

Abstract
Background
Pollen is the male gametophyte of higher plants. Upon pollination, it germinates and develops into a fast-growing cytoplasmic extension, the pollen tube, which ultimately delivers the sperm into the ovary. The biological relevance of its role, and the uniqueness of this kind of cellular organization, have made pollen the focus of many approaches, and it stands today as one of the best-known models in plant cell biology. In contrast, the genetic background of its development has been until recently largely unknown. Some genes involved have been described and a few functional mutants have been characterized, but only to a limited extent and allowing only a limited understanding of the regulatory mechanisms. Yet, being a relatively simple organ (2 or 3 cells), pollen stands as an excellent target for molecular-biology-based approaches.
Recent Progress
Recent studies on Arabidopsis thaliana have characterized the transcriptional profile of pollen grains and microgametogenesis in comparison to sporophytic tissues. They underline the unique characteristics of pollen, not only in terms of a strongly reduced set of genes being expressed, but also in terms of the functions of the proteins encoded and the pathways they are involved in. These approaches have expanded the number of genes with known expression in pollen from a few hundred to nearly eight thousand. While for the first time allowing systems and/or gene-family approaches, this information also expands dramatically the possibility of hypothesis-driven experimentation based on specific gene function predictions. Recent studies reveal this to be the case in, for example, transcriptional regulation, cell-cycle progression and gene-silencing mechanisms in mature pollen.
Key words: Pollen, transcriptome, proteome, microarray, Arabidopsis
INTRODUCTION
Studies on pollen development and pollen-tube growth have their origins almost two centuries ago and were primarily focused on the role of the male gametophyte in sexual plant reproduction. While research in this field has never lost sight of its biological role (McCormick, 2004; Boavida et al., 2005b), the last decades have witnessed an increasing shift towards the use of pollen as a model to study fundamental aspects of cellular differentiation, apical cell growth and cell–cell interaction. But the substantive progress achieved in our understanding of microsporogenesis and cellular processes underlying pollen tube growth (e.g. ion fluxes, cytoskeleton dynamics, cell wall metabolism, etc.; Feijó et al., 2004, Boavida et al., 2005a, b), stood until recently in stark contrast to what was known about the genetic basis underlying these processes. Genetic and functional analysis was in most cases proceeding on a gene-by-gene scale, mostly through mutant screens and forward genetics approaches. Reverse genetic approaches (i.e. starting with a candidate gene up to the definition of a phenotype through a knock-out mutation) or gene functional over-expression have progressed more slowly owing to the lack of large gene expression profiles in pollen. Nonetheless, some of these approaches have made very relevant contributions to our understanding of pollen development and pollen-tube growth (Twell et al., 2006), and suppression subtractive hybridization screens that result in cDNA libraries containing the potential candidate genes are still the method of choice, if no or limited information about the genomic sequence is available for the species under study. Recent examples include studies investigating the effects of cold storage on transcript abundance in Lilium longiflorum pollen (Wang et al., 2004) and the identification of early genes involved in Nicotiana tabacum pollen development (Shary et al., 2006). cDNA-amplified fragment length polymorphism analysis (cDNA-AFLP) represents a further improvement and has been used, for example, to analyse gene expression during micro- and macrosporogenesis in Petunia hybrida (Cnudde et al., 2003).
Another approach that has had a positive impact on the field has been serial analysis of gene expression (SAGE). This method measures transcript abundance by sequencing mRNA-derived, concatemeric 10- to 19-base-pair-long sequence tags. Application to the transcriptome of mature pollen of Arabidopsis thaliana under normal and chilling conditions allowed identification of 4211 unique tags under normal conditions (Lee and Lee, 2003).
A further step to large-scale analyses became possible with the completion of the genome sequence of the dicot model plant Arabidopsis thaliana (Arabidopsis Genome Initiative, 2000), and the development of DNA microarray technology. The underlying principle of this technology is the hybridization of a labelled sample (target), generated from mRNA that has been isolated from a given cell line or tissue, to many thousands of DNA sequences (probes), which are immobilized on a solid surface in an ordered array (Schena et al., 1995). The amount of target bound to a particular probe, typically read out as intensity of fluorescence, serves as an estimate of the abundance of that transcript in the mRNA population. The application of microarray-based transcriptome analyses to pollen biology has subsequently resulted in the quantum leap needed for an established database of gene expression profiles in pollen.
THE POLLEN TRANSCRIPTOME: AS SPECIALIZED AS THE CELL THAT CARRIES IT
A generalized transcriptomic approach to the pollen of Arabidopsis thaliana was firstly made possible by use of the Affymetrix AG GeneChip arrays, which represented approximately 26 % of genes predicted for Arabidopsis (Becker et al., 2003; Honys and Twell, 2003). Both these studies compared the transcriptional profile of mature pollen grains with those of sporophytic tissues, but they differed in the number of genes identified as being expressed in pollen and as being selectively expressed only in this cell type. While our group concluded that 1584 are expressed in pollen and 10 % of these selectively, Honys and Twell identified only 992 as expressed and 40 % as selectively expressed. These discrepancies can be attributed to a number of reasons, namely (1) the methods used for isolation of the pollen samples (we used a further step by which pollen grains were sorted by fluorescence-activated cell sorting); (2) differences in the sporophytic tissue types and Arabidopsis ecotypes used for comparison; and (3) last but not least, differences in the normalization procedures used to compare the values from the different samples (for a detailed discussion see Boavida et al., 2005a). Despite the discrepancies, both studies concurred that the pollen transcriptome exhibits a strongly reduced complexity in comparison with the sporophyte; this conclusion agrees with the concept that pollen stores the limited number of transcripts needed for germination, rapid cell growth and cell–cell signalling. Importantly, while based on a GeneChip covering a quarter of the Arabidopsis genome, these first two studies increased the number of genes identified as being expressed in pollen by more than an order of magnitude.
Further progress was made possible with the release of the Affymetrix ATH1 GeneChip arrays, representing 22 800 transcripts or approximately 73 % of the currently annotated Arabidopsis genes. We have used this type of array to compare the transcriptional profile of cell-sorted, mature pollen grains with those of seedlings, flowers, leaves and siliques (Pina et al., 2005). Of the 6587 genes identified as expressed in pollen in this study, comparisons with the vegetative tissues indicated that 11 % are pollen-selectively expressed and 26 % show higher expression in pollen than in the vegetative tissues (pollen enriched). Just for comparison, seedlings and leaves express each approximately 15 500 genes, with only 4 % and 6 %, respectively, selectively expressed. Thus pollen accumulates a restricted and to a certain extent unique set of transcripts, as can be visualized by hierarchical clustering (Fig. 1A) or principal component analysis (Fig. 1B). Interestingly, gametophytic selection seems to lead to a reduced length of introns in genes highly expressed in pollen in comparison with genes expressed in the sporophyte, a fact that may be interpreted as reflecting an increased efficiency of transcription for these pollen-expressed genes (Seoighe et al., 2005). The fact that this analysis was based on a much more representative database allowed a much more detailed and meaningful bio-informatics analysis of gene families and possible inferences for the functional and mechanistic characterization of pollen. For example, functional classification of pollen-expressed genes based on gene ontology terms in comparison with the sporophytic tissues revealed that signalling, vesicle trafficking, cytoskeleton and membrane transport were proportionally over-represented in pollen (Fig. 2, and statistical analysis in Pina et al., 2005), with a number of pollen-unique genes. Broadly, these functional classes have been assumed from physiology and cell biology to be essential for successful germination, pollen-tube growth and fertilization, and therefore some of these genes are prime-targets for reverse genetics. SAGE analysis of enriched and selectively expressed genes as well as their functional classification (Lee and Lee, 2003) is largely concordant with the microarray analyses.
Fig. 1.
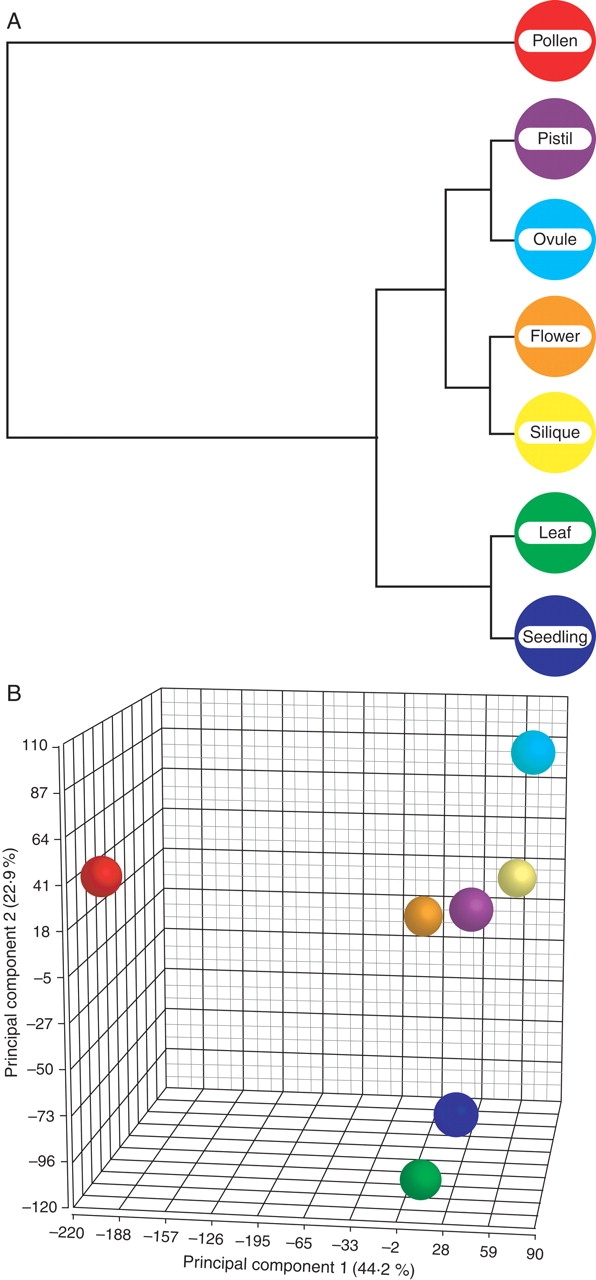
Hierarchical clustering and principal component analysis of transcriptome data for Arabidopsis vegetative tissues and pollen. (A) Hierarchical clustering is used to group similar objects into ‘clusters’, producing a tree (dendrogram) that shows the hierarchy of the clusters. The dendrogram shows a clear separation of a pollen cluster and a vegetative tissues cluster with a further separation of the vegetative tissue cluster (with Pearson's Dissimilarity to calculate row dissimilarity and Ward's method for row clustering). (B) Principal component analysis is an exploratory technique used to describe the structure of high-dimensional data, e.g. derived from microarrays, by reducing its dimensionality. Here, expression values for 22 800 genes in seven tissue/cell types are projected onto the first three principal components. The closer two points are in the plot, the more similar are the tissue/cell types in terms of gene expression. The first principal component separates pollen from the vegetative tissues, while the second and third principal components show a further separation of the vegetative tissues within themselves. Analyses are based on expression values from the study by Pina et al. (2005), reanalysed to include data for pistils and ovules (L.C. Boavida, J.D. Becker and J.A. Feijó, unpubl. res.).
Fig. 2.

Classification of biological activities based on transcriptome and proteome data. A total of 8463 genes represented on the ATH1 GeneChip that were classified into at least one gene ontology category (biological process terms as of September 2003) were regrouped into 14 biological activity classes. The proportion of the genes expressed in each data set assigned to the different activity classes is represented. Transcriptomic data sets are based on Pina et al. (2005) with vegetative tissues representing an average of the distribution found in leaves, seedlings and siliques. Proteomic data originate from the union of proteins identified in three studies (Holmes-Davis et al., 2005; Noir et al., 2005; Sheoran et al., 2006).
Using a different biological bias, Honys and Twell (2004) focused their transcriptomics analysis on the pollen development process (microsporogenesis). They have generated samples enriched for uninuclear microspores, and bicellular, tricellular and mature pollen grains, and analysed them on the ATH1 GeneChip to conclude that generally the post-meiotic stages of pollen development are associated with a strong decline in the diversity of RNA transcripts. The final count for genes expressed in mature pollen was 6587, but the discrepancy between this figure and the one reported by Pina et al. (2005) can be explained by divergences in technical procedures (Boavida et al., 2005a). The study by Honys and Twell (2004) underlined the general idea of a specialization of the pollen transcriptome in terms not only of a reduced diversity of transcripts but also of an increasing functional skew towards cytoskeleton-, cell wall- and signalling-related transcripts. Further, they were the first to do cluster analysis of co-regulated transcription factors in this data set.
The study of expression levels of transcription factors in mature pollen is pivotal towards answering the pending question of the existence and importance of de novo transcription in germinating pollen, and its putative relevance for pollen-tube growth in later stages. Various approaches, including the bio-informatics ‘digestion’ of the transcriptomics data, have shown that non-classical MADS-box genes (MIKC* subgroup) might play a major role as they constitute a class of transcription factors with a high percentage of pollen-enriched transcripts (Hennig et al., 2004; Pina et al., 2005). In accordance with this prediction it was recently shown that MIKC* binding sites are over-represented in late pollen-specific promoters and that the absence or strong reduction of MIKC* proteins and their complexes strongly impairs in vitro pollen germination (Verelst et al., 2007).
Microarray studies produce amounts of data that are difficult to access without prior meaningful processing. Most of the above-mentioned studies provide help in the form of detailed analyses of gene families either as part of the original publication (Honys and Twell, 2004; Pina et al., 2005) or in publications focusing on specific aspects such as the proton–cation exchangers (AtCHX) gene family (Sze et al., 2004) or membrane transporters in general (Bock et al., 2006). Reverse-genetics approaches in particular can by and large benefit from gene-family analysis. Functional redundancy can obscure the phenotypic effect of the mutated gene expressed in a given cell type, a problem potentially circumvented by adequate knowledge of the specific members of a family expressed in a specific cell/tissue. Alternatively, web-based data mining interfaces such as Genevestigator (https://www.genevestigator.ethz.ch/; Zimmermann et al., 2005) have been developed, allowing complex queries of Arabidopsis ATH1 datasets publicly available. This includes the most comprehensive dataset to date with 79 developmental stages (Schmid et al., 2005) plus a large number of biotic and abiotic treatments. Here, potential candidate genes can be further characterized by their expression or non-expression in developmental stages or conditions not tested in the original study. However, caution should be taken if expression values are to be compared because the normalization methods used for datasets included in the Genevestigator database are inadequate for pollen and may lead to over-estimation of the respective expression values.
Given sufficient genome coverage, an analysis of pathways based on gene expression data can potentially extract biologically meaningful indications and thus create novel hypotheses, especially when data on single cell types are available, as for pollen (reviewed by Galbraith and Birnbaum, 2006). The detailed analysis of cell cycle-related transcripts in pollen has shown that pollen might be arrested in G1–S of the cell cycle through an absence of transcripts encoding essential proteins for this transition (CycD, E2F-DP) in combination with an up-regulation of potential repressors (e.g. DEL3; Pina et al., 2005). Unexpectedly, pollen seems to express most transcripts needed for the G2–M transition, an observation that is supported by the results of a GeneChip study profiling three stages of Arabidopsis flower and fruit development (Hennig et al., 2004). The exact role of these transcripts in the pollen context remains to be shown, but it raises the interesting hypothesis that they might act as key players during karyogamy and the first mitosis after fertilization. A second surprising finding of the pathway analysis by Pina et al. (2005) is the apparent inactivation of small RNA pathways in mature pollen, as indicated by an absence of all transcripts known to be involved in the pathway. If confirmed by functional analysis, this would establish mature pollen as the first known organ without an expressed post-transcriptional silencing mechanism, and would open the way to the proposal of new hypotheses in terms of genetic and epigenetic regulation of the male gametophyte and fertilization.
PROTEOMIC APPROACHES TO POLLEN BIOLOGY
Despite the wealth of information rendered by microarrays, the issue of functionality will always depend on confirmation at the protein level. Evolving at a completely different pace, some recent proteomic approaches deserve attention, and allow the first rough comparisons with the transcriptomics' data. Three recent studies have focused on proteome analysis of mature pollen in the Arabidopsis ecotypes Columbia (Holmes-Davis et al., 2005; Noir et al., 2005) and Landsberg erecta (Sheoran et al., 2006). Notably, the three studies identified only 135, 116 and 95 proteins, respectively, which can be explained by the limitations of two-dimensional electrophoresis (2-DE) in detecting low-abundance and integral membrane proteins. The overlap between the three studies is restricted to 18 proteins (Fig. 3), possibly because of different protocols and ecotypes. A total of 262 unique proteins have been identified, and for 22 of these the Arabidopsis ATH1 GeneChip lacks probes for the corresponding transcripts. A comparison with the GeneChip data on mature pollen grains (Honys and Twell, 2004; Pina et al., 2005; Schmid et al., 2005) reveals that for 224 of the remaining 240 proteins the corresponding transcript was detected as expressed, and that nine of the 16 transcripts that were not detected in mature pollen grains are expressed in earlier stages of microgametogenesis, possibly revealing proteins with a very low rate of turnover. This is an important aspect to delve into when a more enlarged view of the proteome becomes available, as it could potentially challenge (or confirm!) some of the prevailing views on the transcriptional status of germinating pollen.
Fig. 3.

Venn diagram of proteins identified in three studies analysing the proteome of Arabidopsis mature pollen (number found in each study is given in brackets).
In short, differences between the proteome and the transcriptome data are obvious when one compares the functional classes to which the proteins can be allocated based on biological-process gene ontologies (Fig. 2). Redox and general metabolism are the highest-represented classes in the pollen proteome and clearly over-represented in comparison with their lower abundance in the pollen transcriptome, as are the classes defence mechanisms and stress, cytoskeleton and translation. By contrast, classes such as transcription and RNA processing, signalling and transport are under-represented. Again technical issues on sensitivity can be invoked to explain these discrepancies rather than biology; all these are highly abundant, soluble proteins. However, one cannot discount the hypothesis that storage of proteins involved in energy metabolism and the cytoskeleton is plausible in the context of rapid germination of pollen grains. The same could be said about the higher abundance of proteins needed for the translation of the mRNA pool stored in mature pollen grains. Experiments with inhibitors of transcription and translation during pollen germination and tube growth in Arabidopsis underline that germination and tube growth are relatively independent of transcription, but not of translation (Honys and Twell, 2004). Proteins involved in signalling are more likely to be involved in interactions during pollen-tube growth along the female reproductive tract, thus offering an explanation for the wide discrepancy apparent in this class.
From this comparison it follows that the potential of proteomics is still grossly hindered by the present technical limitations, but the recent identification of 4200 proteins by shot-gun proteomics on proteins extracted from mature Arabidopsis pollen (M. Grobei and U. Grossniklaus, University of Zürich, pers. com.), and other ongoing efforts, clearly show that the situation may change dramatically in the near future. Proteomics studies on pollen of the monocot model plant Oryza sativa constitute an important complement, in particular the identification of 120 diverse protein species differentially expressed in mature and germinated rice pollen (Dai et al., 2006). Notably these include multiple isoforms of proteins that might originate from alternative splicing events, constituting a new pool of experimental data still not accessible by current transcriptomics approaches.
FUTURE PERSPECTIVES
Linear extrapolation of the 7264 genes that we have proposed to be expressed in pollen from the ATH1 GeneChip (Pina et al., 2005) up to a complete genome coverage would lead us to the assume that Arabidopsis mature pollen carries a load of something in the order of 10 000 different transcripts. This number, and the qualitative analysis of the genes present, should more than fulfil the expectations that we may have achieved sufficient gene expression data to understand the major functional framework for cell growth in pollen. However, the recent discovery of other regulatory levels and genetic interactions calls for caution with this conclusion. With the availability of an Affymetrix Arabidopsis tiling array, which features probes placed at 10-nucleotide intervals across the entire 125-Mb Arabidopsis genome, a largely unbiased interrogation of the transcriptome has come into reach, and will be likely to expand this knowledge. This should allow not only the detection of annotated transcripts not interrogated in previous studies and the potential detection of novel, non-annotated transcripts, but also the detection of splice variants. Considering the emerging major role of alternative splicing in protein diversity, we view this as an important aspect that could not be addressed so far. Other technologies, currently allowing similar in-depth transcriptome analysis though at significantly higher expense, are massively parallel signature sequencing (MPSS) and 454 sequencing (Margulies et al., 2005). A comparison with pollen microarray data sets from other species such as maize (Ma et al., 2006) complements the studies on Arabidopsis pollen and should eventually allow the identification of a common conserved set of pollen-specific transcripts.
Combination of the current pollen data sets with those of other cell types promises to provide valuable insights into a number of biological issues. For one, pollen tubes and root hairs share strictly polar cell expansion, called tip growth (Cole and Fowler, 2006; Campanoni and Blatt, 2007). Thus a bio-informatics, system-level comparison of the pollen transcriptome with that of root hairs could allow the identification of an evolutionarily conserved ‘apical growth signature’, which we hope would include key players in this process. Further clues can be expected from comparisons with other cell types showing polarized cell growth, such as fungal hyphae or fern spores (Salmi et al., 2005).
It should be emphasized that fertilization, despite being one of the most important steps in the plant life cycle, is still poorly profiled in respect of the gene-expression profiles of its major steps. A time-course of pollen–pistil interactions from pollen germination to fertilization in Arabidopsis reveals co-ordinated changes of expression levels for more than 1500 genes, unveiling major changes in transcriptional profiles likely to reveal major functions still uncharacterized (L.C. Boavida, J.D. Becker and J.A. Feijó, unpubl. res.). While so far we have been referring to pollen in general, sequencing of expressed sequence tags of isolated sperm cells from maize and lily has shown that the generative cells of these species carry a diverse complement of transcripts (Engel et al., 2003; Okada et al., 2006). The analysis of such sperm-specific transcripts has, among others, led to the identification of the plasma-membrane protein GCS1 as a critical factor for gamete fusion (Mori et al., 2006). The transcriptional profile of Arabidopsis sperm cells is still lacking, because the isolation of enough of these relatively small sperm cells constitutes a significant experimental challenge. However, recent technological advances should enable us to compare the transcriptional profile of FACS-sorted Arabidopsis sperm cells with those of egg/central cells, isolated by either micro-dissection or laser capture microscopy. Such studies will provide valuable new insights into the genetic basis of sperm–egg and sperm–central-cell interactions.
In short, by virtue of the very specific functions that they must carry out in the plant life cycle, evolution seems to have shaped pollen grains with a reduced, but relatively specific and complex gene expression profile. This has now become unveiled through various transcriptomics approaches and has turned pollen into a powerful model organ for system approaches or hypothesis-driven research based on the presence of specific gene profiles.
ACKNOWLEDGEMENTS
Funding for this work was provided by Fundação para a Ciência e a Tecnologia (SFRH/BPD/31047/2006 to JDB, POCTI/BIA-BCM/60046/2004, POCTI/BIA-BCM/61270/2004).
LITERATURE CITED
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Becker JD, Boavida LC, Carneiro J, Haury M, Feijo JA. Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiology. 2003;133:713–725. doi: 10.1104/pp.103.028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boavida LC, Becker JD, Feijo JA. The making of gametes in higher plants. International Journal of Developmental Biology. (a) 2005;49:595–614. doi: 10.1387/ijdb.052019lb. [DOI] [PubMed] [Google Scholar]
- Boavida LC, Vieira AM, Becker JD, Feijo JA. Gametophyte interaction and sexual reproduction: how plants make a zygote. International Journal of Developmental Biology. (b) 2005;49:615–632. doi: 10.1387/ijdb.052023lb. [DOI] [PubMed] [Google Scholar]
- Bock KW, Honys D, Ward JM, Padmanaban S, Nawrocki EP, Hirschi KD, Twell D, Sze H. Integrating membrane transport with male gametophyte development and function through transcriptomics. Plant Physiology. 2006;140:1151–1168. doi: 10.1104/pp.105.074708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanoni P, Blatt MR. Membrane trafficking and polar growth in root hairs and pollen tubes. Journal of Experimental Botany. 2007;58:66–74. doi: 10.1093/jxb/erl059. [DOI] [PubMed] [Google Scholar]
- Cnudde F, Moretti C, Porceddu A, Pezzotti M, Gerats T. Transcript profiling on developing Petunia hybrida floral organs. Sexual Plant Reproduction. 2003;16:77–85. [Google Scholar]
- Cole RA, Fowler JE. Polarized growth: maintaining focus on the tip. Current Opinion in Plant Biology. 2006;9:579–588. doi: 10.1016/j.pbi.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Dai SJ, Chen TT, Chong K, Xue YB, Liu SQ, Wang T. Proteomic identification of differentially expressed proteins associated with pollen germination and tube growth reveals characteristics of germinated Oryza sativa pollen. Molecular Cell Proteomics. 2007;6:207–230. doi: 10.1074/mcp.M600146-MCP200. [DOI] [PubMed] [Google Scholar]
- Engel ML, Chaboud A, Dumas C, McCormick S. Sperm cells of Zea mays have a complex complement of mRNAs. Plant Journal. 2003;34:697–707. [PubMed] [Google Scholar]
- Feijó JA, Costa SS, Prado AM, Becker JD, Certal AC. Signalling by tips. Current Opinion in Plant Biology. 2004;7:589–598. doi: 10.1016/j.pbi.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Galbraith DW, Birnbaum K. Global studies of cell type-specific gene expression in plants. Annual Review of Plant Biology. 2006;57:451–475. doi: 10.1146/annurev.arplant.57.032905.105302. [DOI] [PubMed] [Google Scholar]
- Hennig L, Gruissem W, Grossniklaus U, Kohler C. Transcriptional programs of early reproductive stages in Arabidopsis. Plant Physiology. 2004;135:1765–1775. doi: 10.1104/pp.104.043182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes-Davis R, Tanaka CK, Vensel WH, Hurkman WJ, McCormick S. Proteome mapping of mature pollen of Arabidopsis thaliana. Proteomics. 2005;5:4864–4884. doi: 10.1002/pmic.200402011. [DOI] [PubMed] [Google Scholar]
- Honys D, Twell D. Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiology. 2003;132:640–652. doi: 10.1104/pp.103.020925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honys D, Twell D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biology. 2004;5:R85. doi: 10.1186/gb-2004-5-11-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Lee DH. Use of serial analysis of gene expression technology to reveal changes in gene expression in Arabidopsis pollen undergoing cold stress. Plant Physiology. 2003;132:517–529. doi: 10.1104/pp.103.020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Morrow DJ, Fernandes J, Walbot V. Comparative profiling of the sense and antisense transcriptome of maize lines. Genome Biology. 2006;7:R22. doi: 10.1186/gb-2006-7-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick S. Control of male gametophyte development. Plant Cell. 2004;16:S142–S153. doi: 10.1105/tpc.016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Kuroiwa H, Higashiyama T, Kuroiwa T. GENERATIVE CELL SPECIFIC 1 is essential for angiosperm fertilization. Nature Cell Biology. 2006;8:64–71. doi: 10.1038/ncb1345. [DOI] [PubMed] [Google Scholar]
- Noir S, Brautigam A, Colby T, Schmidt J, Panstruga R. A reference map of the Arabidopsis thaliana mature pollen proteome. Biochemical and Biophysical Research Communications. 2005;337:1257–1266. doi: 10.1016/j.bbrc.2005.09.185. [DOI] [PubMed] [Google Scholar]
- Okada T, Bhalla PL, Singh MB. Expressed sequence tag analysis of Lilium longiflorum generative cells. Plant and Cell Physiology. 2006;47:698–705. doi: 10.1093/pcp/pcj040. [DOI] [PubMed] [Google Scholar]
- Pina C, Pinto F, Feijo JA, Becker JD. Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiology. 2005;138:744–756. doi: 10.1104/pp.104.057935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmi ML, Bushart TJ, Stout SC, Roux SJ. Profile and analysis of gene expression changes during early development in germinating spores of Ceratopteris richardii. Plant Physiology. 2005;138:1734–1745. doi: 10.1104/pp.105.062851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene-expression patterns with a complementary-DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, et al. A gene expression map of Arabidopsis thaliana development. Nature Genetics. 2005;37:501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- Seoighe C, Gehring C, Hurst LD. Gametophytic selection in Arabidopsis thaliana supports the selective model of intron length reduction. PLoS Genetics. 2005;1:154–158. doi: 10.1371/journal.pgen.0010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shary S, Kumar R, Guha-Mukherjee S. Isolation of pollen early genes and analysis of expression pattern during the development of male gametophyte. Plant Science. 2006;170:417–425. [Google Scholar]
- Sheoran IS, Sproule AT, Olson DJH, Ross ARS, Sawhney VK. Proteome profile and functional classification of proteins in Arabidopsis thaliana (Landsberg erecta) mature pollen. Sexual Plant Reproduction. 2006;19:185–196. [Google Scholar]
- Sze H, Padmanaban S, Cellier F, Honys D, Cheng NH, Bock KW, et al. Expression patterns of a novel AtCHX gene family highlight potential roles in osmotic adjustment and K+ homeostasis in pollen development. Plant Physiology. 2004;136:2532–2547. doi: 10.1104/pp.104.046003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twell D, Oh SA, Honys D. Pollen development, a genetic and transcriptomic view. In: Malho R, editor. The pollen tube. Heidelberg: Springer-Verlag Berlin; 2006. [Google Scholar]
- Verelst W, Saedler H, Münster T. MIKC* MADS-protein complexes bind motifs enriched in the proximal region of late pollen-specific Arabidopsis promoters. Plant Physiology. 2007;143:447–460. doi: 10.1104/pp.106.089805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ML, Hsu CM, Chang LC, Wang CS, Su TH, Huang YJJ, Jiang LW, Jauh GY. Gene expression profiles of cold-stored and fresh pollen to investigate pollen germination and growth. Plant and Cell Physiology. 2004;45:1519–1528. doi: 10.1093/pcp/pch174. [DOI] [PubMed] [Google Scholar]
- Zimmermann P, Hennig L, Gruissem W. Gene-expression analysis and network discovery using Genevestigator. Trends in Plant Science. 2005;10:407–409. doi: 10.1016/j.tplants.2005.07.003. [DOI] [PubMed] [Google Scholar]