200 nm radius around dense bodies (Figure 4E–F, J) appear normal in tweek mutants compared to controls, vesicle counts reveal ~64% less vesicles per unit area (Figure 4A–D, K). tweek boutons also contain more large diameter vesicles than controls: the diameter of synaptic vesicles is typically <50 nm in controls (Figure 4A, L–M), but tweek mutants contain numerous vesicles and cisternae that exceed this diameter (Figure 4B–D, L–M). Hence, loss of Tweek, similar to endocytic mutants AP180/lap, dap160, eps15, and stoned (Chen et al., 1998; Fergestad et al., 1999; Gonzalez-Gaitan and Jackle, 1997; Stahelin et al., 2003; Stimson et al., 2001; Zhang et al., 1998), leads to reduced vesicle density and causes the formation of larger and more heterogeneously-sized vesicles.
Figure 4. Vesicle size and number are altered in tweek mutants.
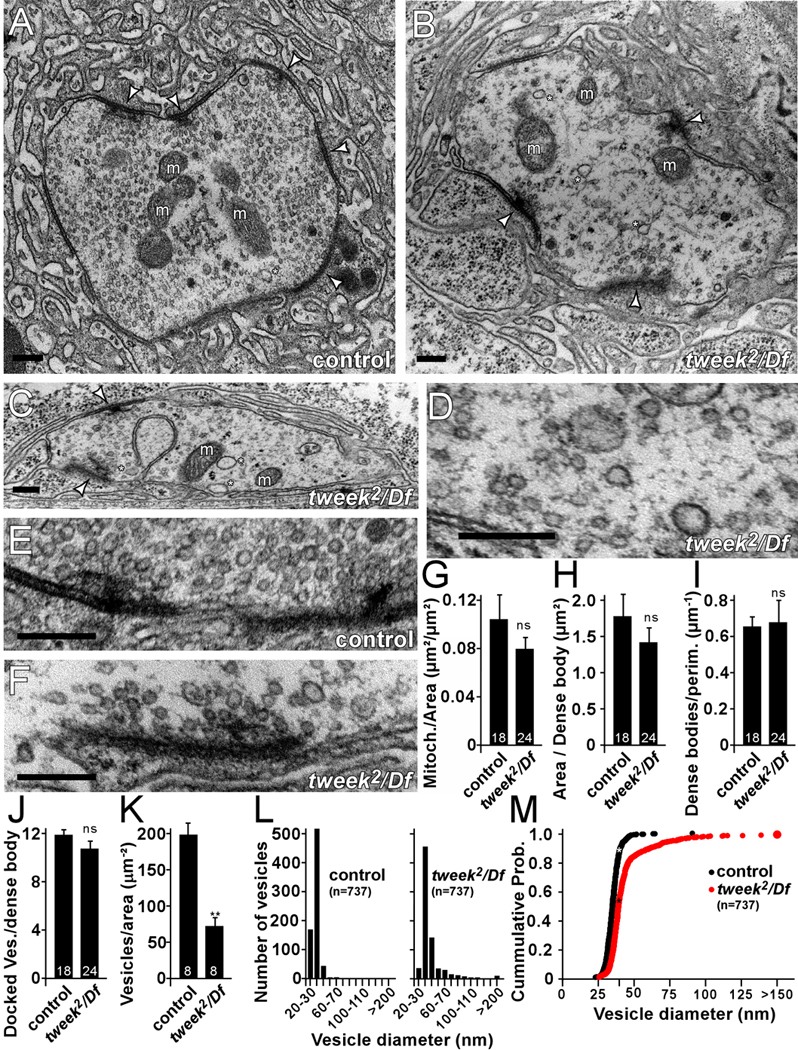
(A–F) Ultrastructure of control (yw; P{y+} FRT40A) (A, E) and tweek2/Df (yw eyFLP; tweek2 P{y+} FRT40A / Df(2L)Exel8036) mutants (B–D, F) NMJ boutons (A–D) and dense bodies (E–F). Note the reduced synaptic vesicle density and the heterogeneity in synaptic vesicle size (asterisks) in the mutants compared to the control. Dense bodies (arrowheads) and mitochondria (m) are marked. Scale bars are 200 nm.
(G–K) Quantification of ultrastructural features: mitochondrial density (G), boutonic area per dense body (H), dense bodies per perimeter bouton (I), number of docked vesicles synaptic in a 200 nm radius around the dense body (J) and synaptic vesicle density (K). ** p<0.01 (t-test), ns, not significant; Error bars: SEM, The number of analyzed sections is indicated in the bars and images were acquired from 8 boutons in 5 different animals.
(L–M) Histograms of synaptic vesicle diameter in controls and tweek2/ Df and cumulative histogram of synaptic vesicle diameters indicating larger vesicles in tweek mutants. 737 vesicle diameters were measured for each genotype.
The fusion of large synaptic vesicles should lead to an increased amount of neurotransmitter released per single vesicle fusion event (Zhang et al., 1998). We therefore recorded spontaneous vesicle fusion events (minis) in current clamp mode (miniature excitatory junctional potentials - mEJPs) (Figure 5A) as well as in voltage clamp mode (miniature excitatory junctional currents - mEJCs) (Figure 5B–C, F). As shown in a cumulative probability plot of mEJP amplitudes (Figure 5A) or by calculating the average mEJP (not shown) and mEJC amplitudes (Figure 5B), mini amplitudes in tweek mutants are larger compared to controls. In addition, we also find a significant reduction in mini frequency (Figure 5C). These data suggest that single vesicle fusions can elicit larger responses in tweek mutants compared to controls. Since GluRIIA and GluRIIC/III immunohistochemical staining of post synaptic glutamate receptors (Marrus et al., 2004; Schuster et al., 1991) do not show differences in receptor cluster size or cluster intensity between tweek and controls (GluRIIC/III cluster intensity in control: 100±8.0% and in tweek1/tweek2: 88.8±1.5, t-test: p=0.29), synaptic vesicles in tweek mutants release abnormally large quantities of neurotransmitters in agreement with an increased vesicle size in the mutants.
Figure 5. Quantal size is increased in tweek mutants.

(A) Cumulative histogram of mEJPs measured from controls (black: yw; P{y+} FRT40A) and tweek1/tweek2 (red: yw eyFLP; tweek1 P{y+} FRT40A / tweek1 P{y+} FRT40A) animals. Note the rightward shift in tweek mutants signifying larger mEJP amplitudes.
(B–C) Average mEJC amplitude (B) and frequency (C) in controls and tweek mutants (tweek2/Df :yw eyFLP; tweek2 P{y+} FRT40A / Df(2L)Exel8036 and tweekc01084/ tweek2: yw eyFLP; tweek2 P{y+} FRT40A / tweekc01084).
(D) Average EJC amplitude in controls and tweek mutants recorded in 1 mM extracellular Ca2+.
(E–F) Sample EJC (E) and mEJC (F) traces recorded from controls and tweek mutants. Recordings were performed for 1 min at 0.1 Hz and all EJC amplitudes were averaged per recording.
(G) Junctional quantal content at 1 mM Ca2+ calculated by dividing the average EJC amplitude by the average mEJC amplitude. ** p<0.01 (t-test), Error bars: SEM, n (the number of animals tested) is indicated in the bars.
(H) Estimation of vesicular membrane added per stimulus in 1 mM extracellular Ca2+ calculated by multiplying the quantal content (control: 189 quanta; tweek2/ Df: 120 quanta) by the average vesicle surface area based on TEM in Figure 4 (control: vesicle radius 16.7 nm; tweek2/ Df: vesicle radius 23.8 nm).
EJP amplitudes are similar in controls and tweek mutants but mini amplitudes are not. Hence, the number of vesicles released per stimulus (quantal content) is likely altered in tweek, possibly as a result of homeostatic regulation (Davis, 2006). To calculate the quantal content we recorded EJC amplitudes in 1 mM Ca2+ (Figure 5D–E) and divided the average EJC amplitude by the average mEJC amplitude (quantal amplitude; Figure 5B). Our data indicate that the quantal content in tweek is reduced by 31–37% compared to controls (Figure 5G). However, as vesicles in tweek are larger, the amount of membrane added per stimulus is comparable or even slightly increased in tweek when compared to controls (Figure 5H). Hence, during stimulation, mutants and controls release similar amounts of synaptic vesicle membrane. Since FM1–43 dye uptake is reduced, these data further support a defect in endocytosis or recycling in tweek mutants.
Proteins required for proper clathrin coat assembly are decreased at tweek NMJs
α-adaptin, AP180/Lap and StonedB are adaptor-like proteins that have been implicated in early steps of vesicle recovery and mutations in the genes that encode these proteins lead to qualitatively similar phenotypes as tweek (Fergestad et al., 1999; Gonzalez-Gaitan and Jackle, 1997; Stimson et al., 2001; Zhang et al., 1998). We therefore assessed the levels of these proteins in tweek mutant boutons (tweek2/ Df(2L)Exel8036, tweek1/tweek2 and for α-adaptin also tweek2/tweekc01084). The levels of these proteins are markedly reduced in tweek NMJ boutons (Figure 6A–C and G and data not shown). This decrease is not due to reduced expression levels of these proteins, as Western Blots using whole larval extracts did not show obvious differences between tweek and controls (Figure 6I and quantification in the legend).
Figure 6. Endocytic adaptor proteins are destabilized at tweek mutant boutons.
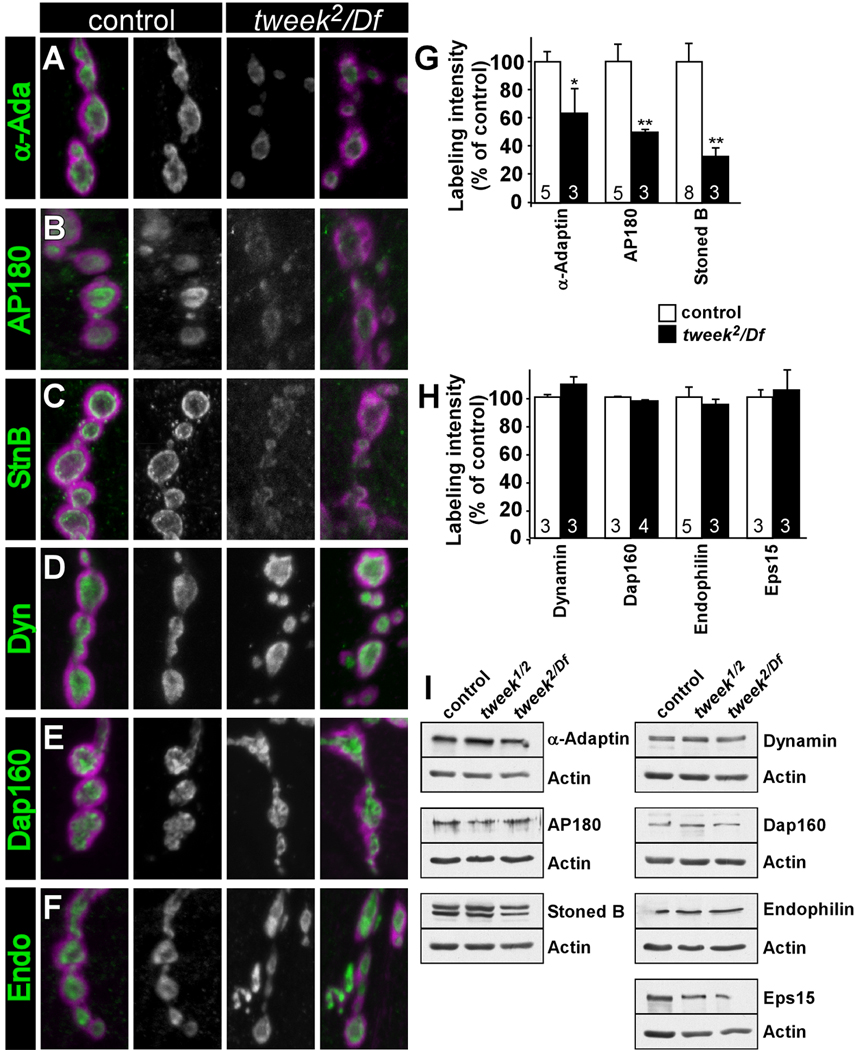
(A–F) Confocal images showing labeling of control (yw; P{y+} FRT40A) (left) and tweek2/Df (yw eyFLP; tweek2 P{y+} FRT40A / Df(2L)Exel8036) (light) larval filets with α-adaptin (A), AP180/Lap (B), StonedB (C), Dynamin (D), Dap160/Intersectin (E) Endophilin (F) (green) and DLG/PSD-95 (magenta). Green channel labeling for control and tweek2/Df is shown in the middle (gray scale).
(G–H) Quantification of boutonic labeling intensity (inside the respective DLG circumscribed areas) for markers shown in (A–F) and for Eps15. Data for tweek1/tweek2 mutants (yw eyFLP; tweek1 P{y+} FRT40A / tweek2 P{y+} FRT40A) is very similar to tweek2/Df mutants (not shown). * p<0.05, ** p<0.01 (t-test), Error bars: SEM, n (the number on animals labeled) is indicated in the bars.
(I) Western Blots of larval extracts of controls, tweek1/tweek2 and tweek2/Df using antibodies against the endocytic proteins tested in (A–H). Protein loading amounts were tested with anti-actin antibodies. Quantification of 3 independent Westerns normalized to actin loading control (values relative to control levels): α-adaptin: control: 100.0±12.7%; tweek1/tweek2: 95.8±19.1%; tweek2/Df(2L)Exel8036: 86.4±20.3%; p:ns. Lap/AP180: control: 100.0±6.4%; tweek1/tweek2: 94.2±8.9%; tweek2/Df(2L)Exel8036: 82.3±21.2%; p:ns. stonedB: control: 100.0±23.0%; tweek1/tweek2: 87.4±15.3%; tweek2/Df(2L)Exel8036: 81.5±2.4%; p:ns.
To assess if tweek mutations affect the localization of other endocytic proteins we labeled NMJs with antibodies against Dynamin (van der Bliek and Meyerowitz, 1991), Dap160, Eps15 (Koh et al., 2004; Majumdar et al., 2006; Marie et al., 2004; Roos and Kelly, 1999) and Endophilin (Verstreken et al., 2002). As shown in Figure 6D–F and H, tweek does not affect the level or localization of these proteins. In summary, Tweek mediates the normal recruitment, retention and/or stability of several endocytic adaptor-like proteins that are intrinsic components of the clathrin coat.
PI(4,5)P2 availability is reduced at tweek NMJs
Several endocytic adaptors have been shown to interact with phosphorylated inositides (e.g. PI(4,5)P2 - reviewed in (Lemmon, 2003; Wenk and De Camilli, 2004). One possibility therefore is that Tweek affects the levels or distribution of synaptic PIs, thought to be critical for vesicle recycling (Cremona et al., 1999; Di Paolo et al., 2004; Micheva et al., 2001; Verstreken et al., 2003). To estimate the PI levels at tweek NMJ boutons we expressed a PI-interacting protein domain fused to EGFP that reports PI(4,5)P2 levels in vivo (Jost et al., 1998; Varnai and Balla, 2006).
To visualize PI(4,5)P2 we cloned an EGFP-fused pleckstrin homology (PH) domain of PLCδ1, known to bind PI(4,5)P2 (Varnai and Balla, 1998), in pUAST and expressed it using the UAS-GAL4 system. To test the specificity of this probe, we also expressed a mutant PLCδ-PHmut-EGFP that cannot bind PI(4,5)P2 (Varnai and Balla, 2006). As shown in Figure S6A, the wild type PLCδ-PH-EGFP probe decorates the membrane in salivary gland cells, whereas the mutant PLCδ-PHmut-EGFP is diffusely present in the cytoplasm and concentrates in the nucleus (Figure S6B). Furthermore, in neurons of the larval ventral nerve cord and in the adult brain, PLCδ-PH-EGFP is present in the neuropil synapses (Figure S6C). In contrast, the mutant PLCδ-PHmut-EGFP is diffusely present in the cytoplasm, labeling mostly the cell bodies and less the neuropil (Figure S6D and data not shown). These data indicate that in Drosophila the PLCδ-PH-EGFP probe is enriched at synapses (Micheva et al., 2001).
To explore possible changes in levels or distribution of PI(4,5)P2 at the NMJ, we used elav-Gal4 to express the PLCδ-PH-EGFP probe in the nervous system of tweek mutants and wild type controls. In wild type controls the probe is present at synaptic boutons and enriched on their membranes (Figure 7A). We also expressed the probe in synj mutants as a positive control to assess the ability of the probe to reveal changes in PI(4,5)P2. Synj is a pre-synaptic phosphoinositide phosphatase, and mouse as well as fly synj mutants show an overall increase in the concentration of PI(4,5)P2 (Cremona et al., 1999; Voronov et al., 2008; L.S. and P.D., unpublished results). In agreement with these observations, the signal produced by PLCδ-PH-EGFP at synj1 mutant synapses is increased (Figure 7B and D). In contrast to synj and to controls, the levels of PLCδ-PH-EGFP fluorescence expressed using elav-Gal4 are decreased in tweek mutants (Figure 7C and D, Figure S7). This difference is not due to an effect of tweek on GFP fluorescence as the levels and localization of untagged GFP, expressed in neurons, is not significantly different in tweek versus controls (Figure 7M and N). Hence, these results suggest that Tweek is required to maintain normal levels or availability of PI(4,5)P2 at Drosophila NMJs.
Figure 7. Synaptic PI(4,5)P2 levels are reduced in tweek mutants.
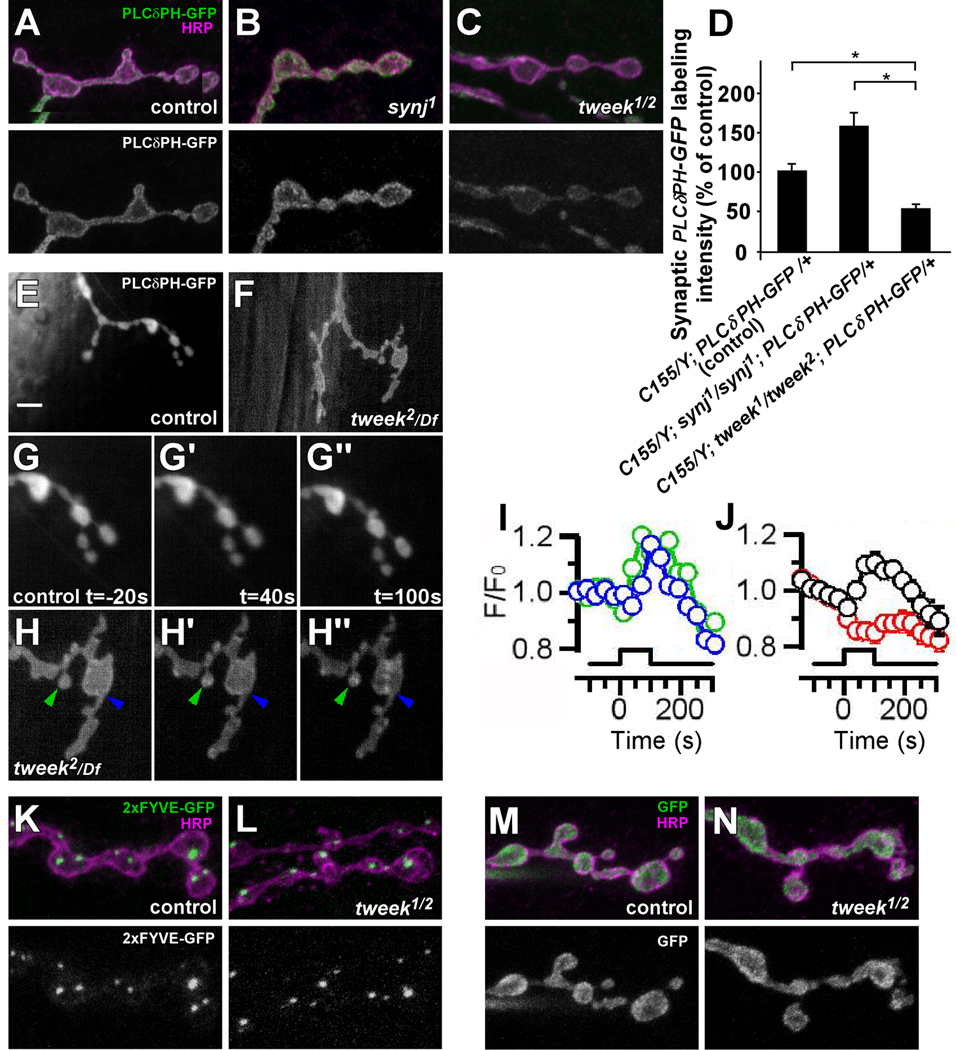
(A–C) Neuronal PI(4,5)P2 levels in boutons of control (elav-GAL4/Y; UAS-PLCδ-PH-EGFP / +), synj1 (elav-GAL4/Y; FRT42D synj1; UAS-PLCδ-PH-EGFP / +) and tweek1/tweek2 (elav-GAL4/Y; tweek1 P{y+} FRT40A / tweek2 P{y+} FRT40A; UAS-PLCδ-PH-EGFP / +) third instar filets were visualized by a PLCδ-PH-EGFP probe (green). Neuronal membranes were counterstained with anti-HRP (magenta). Green channel is separately shown on the bottom. Note increased EGFP levels in synj1 mutants and decreased EGFP levels in tweek mutants.
(D) Quantification of PLCδ-PH-EGFP intensity shown in (A–C) inside the volume demarcated by anti-HRP labeling in the indicated genotypes. * p<0.05 (t-test)
(E–H) Live imaging of PLCδ-PH-EGFP localization before (E, F, G and H) and after 40s (G’, H’) or 100s (G”, H”) of 20 Hz stimulation of (E and G–G”) control (yw /w; FRT40A /+; UAS-PLCδ-PH-EGFP, nsyb-Gal4/+) and (F and H–H”) tweek2/Df (yw /w; tweek2 FRT40A / Df(2L)Exel8036; UAS-PLCδ-PH-EGFP, nsyb-Gal4/+) third instar NMJs. GFP imaging was performed with a CCD camera and G and H are magnifications of E and F respectively. Controls never show PLCδ-PH-EGFP clusters (n= 7 animals) whereas numerous PLCδ-PH-EGFP clusters (green and blue arrowhead) appear in tweek mutants during stimulation. Such clusters are not observed using the PLCδ-PHMUT-EGFP, indicating specificity. Scale bar: 5 µm.
(I–J) Quantification of the fluorescence of PLCδ-PH-EGFP clusters during stimulation (start at t=0, marked by the bar). (I) Green and blue traces show the normalized fluorescence of individual clusters indicated in H–H”, showing that some clusters form early in the stimulus (green) and others form later (blue). Note also that clusters remain for an extended period of time (>100 s) following stimulation before they dissapear (J) Black trace shows the average ± SEM for all clusters formed in the tweek2/Df experiments (23 clusters, 4 animals). The red trace shows the corresponding change in fluorescence in the remainder of the terminal of tweek2/Df, and this trace appears very similar to the one measured from control boutons where also no clusters were observed (G–G”).
(K–L) Neuronal PI(3)P levels in boutons of control (elav-GAL4/Y; UAS-2xFYVE-EGFP / +) or tweek1/tweek2 mutants (elav-GAL4/Y; tweek2 UAS-2xFYVE-EGFP / tweek1 P{y+} FRT40A) third instar filets were visualized with a 2xFYVE-EGFP probe (green). Neuronal membranes were counterstained with anti-HRP (magenta). Green channel is separately shown on the bottom.
(M–N) Neuronal GFP levels in boutons of control (elav-GAL4/Y; UAS-GFP / +) or tweek1/tweek2 mutant (elav-GAL4/Y; tweek2 UAS-GFP / tweek1 P{y+} FRT40A) third instar filets (green). Neuronal membranes were counterstained with anti-HRP (magenta). Green channel is separately shown on the bottom.
To analyze the dynamics of PI(4,5)P2 localization in control and tweek mutants during vesicle cycling we performed live imaging of PLCδ-PH-EGFP driven by the strong neuronal nsyb-Gal4 driver, while stimulating the motor nerve for 100 s at 20 Hz. In controls PLCδ-PH-EGFP distribution during stimulation shows no obvious changes when compared to synapses at rest whereas the PLCδ-PH-EGFP distribution in tweek mutants shows obvious differences (Figure 7E–H). Prior to stimulation the distribution of PLCδ-PH-EGFP in controls and tweek mutants are similar. Note however that the levels of PLCδ-PH-EGFP are decreased when compared to controls (Figure 7E–H). During stimulation, PLCδ-PH-EGFP redistributes and concentrates in presynaptic clusters in tweek mutants, while the mutant PLCvδ-PHmut-EGFP probe does not concentrate in such clusters (Figure 7H, H’ and H” and data not shown). These structures in tweek mutants often persist for ~100 s after cessation of the stimulation (Figure 7I and J). The clusters may represent lingering endocytic intermediates that still harbor PI(4,5)P2. Hence, the data suggest that Tweek not only affects the basal levels of PI(4,5)P2 but also its synaptic distribution during neuronal activity.
To test if Tweek affects the availability of other PIs we also expressed 2xFYVE-EGFP, a marker for PI(3)P in the nervous system (Wucherpfennig et al., 2003). PI(3)P is enriched on endosomes but limited amounts may also be present on the plasma membrane. However, the expression of 2xFYVE-EGFP in controls and tweek mutants is not significantly different (Figure 7K and L), indicating that tweek does not affect the localization or abundance of PI(3)P.
Synaptojanin mutations partially suppress endocytic defects in tweek
To provide additional in vivo evidence that the availability of PI(4,5)P2 is reduced in tweek, we tested vesicle uptake in tweek mutants with reduced PI phosphatase activity by removing one copy of synj. Removal of a single copy of synpatojanin may elevate the reduced PI(4,5)P2 levels observed in tweek mutants providing strong genetic evidence for a direct link of Tweek to vesicle recycling and PIPs. To assess endocytosis in these animals, we stimulated synapses for 5 min with KCl in the presence of FM 1–43, and measured labeling intensity. Using this protocol, dye uptake in synj/+ heterozygotes and controls is indistinguishable, indicating no dominant effect of the synj1 mutation on endocytosis (Figure 8A, D). Interestingly, tweek mutants that lack one copy of synj (tweek1 synj1/ tweek2 or tweek2 synj1/ Df(2L)Exel8036) take up significantly more dye than tweek mutants (tweek1/ tweek2 or tweek2/ Df(2L)Exel8036) (Figure 8B–D). However, dye uptake in tweek mutants that are heterozygous for synj is still less than in controls (eyFLP; FRT40A) or in synj/+. These data indicate that the levels of PI(4,5)P2 can be genetically manipulated to affect FM 1–43 dye uptake.
Figure 8. Removal of a single mutant copy of synaptojanin suppresses endocytic defects in tweek.

(A–D) FM 1–43 dye uptake experiment on synj1/+x controls (yw ey-FLP; FRT42D synj1 / FRT42D), tweek2/Df mutants (yw eyFLP; tweek2 P{y+} FRT40A / Df(2L)Exel8036) and tweek2/Df mutants that lack one functional copy of the synj gene (yw ey-FLP / yw; tweek 2 synj1 / Df(2L)Exel8036). Preparations were stimulated in 90 mM KCl for 5 min, washed and imaged (A–C) and labeling intensity quantified (D). Dye uptake in synj1/+ and yw eyFLP; FRT40A controls is indistinguishable (shown in D). Note the increased FM 1–43 dye uptake in tweek mutants with reduced synj function compared to tweek mutants. To measure PI(4,5)P2 levels in synj1 and synj1/+ animals we expressed the PLCδ-PH-EGFP probe and measured boutonic fluorescence relative to controls: 100±12% for controls (elav-GAL4/Y; P{w+UAS-PLCδ-PH-EGFP} / +); 157±20% p<0.05 for synj1 (elav-GAL4/Y; P{neoFRT}42D synj1; P{w+UAS-PLCδ-PH-EGFP} / +); 117±8% p<0.1 for synj1/+ (elav-GAL4/Y; P{neoFRT}42D synj1/+; P{w+UAS-PLCδ-PH-EGFP} / +).
(E–H) α-adaptin labeling in synj1/+ controls, tweek2/Df mutants and tweek2/Df mutants that lack one functional copy of the synj gene (tweek2 synj1/Df). (H) Quantification of α-adaptin labeling intensity. α-adaptin labeling in synj1/+ and yw eyFLP; FRT40A controls is indistinguishable (shown in D). Note the increased α-adaptin labeling in tweek mutants with reduced synj function compared to tweek mutants. * p<0.05, ** p<0.01 (t-test), Error bars: SEM, n (the number on animals labeled) is indicated in the bars.
We also tested if the removal of one copy of synj in tweek mutants partially restores α-adaptin levels in boutons. While tweek mutants (tweek1/ tweek2, tweek2/ Df(2L)Exel8036 or tweek2/ tweekc01084) show reduced α-adaptin levels in synaptic boutons (Figure 6A and G, and Figure 8F and H), removing one copy of synj (tweek1 synj1/ tweek2, tweek2 synj1/ Df(2L)Exel8036 or tweek2 synj1/ tweekc01084) partially restores α-adaptin levels (Figure 8 E–H). Hence, levels of an endocytic adaptor protein in tweek mutant NMJ synapses can be restored by decreasing phosphoinositide dephosphorylation activity, consistent with the observation that the availability of phosphorylated inositides is reduced in tweek mutants.
Discussion
In an unbiased genetic screen to identify genes that affect neuronal communication we have identified mutants that affect different aspects of synaptic function (Hiesinger et al., 2005; Koh et al., 2004; Verstreken et al., 2003). In the same screen, we also identified tweek, which encodes a very large protein without established protein motifs. Interestingly, a C. elegans tweek homologue lpd-3 which encodes the carboxyterminal end of Tweek, but is most likely part of a much larger gene (including Y47G6A.23 and Y47G6A.29), was previously identified in an RNAi screen for lipid storage defects (McKay et al., 2003). However, the molecular nature underlying the RNAi phenotype in worms has not been investigated. Our data indicate that tweek plays a role in synaptic vesicle recycling likely by regulating PI-lipid signaling at the synapse.
The Tweek protein is unusually large as it encodes a protein of 5,076 amino acids. Interestingly, Tweek does not contain any known motifs, and hence cloning of the gene did not reveal any hints about its potential function or localization. Note that the three CGs that form the tweek transcript are also found clustered in other species. These tweek homologues have most likely been wrongly annotated in almost all species (except human) as separate genes. No full length cDNA has been reported in any species and when expression patterns are available of portions of the tweek homologs, they are localized in the nervous system and fat tissue (Chintapalli et al., 2007; Manak et al., 2006; McKay et al., 2003). In flies, the TWEEK RNA transcript is also expressed in the CNS, but localization of the protein could not be established as we were unable to raise an antibody upon injection of various antigens in 10 animals. Similarly, localization of the protein using a mCherry-tagged genomic construct that can rescue the tweek mutations failed, even when we tried to enhance signal using antibodies against the tag (data not shown). Taken together these data suggest that the protein derived from the tweek locus is not abundant and this may explain why it has not been identified previously through biochemical approaches.
tweek mutants harbor the hallmarks of mutants that affect endocytosis. Electrophysiological analyses show that tweek mutants fail to maintain neurotransmitter release during intense stimulation and internalize less FM 1–43 dye, indicating a smaller vesicle pool. They also display reduced synaptic vesicle numbers and aberrantly large vesicles. We used Synaptophluorin (Poskanzer et al., 2003) to further test membrane recycling following stimulation (0.2 s at 50 Hz), but observed only a slightly slower and statistically insignificant decay in fluorescence quenching in tweek mutants compared to controls. While these data suggest that part of the recycling defect in tweek mutants may occur after newly endocytosed vesicles are acidified, we also did not observe a difference in Synaptophluorin fluorescence decay in dap160 endocytic mutants when compared to controls when stimulated using the same paradigm (Supplemental figure 8). These data suggest that Synaptophluorin may not be able to detect the defects in membrane recycling in these ‘endocytic’ mutants.
While some of the phenotypes we observe in tweek mutants may be recapitulated in other fly mutants (Daniels et al., 2006; Daniels et al., 2004), the tweek mutants are phenotypically much more similar to mutants that affect endocytosis. Indeed, stoned, AP180, dap160, and eps15 mutants all exhibit reduced vesicle numbers (Fergestad et al., 1999; Ferguson et al., 2007; Koh et al., 2004; Majumdar et al., 2006; Marie et al., 2004; Nonet et al., 1999; Stimson et al., 2001; Zhang et al., 1998). These phenotypes combined with the aberrant localization of endocytic adaptor proteins in tweek mutants is consistent with the hypothesis that Tweek at least in part affects vesicle recycling early, when adaptor proteins are recruited to the plasma membrane, and this is bolstered by the dominant genetic interaction with synj. However, we cannot exclude additional roles for the protein at later steps of the vesicle cycle.
Both synaptic vesicle exocytosis and endocytosis are critically dependent on PIs present at synaptic membranes (Di Paolo et al., 2004; Schiavo et al., 1996; Verstreken et al., 2003). PI(4,5)P2 in the plasma membrane is thought to be a major anchor point for endocytic adaptor proteins that link clathrin to this membrane (Cremona et al., 1999; Gaidarov et al., 1996). Reduced availability of PI(4,5)P2 in tweek mutants may lead to reduced coat nucleation at the synapse and therefore provides a rationale for the mislocalization of endocytic adaptor proteins in the mutants. While a biochemical link between Tweek, phosphoinositide availability and adaptor localization awaits further investigation, our model is consistent with the observation that increasing the PI(4,5)P2 levels in tweek mutants, by reducing synj expression, partially restores the localization of the AP2 adaptor. Note that not all PI(4,5)P2 interacting proteins are destabilized at tweek mutant synapses. For example Dynamin binds PI(4,5)P2, yet, it seems to be localized similarly in tweek and controls. Furthermore, the levels of Synaptotagmin, a synaptic vesicle protein known to interact with PI(4,5)P2, appears similar in controls and tweek mutants (data not shown). We surmise that while PI binding may be mediating the function of these proteins (Achiriloaie et al., 1999; Bai et al., 2004; Zoncu et al., 2007), they may require less PI(4,5)P2 than adaptor proteins.
Although a large body of evidence demonstrates a role for PI(4,5)P2 in various cellular processes (Hassan et al., 1998; Horn, 2005; York, 2006), tweek mutant cells do not show signs of major cellular dysfunction beyond a defect to maintain synaptic transmission, indicating that the effect of tweek loss-of-function in neurons is rather specific to synaptic vesicle recycling. We and others have observed a similar remarkably specific phenotype in synj mutants (Cremona et al., 1999; Harris et al., 2000; Van Epps et al., 2004; Verstreken et al., 2003). Although synj mutants lead to an increase in phosphorylated PIs, defects at the NMJ appear to be specific to synaptic vesicle endocytosis. Hence, specific factors locally regulate the availability of specific PI lipids, even within the synapse. As a result, PI phosphatases, kinases and proteins like Tweek that may control PI availability must play important roles during vesicle recycling.
Methods
Genetics and Molecular Biology
Throughout the paper, control animals were isogenized y w; P{y+, ry+}25F P{neoFRT}40A unless otherwise indicated and tweek1 or 2 mutants were (y w P{ey-FLP.N}2; tweek1 P{y+, ry+}25F P{neoFRT}40A and y w P{ey-FLP.N}2; tweek2 P{y+, ry+}25F P{neoFRT}40A. Additional details on tweek alleles and identification of the tweek gene as well as generation of transgenic animals appear in the Supplemental Methods
Immunohistochemistry and Western Blotting
Labeling was performed following standard protocols and fluorescent images (including EGFP) were captured using a Zeiss 510 confocal microscope and imported in Amira 2.2 to adjust their brightness and contrast and then imported in Photoshop 7.0 to assemble them in figures.
Samples for Western Blots were prepared by crushing third instar larvae in modified HL-3 with 0.4% Triton-X-100 and proteinase inhibitors on ice, and then boiling them in sample buffer for 5 min. Western Blots were run and quantified according to standard protocols.
Antibodies and concentrations for immunohistochemistry/Western Blotting: α-adaptin (Gonzalez-Gaitan and Jackle, 1997) 1:500/1:5000; Dynamin (Upstate) 1:200/0.125µg/ml; Eps15 (Koh et al., 2007) 1:5000/1:10000; Endophilin (Verstreken et al., 2002) 1:200/1:5000; Lap (AP180) (Zhang et al., 1998) 1:150/1:2500; Dap160 (Roos and Kelly, 1999) 1:500/1:5000; StonedB (Phillips et al., 2000) 1:200/1:5000. Antibodies for immunohistochemistry: DLG (mouse; 4F3) (Parnas et al., 2001) 1:50; DLG (rabbit) (Kwang Choi, BCM) 1:500; HRP (Jackson Labs), 1:500; GluRIIA (mouse; 8B4) 1:50 (Schuster et al., 1991) and GLURIII/IIC 1:500 (Marrus et al., 2004). Cy3 (Jackson Labs) or Alexa 488 (Invitrogen) conjugated antibodies were used at 1:250. HRP conjugated antibodies (Jackson Labs) were used at 1:2500.
Labeling intensity of endocytic markers was quantified as described (Koh et al., 2004). Briefly, mutant and control samples were labeled together and imaged using identical settings. Pixel intensities inside the boutonic volumes, circumscribed by an independent marker were calculated using Amira 2.2. For quantification of endocytic markers we used DLG labeling as an independent outline of the boutonic volume and subtracted background labeling in the muscle.
For each lipid-GFP probe at least 3 controls and 3 mutant animals were labeled together with a synaptic marker (anti-HRP). GFP fluorescence of controls and mutants was imaged using identical settings, and using the synaptic marker as an outline of the synapse, we measured absolute GFP fluorescence intensity and normalized this to control values.
Electrophysiology and FM 1–43 dye uptake and live imaging
ERGs and current clamp (CC) recordings were performed as described (Verstreken et al., 2003; Verstreken et al., 2002). Two electrode voltage clamp (TEVC) recordings were made in modified HL-3 [(in mM) NaCl 110; KCl 5; NaHCO3 10; HEPES 5; Sucrose 30; Threalose 5; MgCl2 10; pH 7.2; CaCl2 as indicated in the text; for minis, we added 5 µM TTX. Resting membrane potentials were between −55 and −65 mV and were clamped at −65 mV. Voltage errors were <1.5 mV for 100 nA EJCs, input resistances were ≥ 4 MΩ and data was filtered at 1 kHz.
FM 1–43 dye uptake experiments were performed as described in the text and images were captured with a Zeiss MRm or on a Nikon Digital Sight DS2Mb-Wc camera using a 40x Zeiss or Nikon water immersion lens (NA 0.8) and analyzed and quantified as described (Verstreken et al., 2007).
Live imaging of PLCδ-PH-EGFP was performed as described in the supplemental methods
Transmission Electron Microscopy
TEM of PRs was performed as described in (Hiesinger et al., 2005) and TEM of NMJ boutons was performed as described (Verstreken et al., 2003). Data was analyzed using Image-J.
Supplementary Material
Acknowledgements
We are grateful to the Bloomington stock center, the Developmental studies hybridoma bank, VDRC, Kwang Choi, Aaron Di Antonio, Barry Dickson, Bruce Edgar, Marcos Gonzalez-Gaitán, Leonard Kelly, Jeanette Kunz, Cahir O’Kane, and Bing Zhang for reagents. We thank Yuchun He for injections, Shinya Yamamoto, Karen Schulze, Elaine Seto, Nikolaos Giagtzoglou, Hiroshi Tsuda, Gilbert Di Paolo, Jeanette Kunz and members of the Bellen, Verstreken and De Camilli labs for comments. PV was supported by a R.L. Kirchstein NRS award, a Marie Curie Excellence Grant (MEXT-CT-2006-042267), the Research Fund K.U.Leuven, FWO grant G.0747.09 and VIB, CVL was supported by an NRS award and PDC and HJB are HHMI investigators.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achiriloaie M, Barylko B, Albanesi JP. Essential role of the dynamin pleckstrin homology domain in receptor-mediated endocytosis. Mol Cell Biol. 1999;19:1410–1415. doi: 10.1128/mcb.19.2.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock MC, Stowers RS, Leither J, Goodman CS, Pallanck LJ. A genetic screen for synaptic transmission mutants mapping to the right arm of chromosome 3 in Drosophila. Genetics. 2003;165:171–183. doi: 10.1093/genetics/165.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Cao YW, Jiang CL, Jiang T. Molecular cloning and preliminary analysis of a fragile site associated gene. Biomed Environ Sci. 2006;19:392–398. [PubMed] [Google Scholar]
- Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, Di Fiore PP, De Camilli P. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature. 1998;394:793–797. doi: 10.1038/29555. [DOI] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99:179–188. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, DiAntonio A. A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle. Neuron. 2006;49:11–16. doi: 10.1016/j.neuron.2005.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis ME, Patrick RL. Diacylglycerol-induced stimulation of neurotransmitter release from rat brain striatal synaptosomes. J Neurochem. 1990;54:662–668. doi: 10.1111/j.1471-4159.1990.tb01922.x. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Verstreken P, Hiesinger PR, Horne JA, Kostyleva R, Zhou Y, Bellen HJ, Meinertzhagen IA. Endophilin promotes a late step in endocytosis at glial invaginations in Drosophila photoreceptor terminals. J Neurosci. 2003;23:10732–10744. doi: 10.1523/JNEUROSCI.23-33-10732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergestad T, Davis WS, Broadie K. The stoned proteins regulate synaptic vesicle recycling in the presynaptic terminal. J Neurosci. 1999;19:5847–5860. doi: 10.1523/JNEUROSCI.19-14-05847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wolfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, et al. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Gaidarov I, Chen Q, Falck JR, Reddy KK, Keen JH. A functional phosphatidylinositol 3,4,5-trisphosphate/phosphoinositide binding domain in the clathrin adaptor AP-2 alpha subunit. Implications for the endocytic pathway. J Biol Chem. 1996;271:20922–20929. doi: 10.1074/jbc.271.34.20922. [DOI] [PubMed] [Google Scholar]
- Gong LW, Di Paolo G, Diaz E, Cestra G, Diaz ME, Lindau M, De Camilli P, Toomre D. Phosphatidylinositol phosphate kinase type I gamma regulates dynamics of large dense-core vesicle fusion. Proc Natl Acad Sci U S A. 2005;102:5204–5209. doi: 10.1073/pnas.0501412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gaitan M, Jackle H. Role of Drosophila alpha-adaptin in presynaptic vesicle recycling. Cell. 1997;88:767–776. doi: 10.1016/s0092-8674(00)81923-6. [DOI] [PubMed] [Google Scholar]
- Golub T, Caroni P. PI(4,5)P2-dependent microdomain assemblies capture microtubules to promote and control leading edge motility. The Journal of cell biology. 2005;169:151–165. doi: 10.1083/jcb.200407058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Hartwieg E, Horvitz HR, Jorgensen EM. Mutations in synaptojanin disrupt synaptic vesicle recycling. J Cell Biol. 2000;150:589–600. doi: 10.1083/jcb.150.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan BA, Prokopenko SN, Breuer S, Zhang B, Paululat A, Bellen HJ. skittles, a Drosophila phosphatidylinositol 4-phosphate 5-kinase, is required for cell viability, germline development and bristle morphology, but not for neurotransmitter release. Genetics. 1998;150:1527–1537. doi: 10.1093/genetics/150.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V. Where proteins and lipids meet: membrane trafficking on the move. Dev Cell. 2003;4:153–157. doi: 10.1016/s1534-5807(03)00026-1. [DOI] [PubMed] [Google Scholar]
- Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, Cao Y, Zhou Y, Kunz J, Bellen HJ. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121:607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R. Electrifying phosphatases. Sci STKE. 2005;2005:pe50. doi: 10.1126/stke.3072005pe50. [DOI] [PubMed] [Google Scholar]
- Jost M, Simpson F, Kavran JM, Lemmon MA, Schmid SL. Phosphatidylinositol-4,5-bisphosphate is required for endocytic coated vesicle formation. Curr Biol. 1998;8:1399–1402. doi: 10.1016/s0960-9822(98)00022-0. [DOI] [PubMed] [Google Scholar]
- Jung N, Haucke V. Clathrin-mediated endocytosis at synapses. Traffic. 2007;8:1129–1136. doi: 10.1111/j.1600-0854.2007.00595.x. [DOI] [PubMed] [Google Scholar]
- Kasprowicz J, Kuenen S, Miskiewicz K, Habets RL, Smitz L, Verstreken P. Inactivation of clathrin heavy chain inhibits synaptic recycling but allows bulk membrane uptake. J Cell Biol. 2008;182:1007–1016. doi: 10.1083/jcb.200804162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TW, Verstreken P, Bellen HJ. Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron. 2004;43:193–205. doi: 10.1016/j.neuron.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Kuo MT, Wei Y, Yang X, Tatebe S, Liu J, Troncoso P, Sahin A, Ro JY, Hamilton SR, Savaraj N. Association of fragile site-associated (FSA) gene expression with epithelial differentiation and tumor development. Biochem Biophys Res Commun. 2006;340:887–893. doi: 10.1016/j.bbrc.2005.12.088. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Majumdar A, Ramagiri S, Rikhy R. Drosophila homologue of Eps15 is essential for synaptic vesicle recycling. Exp Cell Res. 2006;312:2288–2298. doi: 10.1016/j.yexcr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Manak JR, Dike S, Sementchenko V, Kapranov P, Biemar F, Long J, Cheng J, Bell I, Ghosh S, Piccolboni A, Gingeras TR. Biological function of unannotated transcription during the early development of Drosophila melanogaster. Nat Genet. 2006;38:1151–1158. doi: 10.1038/ng1875. [DOI] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, Ryan TA. The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron. 2007;56:1004–1018. doi: 10.1016/j.neuron.2007.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, Davis GW. Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron. 2004;43:207–219. doi: 10.1016/j.neuron.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TF. Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu Rev Cell Dev Biol. 1998;14:231–264. doi: 10.1146/annurev.cellbio.14.1.231. [DOI] [PubMed] [Google Scholar]
- McKay RM, McKay JP, Avery L, Graff JM. C elegans: a model for exploring the genetics of fat storage. Dev Cell. 2003;4:131–142. doi: 10.1016/s1534-5807(02)00411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Holz RW, Smith SJ. Regulation of presynaptic phosphatidylinositol 4,5-biphosphate by neuronal activity. J Cell Biol. 2001;154:355–368. doi: 10.1083/jcb.200102098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic I, Sorensen JB, Lang T, Krauss M, Nagy G, Haucke V, Jahn R, Neher E. Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J Neurosci. 2005;25:2557–2565. doi: 10.1523/JNEUROSCI.3761-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Holgado AM, Brewer F, Serpe CJ, Norbeck BA, Holleran J, Wei L, Hartwieg E, Jorgensen EM, Alfonso A. UNC-11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol Biol Cell. 1999;10:2343–2360. doi: 10.1091/mbc.10.7.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, Tan LR, Winter CG, Bogart KP, Deal JE, et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet. 2004;36:288–292. doi: 10.1038/ng1312. [DOI] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS. Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron. 2001;32:415–424. doi: 10.1016/s0896-6273(01)00485-8. [DOI] [PubMed] [Google Scholar]
- Phillips AM, Smith M, Ramaswami M, Kelly LE. The products of the Drosophila stoned locus interact with synaptic vesicles via synaptotagmin. J Neurosci. 2000;20:8254–8261. doi: 10.1523/JNEUROSCI.20-22-08254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, Marek KW, Sweeney ST, Davis GW. Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature. 2003;426:559–563. doi: 10.1038/nature02184. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Krishnan KS, Kelly RB. Intermediates in synaptic vesicle recycling revealed by optical imaging of Drosophila neuromuscular junctions. Neuron. 1994;13:363–375. doi: 10.1016/0896-6273(94)90353-0. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Roos J, Kelly RB. The endocytic machinery in nerve terminals surrounds sites of exocytosis. Curr Biol. 1999;9:1411–1414. doi: 10.1016/s0960-9822(00)80087-1. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Gu QM, Prestwich GD, Sollner TH, Rothman JE. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc Natl Acad Sci U S A. 1996;93:13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuske KR, Richmond JE, Matthies DS, Davis WS, Runz S, Rube DA, van der Bliek AM, Jorgensen EM. Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron. 2003;40:749–762. doi: 10.1016/s0896-6273(03)00667-6. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Ultsch A, Schloss P, Cox JA, Schmitt B, Betz H. Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science. 1991;254:112–114. doi: 10.1126/science.1681587. [DOI] [PubMed] [Google Scholar]
- Sladeczek F. Putative role of inositol phospholipid metabolism in neurons. Biochimie. 1987;69:287–296. doi: 10.1016/0300-9084(87)90019-8. [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Long F, Peter BJ, Murray D, De Camilli P, McMahon HT, Cho W. Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J Biol Chem. 2003;278:28993–28999. doi: 10.1074/jbc.M302865200. [DOI] [PubMed] [Google Scholar]
- Stimson DT, Estes PS, Rao S, Krishnan KS, Kelly LE, Ramaswami M. Drosophila stoned proteins regulate the rate and fidelity of synaptic vesicle internalization. J Neurosci. 2001;21:3034–3044. doi: 10.1523/JNEUROSCI.21-09-03034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1939. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek AM, Meyerowitz EM. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature. 1991;351:411–414. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- Van Epps HA, Hayashi M, Lucast L, Stearns GW, Hurley JB, De Camilli P, Brockerhoff SE. The zebrafish nrc mutant reveals a role for the polyphosphoinositide phosphatase synaptojanin 1 in cone photoreceptor ribbon anchoring. J Neurosci. 2004;24:8641–8650. doi: 10.1523/JNEUROSCI.2892-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Balla T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta. 2006;1761:957–967. doi: 10.1016/j.bbalip.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Kjaerulff O, Lloyd TE, Atkinson R, Zhou Y, Meinertzhagen IA, Bellen HJ. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell. 2002;109:101–112. doi: 10.1016/s0092-8674(02)00688-8. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ohyama T, Bellen HJ. FM 1–43 labeling at the Drosophila neuromuscular junction. Methods in Molecular Biology. 2007;440:349–369. doi: 10.1007/978-1-59745-178-9_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, D'Angelo G, Di Campli A, De Matteis MA. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008;27:2457–2470. doi: 10.1038/emboj.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, et al. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc Natl Acad Sci U S A. 2008;105:9415–9420. doi: 10.1073/pnas.0803756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk MR, De Camilli P. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc Natl Acad Sci U S A. 2004;101:8262–8269. doi: 10.1073/pnas.0401874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig T, Wilsch-Brauninger M, Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J Cell Biol. 2003;161:609–624. doi: 10.1083/jcb.200211087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York JD. Regulation of nuclear processes by inositol polyphosphates. Biochim Biophys Acta. 2006;1761:552–559. doi: 10.1016/j.bbalip.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci U S A. 2003;100:10860–10865. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Sebastian R, Nakatsu F, Chen H, Balla T, Ayala G, Toomre D, De Camilli PV. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A. 2007;104:3793–3798. doi: 10.1073/pnas.0611733104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.