

Abstract
VIM-2 is an Ambler class B metallo-β-lactamase (MBL) capable of hydrolyzing a broad-spectrum of β-lactam antibiotics. Although the discovery and development of MBL inhibitors continues to be an area of active research, an array of potent, small molecule inhibitors has yet to be fully characterized for VIM-2. In the presented research, a compound library screening approach was used to identify and characterize VIM-2 inhibitors from a library of pharmacologically active compounds as well as a focused “click” chemistry library. The four most potent VIM-2 inhibitors resulting from a VIM-2 screen were characterized by kinetic studies in order to determine Ki and mechanism of enzyme inhibition. As a result, two previously described pharmacologic agents, mitoxantrone (1,4-Dihydroxy-5,8-bis([2-([2-hydroxyethyl]amino)ethyl]amino)-9,10-anthracenedione) and 4-chloromercuribenzoic acid (pCMB) were found to be active, the former as a non-competitive inhibitor (Ki = K′i = 1.5 ± 0.2 μM) and the latter as a slowly reversible or irreversible inhibitor. Additionally, two novel sulfonyl-triazole analogs from the click library were identified as potent, competitive VIM-2 inhibitors: N-((4-((but-3-ynyloxy)methyl)-1H-1,2,3-triazol-5-yl)methyl)-4-iodobenzenesulfonamide (1, Ki = 0.41 ± 0.03 μM) and 4-iodo-N-((4-(methoxymethyl)-1H-1,2,3-triazol-5-yl)methyl)benzenesulfonamide (2, Ki = 1.4 ± 0.10 μM). Mitoxantrone and pCMB were also found to potentiate imipenem efficacy in MIC and synergy assays employing E. coli. Taken together, all four compounds represent useful chemical probes to further investigate mechanisms of VIM-2 inhibition in biochemical and microbiology-based assays.
Keywords: VIM-2, Inhibitors, beta-lactamase
3. Introduction
Bacterial infections that demonstrate antibiotic resistance are a major public health concern. 1 2, 3 One well-known mechanism of resistance, particularly prevalent in Gram-negative bacteria, involves endogenous β-lactamase enzymes. These enzymes catalyze hydrolysis of β-lactam antibiotics, rendering them inactive. 4 5 Metallo-β-lactamases (MBLs) are Ambler class “B” zinc-dependent enzymes capable of hydrolyzing a broad-spectrum of clinically relevant β-lactam classes, such as penicillins, cephalosporins, and carbapenems. 4 Due to an increase in the rate of infections attributed to bacteria harboring MBLs, there is concern of their growing clinical threat 6 7. Along with IMP-1, another subclass B1 MBL, VIM-2 is considered one of the more clinically relevant MBLs. 8 9 10-12 Pathogenic clinical isolates containing VIM-2 have been found in Europe, Asia, and the Americas. 8
High-throughput screening (HTS) is an effective tool for rapidly identifying novel scaffolds for drug discovery, and more recently aiding chemical probe development. 13 Presented here is the execution of a screening-based effort to discover selective VIM-2 inhibitors. This effort entailed the development & execution of an HTS-compatible VIM-2 nitrocefin assay. To further confirm VIM-2 activity and also identify non-selective compounds, a VIM-2 fluorescence-resonance energy transfer (FRET) inhibition assay and an IMP-1 inhibition assay were employed. Additionally, antibiotic potentiation studies were conducted in E. coli to determine efficacy of four compounds demonstrating biochemical potency. The success of this effort at identifying & characterizing these inhibitors, including the molecular modeling of the competitive inhibitors in the VIM-2 active site, is presented.
4. Results
VIM-2 and IMP-1 enzyme inhibition assays
Three enzymatic assays were executed to identify and characterize VIM-2 inhibitors. The VIM-2 nitrocefin assay (Figure 1a) 14 was screened against a library of pharmacologically active compounds (LOPAC, n = 1280 compounds) as well as a novel click chemistry library enriched in metalloenzyme inhibitors (n = 267 compounds). To confirm the activity of VIM-2 inhibitors found via the nitrocefin assay, a FRET-based CCF2 substrate assay was employed 15 (Figure 1b). Finally, an IMP-1 nitrocefin assay was used to determine the selectivity of potent compounds identified from the screening effort.
Figure 1.
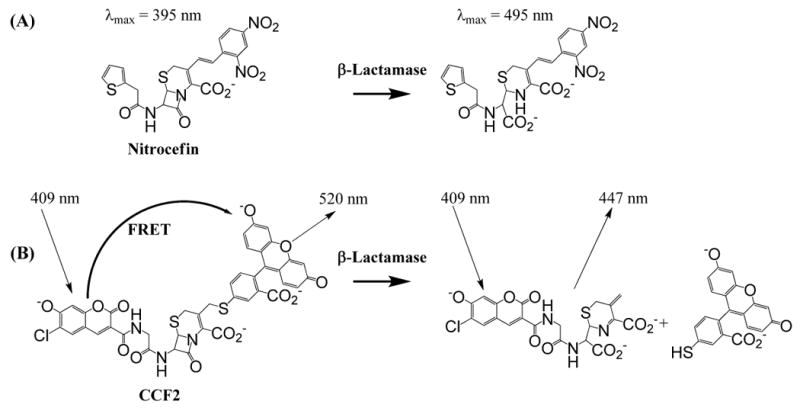
a) Nitrocefin assay principle. A change in absorbance at λ=495 nm is measured after the cephalosporin core of nitrocefin is hydrolyzed by β-lactamase b) CCF2 (FRET) assay principle. In the intact CCF2 substrate, coumarin moiety's donor FRET resulting from λ= 409 nm excitation is efficiently quenched by the acceptor fluorescein moiety; hydrolysis of the β-lactam leads to the increase of a donor fluorescence measured at 447 nm and simultaneous decrease of an acceptor fluorescence at measured at 520 nm.
The enzymology for each assay was optimized to enable its compatibility with high-throughput screening (HTS) techniques 16; final conditions of all assays are presented in Table 1. In summary, it was found that an enzyme concentration of ∼0.1 nM yielded an optimal assay signal in 25 minutes. At this enzyme concentration, the nitrocefin assay window was acceptable when assayed at substrate concentrations of ∼2KM (i.e. 60 μM). The CCF2 assay was able to achieve a comparable assay window at a substrate concentration of ∼1/2 KM (i.e. 10 μM). All enzymatic assays were configured to run as endpoint assays in the presence of high concentrations of zinc (50 μM) and stopped by addition of EDTA at <50% substrate turnover. In these assay conditions, the well-characterized metallo-β-lactamase inhibitor 2-(2-chlorobenzyl) succinic acid (NSC 20707) inhibited IMP-1 with a Ki value (1.6 ± 0.3 μM) comparable to that found in literature (3.3 ± 1.7 μM, 17), and also exhibited modest potency against VIM-2 (IC50 = 33 ± 9 μM) Table 1.
Table 1. Summary of nitrocefin and CCF2 assay parameters, including IC50 values for the positive control inhibitor 2-(2-chlorobenzyl) succinic acid (NSC-20707).
| Enzyme | Format | KM(a), μM | [Enzyme], nM | [Substrate], μM | NSC 20707 IC50(b), μM | S/B(c) | Z′(d) |
|---|---|---|---|---|---|---|---|
| VIM-2 | Nitrocefin | 30 ± 9 | 0.13 | 60 | 33 ± 9 | 3.2 ± 0.1 | 0.75 ± 0.1 |
| IMP-1 | Nitrocefin | 29 ± 4 | 0.1 | 60 | 1.6 ± 0.3 | 3.5 ± 0.1 | 0.81 ± 0.03 |
| VIM-2 | CCF2 | 21 ± 7 | 0.1 | 10 | 50 ± 18 | 4.0 ± 0.1 | 0.79 ± 0.09 |
KM values calculated for IMP-1 and VIM-2 assays ± error reported as the standard deviation of 3 replicates. Reported values for VIM-2 with Nitrocefin is 18 μM11 and IMP-1 with Nitrocefin is 27 μM 56.
IC50 ± error reported as the standard deviation of 16 replicates.
S/B ± error reported as the standard deviation of 24 replicates. The signal to basal (S/B) is defined as the ratio of raw signals from wells with positive and negative controls.
Z′ ± error reported as the standard deviation of 24 replicates. Z'-factor is a measure of assay quality and was calculated according to 32.
LOPAC and Click Chemistry Library screening assays
All LOPAC compounds were tested at a single concentration (14 μM) in triplicate, while click chemistry test compounds were screened as 10-point dose response curves in triplicate. Two different criteria were used to determine activity: in the case of the LOPAC library, a standard inhibition cutoff parameter was calculated (see Experimental section); any compound that yielded an average percent inhibition greater than this cutoff parameter, i.e. ≥8.82% inhibition, was declared active and advanced to dose response experiments. For the click chemistry library screen a compound that yielded an IC50 value less than 10 μM was declared active. The results of two separate artifact assays proved that the inhibitors declared active in the screening assays could not be attributed to intrinsic compound absorbance (see Absorbance artifact assays in Experimental section).
Determination of VIM-2 inhibitor potency, Ki and mechanism
The potency of any compound found active in the LOPAC and click chemistry library screens was determined in the VIM-2 nitrocefin and CCF2 assay formats. The Ki values and mechanism of action for the four most potent compounds (two from LOPAC and two from the click library) were also measured (Figure 2). From the LOPAC screen, mitoxantrone, an anthracenedione antibiotic and antineoplastic, was found to be a pure non-competitive inhibitor of VIM-2 with Ki = K′i = 1.5 ± 0.2 μM. The sulfhydryl reagent 4-chloromercuribenzoic acid (pCMB) 18, 19 was the only other inhibitor of VIM-2 from the LOPAC screen. Rapid dilution experiments with VIM-2 were performed as a first step to characterize its mode of action. 20 The recovery of VIM-2 activity after dilution of pCMB was gradual and only 16.5% of activity was recovered after 15 minutes (data not shown). The near linearity of enzyme recovery over this time period would suggest that this compound acts as a slowly reversible or irreversible inhibitor. Also pCMB was found to have modest potency in the IMP-1 nitrocefin assay (82% inhibition was achieved after 15 minutes of preincubation with IMP-1 and 140 μM pCMB) whereas mitoxantrone was inactive at the range of concentrations tested (Table 2). In contrast to pCMB, mitoxantrone could not be established as an inhibitor in the VIM-2 CCF2 assay due to its interference with the fluorescence emission of CCF2 at 535nm (see FRET artifact assay in Experimental section). The potency of both compounds was not significantly affected by the absence of zinc in the assay buffer (data not shown).
Figure 2. Structures of the four VIM-2 inhibitors found from HTS efforts.

Mitoxantrone and pCMB were found active in LOPAC screening. Compounds 1 and 2 are sulfonyl-triazoles found from click-chemistry library screening.
Table 2. Results of potency and kinetic assays for different VIM-2 inhibitors.
| Compound Name | VIM-2 Ki, μM(a) (mechanism of inhibition) | IC50(b) or Max Inhibition @ Tested Concentration(c) | ||||
|---|---|---|---|---|---|---|
| VIM-2 (Nitrocefin) | VIM-2 (CCF2) | IMP-1 Nitrocefin | TEM-1 Nitrocefin | AmpC Nitrocefin | ||
| Mitoxantrone | 1.5 + 0.2 (noncompetitive) |
0.63 ± 0.04 μM (67% @ 44 μM) |
ND(d) | >56 (0% @ 56 μM) |
>25 (0% @ 25 μM) |
>25 (0% @ 25 μM) |
| pCMB | (Slowly reversible or irreversible) | NA(e) (80% @ 15 μM) |
NA (83% @ 36 μM) |
NA (82% @ 140 μM) |
>25 (0% @ 25 μM) |
>25 (0% @ 25 μM) |
| 1 | 0.41 ± 0.03 (competitive) |
3.3 ± 0.4 μM (79% @ 56 μM) |
1.3 ± 0.1 μM (76% @ 44 μM) |
>56 (0% @ 56 μM) |
>25 (0% @ 25 μM) |
>25 (0% @ 25 μM) |
| 2 | 1.41 ± 0.12 (competitive) |
7.3 ± 1.9 μM (74% @ 56 μM) |
4.1 ± 0.3 μM (90% @ 44 μM) |
>56 (0% @ 56 μM) |
>25 (0% @ 25 μM) |
>25 (0% @ 25 μM) |
Ki ± error reported as the standard deviation of 4 replicates.
IC50 ± error reported as the standard deviation of 3 replicates.
In parenthesis maximum inhibition achieved at the indicated concentration.
ND - Not determined due to assay artifact.
NA – Not applicable due to the nature of inhibition.
Two click chemistry library compounds were identified from the VIM-2 nitrocefin screen. These sulfonamide-1,2,3-triazole analogs (sulfonyl-triazoles), N- ((4-((but-3-ynyloxy)methyl)- 1H-1,2,3-triazol-5-yl)methyl)- 4-iodobenzenesulfonamide (compound 1) and 4-iodo-N-((4-(methoxymethyl)-1H-1,2,3-triazol-5-yl)methyl)benzenesulfonamide (compound 2) were found to be competitive inhibitors of VIM-2 (Table 2). They also demonstrated comparable potency in the VIM-2 nitrocefin and CCF2 assays and their potency was not significantly affected by the absence of zinc in the assay buffer. Both compounds were found to be inactive in the IMP-1 inhibition assay.
All four metallo-β-lactamase inhibitors were tested for potency against serine β-lactamases from Ambler class A (TEM-1) and C (AmpC). None of the tested compounds exhibited inhibitory activity against either TEM-1 or AmpC at the highest concentration tested 25 μM (Table 2).
MIC assays
Mitoxantrone, pCMB, as well as compounds 1 and 2 were further tested for their inherent antimicrobial activity. Minimal inhibitory concentration (MIC) values were measured in both resistant (BL21/VIM-2) and non-resistant (BL21) E. coli, with imipenem as a positive control. The sulfonyl-triazoles, 1 and 2, exhibited no efficacy at the highest concentrations tested (60 μg/mL). In contrast, mitoxantrone exhibited antibacterial activity (MIC = 8.4 μg/mL) against both resistant (BL21/VIM-2) and non-resistant (BL21) E.coli (Table 3). Similarly, pCMB demonstrated antibacterial activity (MIC = 17.9 μg/mL) against resistant (BL21/VIM-2) and non-resistant (BL21) E.coli. For comparison, the MIC of imipenem against resistant (BL21/VIM-2) and non-resistant (BL21) E. coli was 1.9 μg/mL and 0.2 μg/mL, respectively.
Table 3. Results of VIM-2 inhibitor MIC and inhibitor plus imipenem MIC potentiation assays.
All experiments were repeated at least on three different days and MIC values were determined as per CLSI guidelines (11). The E. coli strain ATCC 25922 was used as a quality control reference.
| Compound Name | Compound MIC (μg/mL) | Imipenem MIC @ 50 μM of compound, μg/mL | |
|---|---|---|---|
| BL21 | BL21+VIM-2 | BL21+VIM-2 | |
| Mitoxantrone | 8.4 | 8.4 | 0.2 |
| pCMB | 17.9 | 17.9 | 0.2 |
| 1 | > 60 | > 60 | 1.9 |
| 2 | > 60 | > 60 | 1.9 |
| Imipenem | 0.2 | 1.9 | - |
Inhibitor/Antibiotic Synergy Assays
The four compounds were also tested for their ability to reverse the resistance of VIM-2 expressing E. coli to imipenem challenge. In nonresistant E. coli (BL21) the MIC for imipenem was 0.2 μg/mL; in resistant E. coli (BL21/VIM-2) the MIC was ∼9 fold less potent (i.e. 1.9 μg/mL). To assess potentiation of imipenem efficacy by a single dose of inhibitor, imipenem MIC values were calculated in the presence of 50 μM of each of the four inhibitors. Mitoxantrone and pCMB were able to completely reverse the effects of VIM-2 on imipenem potency, while the click chemistry sulfonyl-triazoles did not demonstrate potentiation of imipenem (Table 3).
Mitoxantrone and pCMB were further tested against resistant E. coli (BL21/VIM-2) using a checkerboard microdilution method. 21-23 Ranges of pCMB and mitoxantrone concentrations that encompassed their respective MICs were tested for potentiation of imipenem efficacy. As determined by methods described previously 23 both pCMB and mitoxantrone exhibited synergy with imipenem when present at concentrations as low as 2.2 μg/mL and 2.1 μg/mL, respectively (Table 4).
Table 4. Activity of Imipenem in Combination with pCMB and Mitoxantrone vs. BL21/VIM2 E. coli.
Results obtained from checkerboard microdilution experiment. Shaded values are combination of Imipenem and inhibitor that were found to be synergistic.
| Imipenem + pCMB | Imipenem + Mitoxantrone | |||
|---|---|---|---|---|
| Imipenem MIC, μg/mL | p-CMB, μg/mL | Imipenem MIC, μg/mL | Mitoxantrone, μg/mL | |
| MIC of Imipenem in Presence of Inhibitor, μg/mL | <0.1 | 35.8 | <0.1 | 66.9 |
| <0.1 | 17.9 | <0.1 | 33.5 | |
| 0.1 | 8.9 | <0.1 | 16.7 | |
| 0.1 | 4.5 | <0.1 | 8.4 | |
| 0.5 | 2.2 | 0.2 | 4.2 | |
| 0.9 | 1.1 | 0.5 | 2.1 | |
| 1.9 | 0.6 | 0.5 | 1.0 | |
| 1.9 | 0.3 | 1.9 | 0.5 | |
| 1.9 | 0.1 | 1.9 | 0.3 | |
| MIC alone, μg/mL | 1.9 | 17.9 | 1.9 | 8.4 |
Determination of bacteriostatic/bactericidal properties of pCMB and mitoxantrone
“Time kill” experiments were performed to assess bacteriostatic/bactericidal properties of pCMB and mitoxantrone. 23 Both pCMB and mitoxantrone were tested against resistant (BL21/VIM-2) and non-resistant (BL21) E. coli over a period of 24 hours at concentrations of 2.2 and 2.1 μg/mL, respectively. The effect of 2.1 μg/mL mitoxantrone was virtually indistinguishable from that of the uninhibited growth control in both imipenem-resistant and non-resistant E. coli (data not shown). Although 2.2 μg/mL of pCMB exhibited slight growth inhibition for the first 6 hours of the experiment, this was followed by growth to the level of the uninhibited control.
Modeling and Docking with VIM-2
Docking studies were performed in an attempt to bring insight in to the mechanism of VIM-2 inhibition (Figure 3). The PDB structure 1KO3 for VIM-2 was used for the modeling studies, since this structure was of high resolution (1.9 Å) and provided the most open conformation for the active site. The ligand induced structure of VIM-2 with bound inhibitor phenylC3SH (PDB 2YZ3) was used for modeling comparison. To interrogate inhibitor binding requirements in the VIM-2 active site, attempts were made to dock all four compounds into the VIM-2 active site from the native (PDB 1KO3) and inhibitor bound (PDB 2YZ3) structures. The inhibitors from the LOPAC screen failed to dock rationally into either structure. However, docking studies with the two inhibitors from the click chemistry library proved more fruitful: both compounds were predicted to bind to the VIM-2 active site through the sulfonyl group as a zinc binding group (Figure 3).
Figure 3.
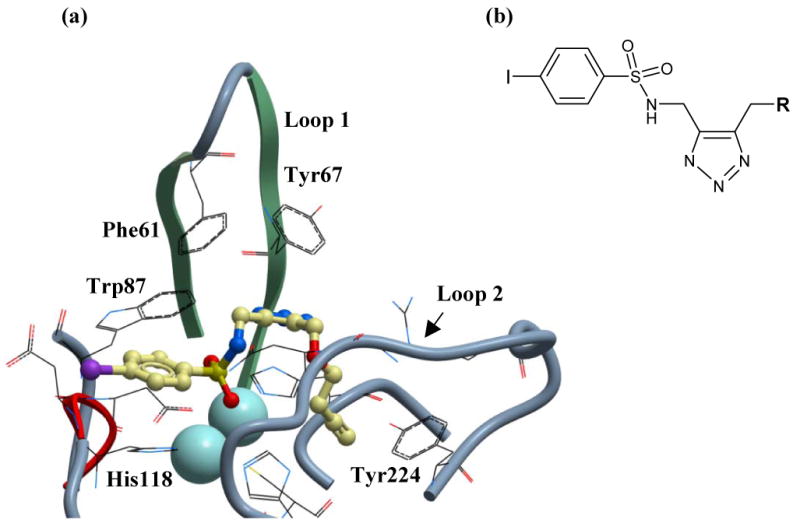
(a) Proposed mode of binding of sulfonyl-triazole compound 1 to VIM-2. VIM-2 is depicted in ribbon form and the two loops that make key interactions with active site binders are indicated. Compound 1 is positioned as a binder of the binuclear zinc cluster through the sulfonyl group and shown as a stick model, with colors as follows: Carbon is yellow, oxygen is red, nitrogen is blue, iodine is purple and sulfur is yellow. The sulfonamide group is surface exposed, whilst the propargyl group (R) buries into a hydrophobic pocket. Highlighted residues are implicated in key binding interactions with compound 1. (b) Click sulfonyl-triazole chemotype identified as selective VIM-2 inhibitors. The chemotype is discussed in text.
5. Discussion
Screening for VIM-2 Inhibitors
Several structural and phylogeny studies show that the MBLs are members of an ancient metalloenzyme superfamily 12. The active site for this enzyme class is formed by a shallow cleft containing one or two Zn2+ cofactor ions and two flexible loops. 24, 25, 26 The plasticity of these loops is thought to enable this enzyme class to hydrolyze a broad spectrum of β-lactam antibiotic substrates. Recently, the importance of these loops for inhibitor recognition in VIM-2 was demonstrated by solving the structure of this enzyme with a potent mercaptocarboxylate inhibitor 26 (PDB 2YZ3). The first loop (Loop1: Phe61 to Ala64) makes key aromatic and hydrophobic interactions with the bound inhibitor through residues Phe61 and Tyr67, while the other loop (Loop2: Ile223-Trp242) undergoes a ligand-induced side chain reorientation of Asn233 to form a salt bridge with the carboxylate of the inhibitor.
The potency of the well-characterized and potent IMP-1 inhibitor NSC20707 in the VIM-2 assay serves as an example of how compounds bearing zinc-binding moieties can be useful MBL inhibitors. Although the VIM-2 mercaptocarboxylate inhibitor described above was not available for this study, it was anticipated that NSC20707 (which has a zinc-chelating succinate moiety) would inhibit VIM-2 activity. Since its succinate group binds to the binuclear zinc cluster in IMP-1, 27 and IMP-1 and VIM-2 share this zinc cluster, it was not surprising that NSC20707 demonstrated modest potency in both the nitrocefin (IC50 = 33 ± 9 μM) and CCF2 (IC50 = 50 ± 18 μM) VIM-2 enzymatic assays (Table 1).
Click chemistry library composition
The principles and benefits of click chemistry are described in detail elsewhere 28. The goal of click chemistry is to use robust, high yielding and clean reactions to access diverse compound libraries. This rapid and reliable approach, in turn, reduces the amount of time taken to produce biologically active molecules. The click chemistry library employed here was specifically designed to probe the active site of VIM-2 via compounds containing moieties that have demonstrated activity against a variety of metalloenzymes (Figure 4). These include known and putative zinc-binding fragments, such as hydroxamates, amides, ureas, thiadiazoles, carboxylates and the sulfonyl-triazoles 1 and 2. The sulfonamide moiety of inhibitors 1 and 2 is known to be a zinc binding group 29 and has been used in the design of inhibitors of the metalloenzyme, carbonic anhydrase. 30 However, in order to avoid the discovery of non-specific zinc chelators, the VIM-2 nitrocefin screens were conducted in the presence of high concentrations (50 μM) of zinc. Consequently, this high concentration of zinc served to abrogate inhibition derived solely from zinc-binding. Despite the presence of zinc binding groups, the activity of 1 and 2 appears independent of the concentration of zinc in the assay buffer; this pharmacology will be further characterized in future studies.
Figure 4. Click chemistry strategy to synthesize selective VIM-2 inhibitors.
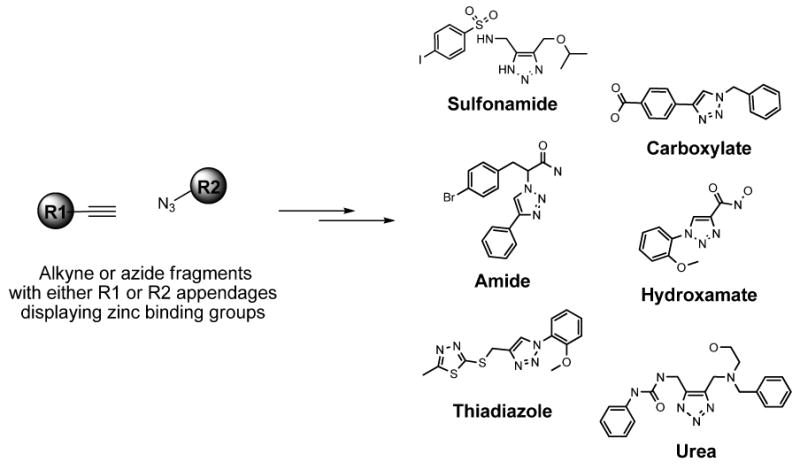
Representative N-substituted and N-unsubstituted 1,2,3-triazoles from alkyne and azide fragments with known or putative zinc binding groups are shown including hydroxamates, amides, ureas, thiadiazoles, carboxylate and sulfonamides.
Assay development
Typical of screening assay development described elsewhere 16 the enzymatic assays described here were optimized to meet HTS-specific criteria. Since HTS assays are usually run against large compound libraries (n=105-106 compounds), they should be amenable to automation, economically feasible and robust without sacrificing the desired pharmacology being measured. Therefore, all enzymatic assays were miniaturized and run in 1536-well microtiter plates as homogeneous, endpoint (rather than kinetic) experiments and configured to use sparing amounts of reagents. Unfortunately, previously described VIM-2 inhibitors were not available for this study; 31 however, the IMP-1 inhibitor NSC20707 served as a useful positive control for both the VIM-2 and IMP-1 assays, allowing the evaluation of assay performance (i.e. Z-factor 32).
Choice of CCF2 VIM-2 assay as a counterscreen
Although more commonly used for mammalian reporter gene assays, 15 the use of the CCF2 substrate in microbiology applications has been previously described. 33, 34 In the present study, the KM value of CCF2 for VIM-2 was determined to be 21 ± 7 μM. This result is comparable to that previously measured for the enzyme in the nitrocefin format (18 μM). 11 Additionally the VIM-2 CCF2 assay yielded reproducible, high quality data that were comparable to those obtained from the nitrocefin assay (Table 1). However, the CCF2 assay is prone to FRET artifact, as evidenced by its inability to detect inhibition of VIM-2 by the colored compound mitoxantrone. Taken together, these results suggest that the CCF2 and nitrocefin substrates may complement each other for inhibition studies.
The LOPAC library gauges assay compatibility for HTS
The LOPAC library was screened primarily to validate the compatibility of the VIM-2 nitrocefin assay to a larger HTS effort. In this regard the VIM-2 nitrocefin assay appears to be HTS-compatible as demonstrated by high assay Z-factor (i.e. >0.5) and a low percentage of potent compounds (∼0.1%, i.e. <1% overall) resulting from the LOPAC screening effort. 32 Also important to note is that none of the compounds that appeared active in LOPAC screening could be attributed to their interference with the measured absorbance; this result signifies that this type of absorbance artifact may be negligible in a larger-scale HTS campaign. Although the CCF2 VIM-2 and nitrocefin IMP-1 assays were not subjected to LOPAC screening for this study, their high Z′-factor suggest that they may also be suitable for HTS.
Since the LOPAC library consists of a diverse set of 1280 well-known chemical probes to a variety of eukaryotic and prokaryotic targets, it is reasonable to assume that a small number of these compounds would be found active in any given screening effort. In the context of the enzymatic assays presented here, the confirmation of pCMB as an inhibitor of VIM-2 and IMP-1 represents an example of this utility. In contrast, mitoxantrone was not expected to be a potent inhibitor of VIM-2. Although its exact mechanism of action at this point is unclear its identification confirms another utility of the LOPAC, viz. to probe well-characterized compounds for activity in an alternative pharmacology.
Characterization of the VIM-2 competitive inhibitors
The primary purpose of the VIM-2 CCF2 and IMP-1 nitrocefin assays was to confirm VIM-2 inhibition and selectivity of compounds found active in the VIM-2 nitrocefin assay, respectively. Their utility has been demonstrated here by characterizing a novel class of selective click-chemistry derived sulfonyl-triazole VIM-2 inhibitors. In particular, compound 1 has comparable potency (Table 2, Ki = 0.41 ± 0.03 μM) to a previously reported mercaptocarboxylate class of VIM-2 inhibitor, i.e. rac-2-ω-phenylpropyl-3-mercaptopropionic acid (PhenylC3SH, Ki = 0.22 μM). 31 In contrast to the mercaptocarboxylates, the two sulfonyl-triazoles described here demonstrate complete selectivity for VIM-2.
Although the sulfonyl-triazoles are potent in vitro, the MIC results show that they do not have intrinsic antibacterial properties nor potentiate the in situ potency of imipenem. From analysis of the MIC studies it can be assumed that the lack of efficacy for sulfonyl-triazoles is due to their inability to reach and/or remain in the E. coli periplasm in sufficient concentration to abrogate imipenemase activity. It is currently unclear whether this is because of a bacterial resistance mechanism or whether they are perhaps interacting with off-target entities. Future studies with hyperpermeable and efflux deficient strains will address this issue.
Biochemical characterization of VIM-2 LOPAC inhibitors
Mitoxantrone was found to be a potent, non-competitive inhibitor of VIM-2 with a Ki of 1.5 ± 0.2 μM. When tested with IMP-1, TEM-1, and AmpC it did not exhibit any inhibitory activity, (Table 2) which indicates that its interaction with VIM-2 is specific. In the absence of structural studies it is difficult to predict the mode of binding to VIM-2 by this compound. Further structural and mechanistic investigations, such as mutual exclusivity studies, 20 will be required to pinpoint the mode of binding.
In contrast to mitoxantrone, pCMB demonstrated potency against IMP-1. (Table 2) This cysteine-reactive reagent, which forms covalent or slowly reversible electrostatic bonds with sulfhydryl groups, is widely used for enzyme characterization, including beta-lactamases. 19, 35-37 38 With this in mind, a mechanism of action can be proposed for VIM-2 and IMP-1 inhibition. Both enzymes share conserved binuclear zinc binding motifs, whereby three histidine residues coordinate one zinc ion; cysteine (i.e. Cys221), aspartate and another histidine coordinate the other ion. 10 It has been shown 12 that once oxidized, Cys221 loses its ability to participate in coordination of the second zinc ion, which deleteriously affects enzyme activity. 24, 39 Therefore it is reasonable to assume that inhibition of VIM-2 and IMP-1 occurs as a result of pCMB reacting with the corresponding cysteine residues. Thus the fact that pCMB exhibited no inhibitory activity against TEM-1 and AmpC (Table 2) is in agreement with literature findings that cysteine residues are not involved in catalysis by these enzymes. 40-42
Bacterial characterization of VIM-2 LOPAC inhibitors
When tested alone mitoxantrone and pCMB exhibited antibacterial activity against resistant (BL21/VIM-2) and non-resistant (BL21) E. coli (MIC = 8.4 and 17.9 μg/mL, respectively, Table 3). Mitoxantrone is a well-characterized chemotherapeutic 43 and is known to have antibacterial and antiviral activity. 44 This has been attributed to its affinity towards double and single-stranded RNA and DNA. 45 Similarly, the antibacterial action of pCMB may be attributed to its ability to modify functionally important cysteine residues in many proteins. For example, pCMB was shown to irreversibly dissociate bacterial ribosomes, thereby inhibiting protein synthesis. 36
At concentrations below the MIC for these compounds checkerboard experiments showed that mitoxantrone at 2.1 μg/mL or pCMB at 2.2 μg/mL can restore the potency of imipenem against resistant E. coli (BL21/VIM-2), (Table 4) At these lower concentrations no toxicity to E. coli cells was observed. However, as MIC experiments measure a single timepoint, time-kill experiments were performed to study the kinetics of growth inhibition induced by these compounds. Although at 2.2 μg/mL pCMB slightly inhibited the growth at 6 hours, the reduction of colony forming units as compared to the negative control was less than three log units, which is not sufficient to classify this compound as being bactericidal or bacteriostatic. Conversely, mitoxantrone at 2.1 μg/mL had no effect on E. coli growth. As a result, time-kill experiments showed conclusively that pCMB and mitoxantrone are not bactericidal or bacteriostatic at 2.2 and 2.1 μg/mL, respectively and therefore in synergy experiments must act primarily by inhibiting VIM-2 in situ.
Docking studies with competitive inhibitors
To interrogate inhibitor binding requirements in the VIM-2 active site, attempts were made to dock all four compounds into the active site of VIM-2 from the native (PDB 1KO3) and inhibitor bound (PDB 2YZ3) structures. Important to note, the active site from the inhibitor-bound structure (PDB 2YZ3) is too small to accommodate these compounds since Asn233, the residue that forms a salt bridge with the inhibitor, occludes part of this hydrophobic cavity. This orientation of Asn233 and surrounding residues on Loop2 determine the size and shape of the hydrophobic cavity.
The inhibitors from the LOPAC screen failed to dock rationally into either structure. Although mitoxantrone has the ability to complex zinc through its alcohol, carbonyl, or amine groups, a realistic docking result was not achieved. This is consistent with its mode of action as a non-competitive VIM-2 inhibitor. Although pCMB is small and fits easily into the VIM-2 active site its mechanism of action is believed to be through formation of a covalent or slowly reversible electrostatic bond with sulfhydryl groups. As such, modeling studies with standard docking protocols would not be expected to identify a realistic ligand-protein docking pose for this class of inhibitor.
Docking studies with the two inhibitors from the click chemistry library proved more fruitful (Figure 3). Compounds 1 and 2 are predicted to bind to VIM-2 by exploiting the sulfonyl moiety as a binder of the active site binuclear zinc cluster. One zinc atom is chelated by both oxygen atoms, with distances of 2.5 Å and 2.9 Å away from the metal. Additionally, one oxygen of the sulfonyl group is located between the zinc cluster with distances of 2.5 Å and 3.0 Å from the metals. As comparison, the free thiol of the VIM-2 inhibitor PhenylC3SH, in PDB 2YZ3, is 2.2 Å away from one zinc in the binuclear cluster. The oxygen to zinc binding distances observed in the current docking studies is longer than expected (< 2.5 Å), but this is likely a consequence of the slight loss in atomic detail when performing molecular docking. The predicted mode of binding for 1 orients the molecule such that the arylsulfonamide is solvent exposed while the alkoxyl group (R) buries into a hydrophobic subpocket at the base of the active site. From these studies, it appears that compound 1, which displays the larger propargyl group, best exploits this hydrophobic cavity. In the docking model the propargyl is 3.3 Å away from Tyr224, but it is likely that the aromatic side chain of this residue will orient to form a better hydrophobic interaction with the propargyl. Furthermore the ether oxygen of 1 is 2.3 Å from the backbone amide hydrogen of Asn233 and likely forms a hydrogen bond.
Modeling studies also implicate the arylsulfonamide and triazole moieties as potentially being involved in aromatic interactions with four residues that ring the active site, namely His118, Trp87 and Phe61 and Tyr67 in Loop1. Both arylsulfonamides 46 and triazoles 47 can make strong interactions with aromatic groups through pi-stacking (edge or face) as well as other hydrophobic modes of binding. The triazole moiety is 2.7 Å from Trp87, while the benzyl group of the sulfonamide is 3.0 Å from His118. Similarly, the triazole group is 3.2 Å from Tyr67 and 5.6 Å from Phe61. However, as compounds were docked in to a rigid representation of the native structure the importance of all four aromatic residues for inhibitor binding cannot be fully appreciated, since ligand-induced changes will be required for the correct orientation of these residues. This is particularly relevant for Phe61 and Tyr67 as the loop to which they belong is known to be highly flexible. 26
SAR of the sulfonyl-triazole scaffold
In order to investigate structure-activity relationship (SAR) amongst the sulfonyl-triazoles, an in silico study was performed by retrieving VIM-2 screening results of click chemistry library compounds containing this scaffold (Table 5). Among the group of structural analogs, some modest inhibition was observed for three of the compounds at the highest test concentration (56 μM). Of course, only 1 and 2 demonstrated potency against VIM-2; however inspection of all compounds may suggest that steric hindrance prevents inhibition of the VIM-2 active site. For example, compound 1 differs from 2 by the presence of an alkyne on its alkoxyl group. Compound 3 is closely related but displays a bulkier alkoxy group. Presumably, the larger size of this dioxolane moiety is detrimental to inhibition of VIM-2. Compounds 4 to 6 display even bulkier R groups which is consistent with them being inactive against VIM-2.
Table 5. SAR of sulfonyl-triazoles within the click-chemistry library.
| ID | Scaffold | R | IC50 or Max Inhibition(a) |
|---|---|---|---|
| 1 | 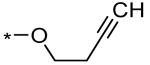 |
3.3 ± 0.4 μM (79% @ 56 μM) | |
| 2 | 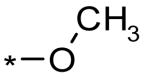 |
7.3 ± 1.9 μM (74% @ 56 μM) | |
| 3 | 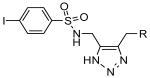 |
 |
>56 μM (31% @ 56 μM) |
| 4 | 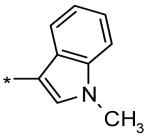 |
>56 μM (11% @ 56 μM) | |
| 5 | 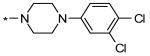 |
>56 μM (5% @ 56 μM) | |
| 6 | 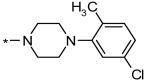 |
>56 μM (0% @ 56 μM) |
IC50 ± error reported as the standard deviation of 3 replicates or in parenthesis maximum inhibition achieved at the indicated concentration in the nitrocefin assay for VIM-2.
As represented in Figures 2 and 3b and Table 5, the sulfonyl-triazole class represents a novel scaffold for further development of more potent and/or selective VIM-2 inhibitors. Furthermore they contain “click chemistry” moieties. Since click-chemistry compounds (by definition) have chemical features which facilitate rapid study of SAR, 28, 48 our research has been directed to improvement of sulfonyl-triazole potency and efficacy. Based on the results of the studies presented here, a medicinal chemistry effort is being pursued to improve the potency for this chemotype against VIM-2 by further elaborating structure activity relationships (SAR) around the arylsulfonamide and R substituents. In particular, the importance of the substitution pattern around the arylsulfonamide (which is predicted to be solvent exposed) as well as the hydrophobicity of the R position (which is predicted to bind in a deeper hydrophobic cavity) merits further exploration.
6. Conclusion
In conclusion, a repertoire of HTS-compatible assays was used to aid the identification of VIM-2 inhibitors. These assays identified a novel class of competitive and selective VIM-2 inhibitors, represented here by click chemistry library compounds 1 and 2. The same assays were also used to identify and characterize two well-known pharmacologic agents from the LOPAC library, mitoxantrone and pCMB, which appear to be non-competitive and irreversible / slowly reversible VIM-2 inhibitors, respectively. The click chemistry library inhibitors did not show efficacy in MIC assays, whereas the LOPAC inhibitors were able to restore efficacy of β-lactam antibiotic imipenem via VIM-2 inhibition in situ. Further studies will lead to a better understanding of their exact mechanism of action, and aim to improve the efficacy of the competitive click-chemistry derived inhibitors described here.
7. Experimental
Sources of screening compounds
The inhibition control compound NSC 20707 (2-(2-chlorobenzyl) succinic acid) was obtained from the Drug Synthesis and Chemistry Branch of National Cancer Institute (MD, USA). The library of pharmacologically active compounds (LOPAC) used for screening was purchased from Sigma-Aldrich (St. Louis, MO). The 267-member click chemistry library was from an in-house compound collection. The methods for click chemistry syntheses presented below represent generalized protocols and more detailed syntheses will be presented in a future publication.
General synthetic procedures for “click chemistry” compounds
Reagents and solvents were purchased from commercial sources and were used as received. Reaction progress was monitored by TLC using Merck silica gel 60 F-254 with detection by UV. Silica gel 60 (Merck 40–63 μm) was used for column chromatography. 1H NMR and 13C NMR spectra were recorded with Bruker AMX-300 spectrometers. Proton magnetic resonance (1H NMR) spectra were recorded at 300 MHz. Data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t= triplet, q = quartet, quin = quintet, sep = septet, m = multiplet, b = broad), coupling constant, J (Hz) and integration. Carbon magnetic resonance (13C NMR) spectra were recorded at 75 MHz. Data for 13C NMR are reported in terms of chemical shifts (ppm). HPLC homogeneities were determined using an Agilent 1100 LC/MSD with an Agilent 1100 SL mass spectrometer. System A: Zorbax 4.6 mm × 30 mm, SB-C18 reverse phase column, preceded by a Phenomenex C18 guard column, eluting with 10-100 % MeCN (+ 0.05 % TFA) in 0.05 % TFA, linear gradient over 10 min then isocratic for 5 min, 0.5 mL/min flow rate with UV detection at 254 nm. System B: Zorbax 4.6 mm × 150 mm, SB-C18 reverse phase column, preceded by a Phenomenex C18 guard column, eluting with 10-100 % MeOH (+ 0.05 % TFA) in 0.05 % TFA, linear gradient over 10 min then isocratic for 10 min, 0.5 mL/min flow rate with UV detection at 254 nm.
Synthesis of the sulfonamide derivatives from the corresponding sulfonylchlorides
To a solution of the sulfonylchloride in THF/H2O (5 mL/mmol of sulfonylchloride) was added K2CO3 (3.0 equiv) followed by 4-chlorobut-2-yn-1-amine hydrochloride (1.3 equiv) at 0 °C. The reaction was stirred at 0 °C for 1 h before allowed to warm up to room temperature. The reaction mixture was diluted with EtOAc (5 mL/mmol of acid) and washed twice with 1 M HCl (20 mL/mmol of acid), NaHCO3 (6 mL/mmol of acid) and brine (6 mL\mmol of acid). The organic portion was dried over MgSO4 before being concentrated under reduced pressure to furnish the crude product. Purification by crystallization from Hex/EtOAc afforded the corresponding sulfonamide in almost quantitative yield as a white solids.
4-Iodo-N-(4-chlorobut-2-ynyl)benzenesulfonamide
1H NMR (300 MHz, DMSO-d6): δ = 7.96 (d, J = 8.7, 2H), 7.52 (d, J = 8.7, 2H), 4.22 (t, J = 6.0, 1H), 3.80 (dt, J = 6.0, 1.8, 2H), 3.70 (s, 2H); LC-MS m/z (%): 391.9 (100) [M+Na]+ (calc. 391.90).
Synthesis of the azide derivatives from the corresponding chlorides
To a solution of N-(4-chlorobut-2-ynyl)arylsulfonamide in DMF (2 mL/mmol of chloride) was added NaN3 (2.0 equiv). The resulting suspension was stirred at room temperature for 12 h before being quenched by the addition of saturated NaCl (10 mL/mmol of chloride). The reaction mixture was extracted two times with Et2O (10 mL/mmol of chloride) and the combined organic layers were dried over MgSO4 before being concentrated under reduced pressure at room temperature to furnish the crude product. The crude product was used for the next reaction without further purification.
Thermally induced Banert Cascade; procedure for the synthesis of NH-triazole-arylsulfonamides
To a solution of the azide (1 mL/mmol of azide) in dioxane was added the nucleophile (1 mL/mmol of azide; 2.5 equiv. for a solid nucleophile). The reaction was stirred at 75 °C for 4 h. Subsequently the reaction was cooled to room temperature and the solvent concentrated under reduced pressure to furnish the crude product. The title compounds were purified by flushing through a short pad of silica gel (50-100% EtOAc in hexanes to 10% MeOH in EtOAc with 0.5% NEt3) and obtained as yellow oils. Solid compounds were purified by crystallization from Et2O/hexane.
4-Iodo-N-((4-((but-3-ynyloxy)methyl)-1H-1,2,3-triazol-5-yl)methyl)benzenesulfonamide (1)
1H NMR (300 MHz, DMSO-d6): δ = 8.17 (s, 1H), 7.96 (d, J = 8.1, 2H), 7.52 (d, J = 8.1, 2H), 4.47 (s, 2H), 4.08 (s, 2H), 3.44 (t, J = 6.9, 2H), 2.77 (t, J = 2.4, 1H), 2.37 (dt, J = 6.9, 2.4, 2H); 13C NMR (75 MHz, DMSO-d6): δ = 139.9, 137.9, 100.4, 81.7, 71.9, 67.7, 19.0; LC-MS m/z (%): 447.2 (100) [M+H]+ (calc. 447.00), 469.1 (55) [M+Na]+ (calc. 468.98); Yield: 92%.
4-Iodo-N-((4-(methoxymethyl)-1H-1,2,3-triazol-5-yl)methyl)benzenesulfonamide (2)
1H NMR (300 MHz, DMSO-d6): δ =8.25 (s, 1H), 7.95 (d, J = 8.4, 2H), 7.53 (d, J = 8.4, 2H), 4.33 (s, 2H), 4.01 (s, 2H), 3.15 (s, 3H); 13C NMR (75 MHz, DMSO-d6): δ = 139.9, 137.9, 128.2, 100.3, 63.4, 57.4, 45.5; LC-MS m/z (%): 409.1 (100) [M+H]+ (calc. 408.98), 431.0 (40) [M+Na]+ (calc. 430.97), 839.0 (15) [2M+Na]+ (calc. 838.94); Yield: 94%.
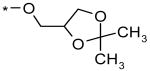
4-Iodo-N-((4-(((2,2-dimethyl-1,3-dioxolan-4-yl)methoxy)methyl)-1H-1,2,3-triazol-5-yl)methyl)benzenesulfonamide (3)
1H NMR (300 MHz, DMSO-d6): δ =7.95 (d, J = 8.4, 2H), 7.53 (d, J = 8.4, 2H), 4.78 (s, 1H), 4.01-3.94 (m, 5H), 3.65-3.60 (m, 2H), 1.30 (s, 3H), 1.25 (s, 3H); LC-MS m/z (%): 509.1 (100) [M+H]+ (calc. 509.04), 431.0 (55) [M+Na]+ (calc. 531.02); Yield: 74%.
4-Iodo-N-((4-((1-methyl-1H-indol-3-yl)methyl)-1H-1,2,3-triazol-5-yl)methyl)benzenesulfonamide (4)
1H NMR (300 MHz, DMSO-d6): δ =8.17 (t, J = 5.4, 1H), 7.91 (d, J = 8.1, 2H), 7.52-7.34 (m, 4H), 7.14-6.95 (m, 3H), 3.99-3.95 (m, 4H), 3.78 (s, 3H); LC-MS m/z (%): 508.1 (100) [M+H]+ (calc. 508.03), 530.1 (55) [M+Na]+ (calc. 530.01); Yield: 65%.
N-((4-((4-(3,4-dichlorophenyl)piperazin-1-yl)methyl)-1H-1,2,3-triazol-5-yl)methyl)-4-iodobenzenesulfonamide (5)
1H NMR (300 MHz, DMSO-d6): δ = 8.30 (s, 1H), 7.95 (d, J = 8.4, 2H), 7.54 (d, J = 8.4, 2H), 7.25 (d, J = 9.0, 1H), 7.10 (d, J = 3.0, 1H), 6.91 (dd, J1 = 9.0, J2 = 3.0, 1H), 4.11 (s, 2H), 3.53 (s, 2H), 3.10 (s, 4H), 2.41 (s, 4H); LC-MS m/z (%): 607.2 (100) [M+H]+ (calc. 606.99); Yield: 69%.
N-((4-((4-(5-chloro-2-methylphenyl)piperazin-1-yl)methyl)-1H-1,2,3-triazol-5-yl)methyl)-4-iodobenzenesulfonamide (6)
1H NMR (300 MHz, DMSO-d6): δ = 8.34 (s, 1H), 7.96 (d, J = 8.7, 2H), 7.55 (d, J = 8.7, 2H), 7.17 (d, J = 8.4, 1H), 7.01-6.95 (m, 2H), 4.12 (s, 2H), 3.55 (s, 2H), 2.78 (s, 4H), 2.44 (s, 4H), 2.19 (s, 3H); LC-MS m/z (%): 587.1 (100) [M+H]+ (calc. 587.05); Yield: 68%.
IMP-1 and VIM-2 nitrocefin kinetic assays
All reagents were obtained from Invitrogen (USA) unless noted below. IMP-1, VIM-2, and Nitrocefin (BD Diagnostic Systems, USA) working solutions were prepared in buffer containing 50mM HEPES, 50μM ZnSO4 Sigma-Aldrich (St. Louis, MO), 0.05% Brij 35 Sigma-Aldrich (St. Louis, MO). Kinetic assays were conducted by incubating the range of substrate concentrations (10-100 μM) with 0.1 nM enzyme at room temperature. Determinations of inhibition constants and modalities were conducted by incubating the range of substrate concentrations (10-100 μM) with 0.1 nM enzyme at room temperature in presence of varying concentrations of inhibitors. Absorbance was measured by a Tecan Safire2 monochromator microplate reader at 495 nm. Initial velocities were obtained from plots of absorbance at 495 nm versus time, using data points from only the linear portion of the hydrolysis curve. Substrate hydrolysis was continuously monitored.
VIM-2 CCF2 kinetic assays
VIM-2 and CCF-2 (Invitrogen, USA) working solutions were prepared in buffer containing 50mM HEPES, 50 μM ZnSO4, 0.05% Brij 35, pH 7.1. Kinetic assays were conducted by incubating the range of substrate concentrations (5-50 μM) with 0.1 nM enzyme at room temperature. Fluorescence resonance energy transfer (FRET) was measured by a Tecan Safire2 using excitation = 409 nm and emission = 447 nm. Initial velocities were obtained from plots of fluorescence at 447 nm versus time, using data points from only the linear portion of the hydrolysis curve. Substrate hydrolysis was continuously monitored.
Rapid dilution experiments
The rapid dilution technique is described in detail elsewhere. 20 VIM-2 was incubated for 30 minutes at room temperature with or without 24 μM pCMB. This high concentration of pCMB corresponds to 10× of its IC50 in the VIM-2 nitrocefin assay (Table 2) and resulted in 90% inhibition. A rapid 1:100 dilution was made directly into 60 μM nitrocefin solution (the solution's composition was identical to the VIM-2 nitrocefin microtiter plate assay described below). Absorbance readings were initiated immediately after the addition of corresponding high and low concentration mixtures. Absorbance was measured on a Safire2 (Tecan, USA) monochromator microplate reader at 495 nm for 30 minutes at 1 minute intervals. Absorbance at 495nm was plotted vs. time. Percent activity in test wells was calculated according to the following equation:
Where “Test” is defined as the absorbance measured from wells containing Nitrocefin, VIM-2, and pCMB; “Positive Control” is defined as the absorbance measured from wells containing Nitrocefin and VIM-2; and “Negative Control” is defined as the absorbance measured from wells containing Nitrocefin alone.
Determination of kinetic parameters
Initial velocities were determined from linear portions of plots of fluorescence at 447nm or absorbance at 495nm versus time. Initial velocities were used to determine kinetic parameters utilizing GraphPad Prism version 5.01 (GraphPad Software, Inc., La Jolla, CA). KM values were determined by non-linear regression analysis using the one site hyperbolic binding model 49 and additionally evaluated by linear analysis. All Ki values were determined by non-linear regression (hyperbolic equation) analysis using the mixed inhibition model which allows for simultaneous determination of mechanism of inhibition. 49 Mechanism of inhibition was determined using the “alpha” parameter derived from a mixed-model inhibition by GraphPad Prism. Kinetic responses over a range of substrate concentrations and with several inhibitor concentrations were analyzed with Hanes-Woolf reciprocal plots. The mechanism of inhibition determined by this analysis (see supplementary information) agreed with non-linear regression (hyperbolic equation) analysis using the mixed inhibition model in GraphPad Prism.
Nitrocefin microtiter plate assays
The nitrocefin assay began by dispensing 2.5μL of 0.26 nM VIM-2 (or 0.2nM IMP-1) to the appropriate wells of a 1536-well microtiter plate (Greiner, USA). Following enzyme addition, 28nL of controls or test compounds were added to the appropriate wells. The plates were then incubated at room temperature for 15 minutes. To start the reaction, 2.5μL of 120μM nitrocefin was dispensed. Final concentrations of 60μM for substrate and enzyme were either 0.13 nM (VIM-2) or 0.1 nM (IMP-1). The plate was then incubated for 25 minutes at RT and the reaction stopped by the addition of 5μL of 500mM EDTA (Invitrogen, USA). Immediately after EDTA addition absorbance readings at 495nm were performed using a Viewlux multipurpose plate reader (Perkin-Elmer, Finland).
FRET (CCF2) microtiter plate assays
The FRET assay began by dispensing 1.25μL of 0.2 nM VIM-2 to the appropriate wells of a 1536-well microtiter plate (Greiner, USA). Following enzyme addition, 11nL of positive control or test compound was added. The plates were then incubated at room temperature for 15 minutes. To start the reaction, 1.25μL of 20μM CCF2-FA substrate (Invitrogen, USA) was dispensed. Final concentrations of substrate and enzyme were 10μM and 0.1nM, respectively. The plate was then incubated for 25 minutes at RT and the reaction stopped by the addition of 2.5μL of 500mM EDTA. Immediately after EDTA addition, fluorescence readings were performed in ratiometric mode using the Viewlux plate reader with an excitation wavelength of 409nm and emission wavelengths of 460nm and 535nm.
Assay quality control
Three quality control parameters were calculated during the screening on a per-plate basis: (a) the signal-to-basal ratio (S/B); (b) the coefficient for variation [CV; CV = (standard deviation/mean) × 100] for all compound test wells; and (c) Z- or Z′-factor [Z′-factor = 1 − [[3 × (σp + σn)]/(μp − μn)], where σ is the standard deviation and μ the mean for positive (p) and negative (n) controls]. 50
Screening assay protocols
The nitrocefin LOPAC HTS assays were performed identical to the nitrocefin microtiter-plate based assays described above. All compounds were tested at 14 μM final concentration in triplicate. Potency (dose-response) screening protocols were identical to the corresponding microtiter plate assays described above, except that each test compound was assayed in triplicate using ten 1:3 serial dilutions, starting at a nominal test concentration of 56 μM. For each compound, either raw absorbance (nitrocefin assay) or ratio (CCF2 assay) data were fitted with a four parameter equation describing a sigmoidal dose-response curve with adjustable baseline using GraphPad Prism version 5.01 suite of programs. The IC50 values were generated from fitted curves by solving for X-intercept at the 50% inhibition level of Y-intercept. Assays were run in the presence and absence of 50 μM ZnS04.
A standard mathematical algorithm 51 was used to determine inhibitory compounds (“hits”) in the VIM-2 nitrocefin assay. Two values were calculated: (a) the average percent inhibition of all compounds tested; and (b) three times their standard deviation. The sum of these two values was used as a cutoff parameter, i.e., any compound that exhibited a % inhibition greater than the cutoff parameter was declared active.
Absorbance artifact assays
Two variants of this assay were executed. In the first variant, the nitrocefin screening protocol was followed with one exception: compounds were added after the reaction was quenched by addition of 500mM EDTA. In the second variant, 5.0 μL of buffer containing 50mM HEPES, 50μM ZnSO4, 0.05% Brij 35, pH 7.1 and 5.0 μL of 500mM EDTA was added to each well of a 1536-well clear-bottom black plate; 28nL of test compounds were added next. Absorbance was measured at 495nm using the Viewlux plate reader (Perkin-Elmer, Finland). Any compound that exhibited a raw absorbance value greater than the average absorbance of all compounds tested was considered an absorbance artifact.
FRET artifact assays
Two variants of this assay were executed. In the first variant, the CCF2 screening protocol was followed with one exception: compounds were added after reaction was quenched by addition of EDTA. In the second variant, 2.5 μL of buffer containing 50mM HEPES, 50μM ZnSO4, 0.05% Brij 35, pH 7.1 and 2.5 μL of 500mM EDTA was added to each well of 1536-well solid white plate; 11 nL of test compound was added next. Fluorescence readings were performed in ratiometric mode using the Viewlux plate reader with an excitation wavelength of 409nm and emission wavelengths of 460nm and 535nm. Any compound that exhibited a 460nm/535nm ratio value greater than the average ratio of all compounds tested was considered FRET assay artifact.
TEM-1 and AmpC Nitrocefin assays
AmpC (Cephalosporinase, from E.cloacae) was from Sigma (USA). The nitrocefin assay began by dispensing 20μL of 0.3 nM TEM-1 in assay buffer (or 4.0 nM AmpC) (PBS pH 7.4, 0.05% Brij or PBS pH 7.4, 5% glycerol, for TEM-1 and AmpC, respectively) to the appropriate wells of a 384-well microtiter plate (Greiner, USA). Following enzyme addition, 100nL of controls or test compounds were added to the appropriate wells. The plates were then incubated at room temperature for 15 minutes. To start the reaction, 20μL of 200μM nitrocefin was dispensed. Final concentrations of substrate and enzyme were 100μM and 0.15 nM (TEM-1) or 2.0 nM (AmpC), respectively. The plate was then incubated for 25 minutes at RT and the reaction stopped by the addition of 10μL of 50μM potassium clavulanate in assay buffer (PBS pH 7.4, 0.05% Brij) in case of TEM-1 assay. In case of the AmpC assay reaction was not quenched. Absorbance readings at 495nm were performed using Envision multipurpose plate reader (Perkin-Elmer, Finland).
Transformation of E. coli with VIM-2 plasmid
The plasmid (pET-9a-based) containing the VIM-2 gene has been described in detail elsewhere. 11 Competent E. coli BL21 (DE3) (Novagen, USA) was transformed with the plasmid using standard techniques and selected on LB agar + kanamycin (30μg/mL). Transformed bacteria were aliquoted as frozen glycerol stocks at -80°C and used directly in experiments by inoculating a starter culture in LB or Iso-Sensitest broth. The stock vials were discarded after inoculation.
MIC assays
Minimal inhibitory concentration (MIC) assays were conducted using two-fold serial broth dilution method as recommended by Clinical and Laboratory Standards Institute (CLSI). 52 All testing was performed in 14 mL tubes (BD, USA) in 2 mL final volume. E. coli BL21 were grown in LB broth (Fisher Scientific, USA) with (transformed BL21-VIM-2 control strain) or without (untransformed BL21 control strain) kanamycin (30 μg/mL) at 37°C for 5 hours. Imipenem (Fisher Scientific, USA) or inhibitor was titrated in Iso-Sensitest broth (Oxoid, UK) using 10 point two-fold serial dilutions immediately prior to testing. Each tube was inoculated with 1mL of bacterial inoculums of 5 × 105 CFU/mL. The tubes were shaken for at least 18 hours at 37°C under aerobic conditions. MIC was determined as per CLSI 53. The E. coli strain ATCC 25922 was used as a quality control reference.
Inhibitor/antibiotic synergy testing
The combined effect of simultaneous application of inhibitor and Imipenem was determined using a “checkerboard” method. 21 All testing was performed in non-treated clear 96 well plates (Corning, USA) in 0.2 mL final volume. E. coli were grown in LB broth with (transformed BL21-VIM-2 control strain) or without (untransformed BL21 control strain) kanamycin (30 μg/mL) at 37°C for 5 hours. Imipenem was titrated in Iso-Sensitest broth using 8 point two-fold serial dilutions immediately prior to testing. Inhibitor was titrated in Iso-Sensitest broth using 9 point two-fold serial dilutions. Imipenem and inhibitor were serially diluted down columns and rows, respectively. This resulted in concentrations of each agent that ranged from at least 4× down to 0.25× of respective MIC values. Each well was then inoculated with 0.1 mL of bacterial inoculums of 1 × 106 CFU/mL. The plates were shaken for at least 18 hours at 37°C under aerobic conditions. MIC was determined as per CLSI. 53 The beta-lactamase resistant E. coli strain ATCC 35218 was used as a quality control reference. Results of synergy testing were interpreted using a Total Fractional Inhibitory Concentration (ΣFIC) method as described previously 23, 54, where:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
The effect of combined imipenem/inhibitor application was considered synergistic when the ΣFIC was ≤ 0.5, indifferent when the ΣFIC was >0.5 and <2, and antagonistic when the ΣFIC was ≥ 2.
Bacteriostatic / bactericidal assays
“Time-kill” assays were performed by the broth macro dilution method as described previously. 21 Briefly, borosilicate glass tubes containing 5 mL of Cation Adjusted Mueller Hinton Broth (CAMHB, USA) along with the appropriate test and control compounds were inoculated with 5 mL of log phase growth bacteria adjusted to 1×106 cfu/mL. Aliquots of the mixture were removed at various time intervals including 0, 2, 4, 6-8, and 24 hours, diluted in 0.9% NaCl solution, and subsequently plated on 100 mm plates containing Mueller-Hinton Agar (Teknova, USA). Following a 24 hour incubation at 37°C viable colonies were enumerated and plotted as log10 cfu/mL vs. time. As per CLSI 21, bactericidally active compounds achieved >3 log10 cfu/mL reduction of the total count in the original inoculums; a compound maintaining that same reduction over 24 hours was considered bacteriostatic. The E. coli strain ATCC 25922 was used as a QC reference.
Molecular modeling
Molecular modeling and docking was performed with the ICM (Internal Coordinate Mechanics) software (Molsoft, USA). The coordinates of the protein were taken from the RCSB Protein Data Bank. Hydrogen and missing heavy atoms were added to the receptor structure followed by local minimization to resolve clashes and to correct chemistry, using a conjugate gradient algorithm and analytical derivatives in internal coordinate. 55 Water molecules were replaced by a continuous dielectric, and the orientations of asparagine and glutamine side chains as well as the tautomeric state of histidine residues were optimized.
Supplementary Material
Acknowledgments
The National Institutes of Health Grant# R21NS059451 supported the research efforts of D. Minond, S.A.Saldanha, P. Subramanian, M. Spaargaren, T. Spicer, B. Sharpless, J.R.Fotsing, T.Weide, and V.Fokin. Pierre Baillargeon and Lina DeLuca (Lead Identification Division, Translational Research Institute, Scripps Florida) are thanked for their assistance with compound management. Prof. Hugh Rosen (The Scripps Research Institute) and Dr. Lynn L. Silver (LL Silver Consulting, LLC) are thanked for their helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lowy FD. J Clin Invest. 2003;111:1265. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush K. Expert Rev Anti Infect Ther. 2004;2:165. doi: 10.1586/14787210.2.2.165. [DOI] [PubMed] [Google Scholar]
- 3.Bush K. Clin Microbiol Infect. 2004;10 4:10. doi: 10.1111/j.1465-0691.2004.1005.x. [DOI] [PubMed] [Google Scholar]
- 4.Jacoby GA, Munoz-Price LS. N Engl J Med. 2005;352:380. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 5.Bonomo RA, Szabo D. Clin Infect Dis. 2006;43 2:S49. doi: 10.1086/504477. [DOI] [PubMed] [Google Scholar]
- 6.Livermore DM. Ann Med. 2003;35:226. doi: 10.1080/07853890310001609. [DOI] [PubMed] [Google Scholar]
- 7.Cornaglia G, Akova M, Amicosante G, Canton R, Cauda R, Docquier JD, Edelstein M, Frere JM, Fuzi M, Galleni M, Giamarellou H, Gniadkowski M, Koncan R, Libisch B, Luzzaro F, Miriagou V, Navarro F, Nordmann P, Pagani L, Peixe L, Poirel L, Souli M, Tacconelli E, Vatopoulos A, Rossolini GM. Int J Antimicrob Agents. 2007;29:380. doi: 10.1016/j.ijantimicag.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Walsh TR, Toleman MA, Poirel L, Nordmann P. Clin Microbiol Rev. 2005;18:306. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bebrone C. Biochem Pharmacol. 2007;74:1686. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 10.Poirel L, Naas T, Nicolas D, Collet L, Bellais S, Cavallo JD, Nordmann P. Antimicrob Agents Chemother. 2000;44:891. doi: 10.1128/aac.44.4.891-897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Docquier JD, Lamotte-Brasseur J, Galleni M, Amicosante G, Frere JM, Rossolini GM. J Antimicrob Chemother. 2003;51:257. doi: 10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Saez I, Docquier JD, Rossolini GM, Dideberg O. J Mol Biol. 2008;375:604. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Pereira DA, Williams JA. British Journal of Pharmacology. 2007;152:53. doi: 10.1038/sj.bjp.0707373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Callaghan CH, Morris A, Kirby SM, Shingler AH. Antimicrob Agents Chemother. 1972;1:283. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Science. 1998;279:84. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- 16.Schroter T, Minond D, Weiser A, Dao C, Habel J, Spicer T, Chase P, Baillargeon P, Scampavia L, Schurer S, Chung C, Mader C, Southern M, Tsinoremas N, LoGrasso P, Hodder P. J Biomol Screen. 2008;13:17. doi: 10.1177/1087057107310806. [DOI] [PubMed] [Google Scholar]
- 17.Moloughney JG, J DT, Toney JH. FEMS Microbiol Lett. 2005;243:65. doi: 10.1016/j.femsle.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 18.Means GE, Feeney RE. Chemical Modification of Proteins. San Francisco: Holden-Day, Inc.; 1971. [Google Scholar]
- 19.Belyaeva T, Leontieva E, Shpakov A, Mozhenok T, Faddejeva M. Cell Biol Int. 2003;27:887. doi: 10.1016/s1065-6995(03)00161-6. [DOI] [PubMed] [Google Scholar]
- 20.Copeland RA. Evaluation of enzyme inhibitors in drug discovery A guide for medicinal chemists and pharmacologists. New York: Wiley Interscience; 2005. [PubMed] [Google Scholar]
- 21.NCCLS. Institute, C. a. L. S., Ed.; NCCLS: Wayne, 1999; Vol. 1.
- 22.Bajaksouzian S, Visalli MA, Jacobs MR, Appelbaum PC. Antimicrob Agents Chemother. 1997;41:1073. doi: 10.1128/aac.41.5.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonapace CR, Bosso JA, Friedrich LV, White RL. Diagn Microbiol Infect Dis. 2002;44:363. doi: 10.1016/s0732-8893(02)00473-x. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Fast W, Valentine AM, Benkovic SJ. Curr Opin Chem Biol. 1999;3:614. doi: 10.1016/s1367-5931(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 25.Salsbury FR, Jr, Crowley MF, Brooks CL., 3rd Proteins. 2001;44:448. doi: 10.1002/prot.1110. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J, Shibata N, Arakawa Y, Kurosaki H. J Med Chem. 2007;50:6647. doi: 10.1021/jm701031n. [DOI] [PubMed] [Google Scholar]
- 27.Toney JH, Hammond GG, Fitzgerald PM, Sharma N, Balkovec JM, Rouen GP, Olson SH, Hammond ML, Greenlee ML, Gao YD. J Biol Chem. 2001;276:31913. doi: 10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- 28.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed Engl. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 29.Scozzafava A, Supuran CT. J Med Chem. 2000;43:3677. doi: 10.1021/jm000027t. [DOI] [PubMed] [Google Scholar]
- 30.Innocenti A, Maresca A, Scozzafava A, Supuran CT. Bioorg Med Chem Lett. 2008;18:3938. doi: 10.1016/j.bmcl.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 31.Jin W, Arakawa Y, Yasuzawa H, Taki T, Hashiguchi R, Mitsutani K, Shoga A, Yamaguchi Y, Kurosaki H, Shibata N, Ohta M, Goto M. Biol Pharm Bull. 2004;27:851. doi: 10.1248/bpb.27.851. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 33.Nord O, Gustrin A, Nygren PA. FEMS Microbiol Lett. 2005;242:73. doi: 10.1016/j.femsle.2004.10.047. [DOI] [PubMed] [Google Scholar]
- 34.Charpentier X, Oswald E. J Bacteriol. 2004;186:5486. doi: 10.1128/JB.186.16.5486-5495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Appelbaum PC, Philippon A, Jacobs MR, Spangler SK, Gutmann L. Antimicrob Agents Chemother. 1990;34:2169. doi: 10.1128/aac.34.11.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ranu RS, Kaji A. J Bacteriol. 1971;107:53. doi: 10.1128/jb.107.1.53-60.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Castillo MC, Islas MI, de Nader OM, de Ruiz-Holgado AP. Rev Latinoam Microbiol. 2001;43:70. [PubMed] [Google Scholar]
- 38.Marumo K, Takeda A, Nakamura Y, Nakaya K. Microbiol Immunol. 1995;39:27. doi: 10.1111/j.1348-0421.1995.tb02164.x. [DOI] [PubMed] [Google Scholar]
- 39.Spencer J, Walsh TR. Angew Chem Int Ed Engl. 2006;45:1022. doi: 10.1002/anie.200502984. [DOI] [PubMed] [Google Scholar]
- 40.Minasov G, Wang X, Shoichet BK. J Am Chem Soc. 2002;124:5333. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 41.Maveyraud L, Pratt RF, Samama JP. Biochemistry. 1998;37:2622. doi: 10.1021/bi972501b. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Minasov G, Roth TA, Prati F, Shoichet BK. J Am Chem Soc. 2006;128:2970. doi: 10.1021/ja056806m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shenkenberg TD, Von Hoff DD. Ann Intern Med. 1986;105:67. doi: 10.7326/0003-4819-105-1-67. [DOI] [PubMed] [Google Scholar]
- 44.Koeller J, Eble M. Clin Pharm. 1988;7:574. [PubMed] [Google Scholar]
- 45.White RJ, Durr FE. Invest New Drugs. 1985;3:85. doi: 10.1007/BF00174154. [DOI] [PubMed] [Google Scholar]
- 46.Ala PJ, Gonneville L, Hillman M, Becker-Pasha M, Yue EW, Douty B, Wayland B, Polam P, Crawley ML, McLaughlin E, Sparks RB, Glass B, Takvorian A, Combs AP, Burn TC, Hollis GF, Wynn R. J Biol Chem. 2006;281:38013. doi: 10.1074/jbc.M607913200. [DOI] [PubMed] [Google Scholar]
- 47.Kocalka P, Andersen NK, Jensen F, Nielsen P. Chembiochem. 2007;8:2106. doi: 10.1002/cbic.200700410. [DOI] [PubMed] [Google Scholar]
- 48.Kolb HC, Sharpless KB. Drug Discov Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 49.Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. New-York: John Wiley and Sons, Inc; 2000. [Google Scholar]
- 50.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 51.Hodder P, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, Culberson C, Bleicher L, Cosford ND, Bayly C, Suto C, Varney M, Strulovici B. Anal Biochem. 2003;313:246. doi: 10.1016/s0003-2697(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 52.CLSI. Institute, C. a. L. S., Ed.; CLSI: Wayne, 2006; Vol. 2.
- 53.CLSI. Institute, C. a. L. S., Ed.; CLSI: Wayne, 2006; Vol. 1.
- 54.Orhan G, Bayram A, Zer Y, Balci I. J Clin Microbiol. 2005;43:140. doi: 10.1128/JCM.43.1.140-143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Totrov M, Abagyan R. Proteins. 1997 1:215. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 56.Laraki N, Franceschini N, Rossolini GM, Santucci P, Meunier C, de Pauw E, Amicosante G, Frere JM, Galleni M. Antimicrob Agents Chemother. 1999;43:902. doi: 10.1128/aac.43.4.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.