

Abstract
The Escherichia coli Hsp40 DnaJ and Hsp70 DnaK cooperate in the binding of proteins at intermediate stages of folding, assembly, and translocation across membranes. Binding of protein substrates to the DnaK C-terminal domain is controlled by ATP binding and hydrolysis in the N-terminal ATPase domain. The interaction of DnaJ with DnaK is mediated at least in part by the highly conserved N-terminal J-domain of DnaJ that includes residues 2–75. Heteronuclear NMR experiments with uniformly 15N-enriched DnaJ2–75 indicate that the chemical environment of residues located in helix II and the flanking loops is perturbed on interaction with DnaK or a truncated DnaK molecule, DnaK2–388. NMR signals corresponding to these residues broaden and exhibit changes in chemical shifts in the presence of DnaK(MgADP). Addition of MgATP largely reversed the broadening, indicating that NMR signals of DnaJ2–75 respond to ATP-dependent changes in DnaK. The J-domain interaction is localized to the ATPase domain of DnaK and is likely to be dominated by electrostatic interactions. The results suggest that the J-domain tethers DnaK to DnaJ-bound substrates, which DnaK then binds with its C-terminal peptide-binding domain.
Genetic and biochemical evidence suggests that DnaJ functionally and physically interacts with DnaK even though no stable binary complex of these two proteins has been observed. DnaJ and DnaK cooperate in many cellular processes, including DNA replication (1–3), protein export (4), and stress response (5, 6). DnaJ and DnaK promote folding of denatured or partially unfolded proteins (7, 8) as well as newly synthesized polypeptides (9). DnaJ can act as a chaperone on its own (6, 8), or it can work together with DnaK (10). DnaK possesses a weak intrinsic ATPase activity (2), and ATP binding and hydrolysis are associated with conformational changes in DnaK that regulate substrate binding and release (11, 12). DnaJ stimulates ATP hydrolysis by DnaK (13), and DnaK(MgADP) has the highest affinity toward polypeptide substrates (14, 15).
The interaction between DnaJ and DnaK appears to be bimodal. A truncated DnaJ molecule consisting of the J-domain and Gly/Phe region stimulates the ATPase activity of DnaK and supports bacteriophage λ DNA replication, although a higher concentration of the truncated protein is required (16, 17). The J-domain by itself neither stimulates the ATPase activity of DnaK nor supports replication of bacteriophage λ DNA. However, the J-domain plus a peptide added in trans act synergistically to stimulate DnaK’s ATPase activity 200-fold (17).
Point mutations in the J-domains of DnaJ and DnaJ homologs abolish the ability of these proteins to function with Hsp70 proteins. These mutations include H33Q in DnaJ (4, 16) and two mutations (18) of sec63p, P156N and D157A, which substitute residues in the conserved tripeptide HPD of the J-domain. An insertion of one amino acid between P34 and D35 in DnaJ also abolishes activity in a bacteriophage λ DNA replication assay (19). Because the tripeptide has no obvious structural role (20) and because there is a high degree of conservation in the HPD segment, the HPD tripeptide could mediate specific interactions between Hsp40 and Hsp70 proteins.
The J-domain-Hsp70 interaction can be subverted by viruses. The J-domain of polyomavirus T antigens is required for viral DNA replication and tissue transformation, possibly by mediating interactions with Hsp70. Simian virus 40 (SV40) large T antigen associates with the constitutively expressed Hsc70 (21), and certain J-domain mutants defective in tissue transformation fail to bind Hsc70 (22). Chimeras constructed by substituting the J-domain of DnaJ with the J-domain from any of several polyomavirus T antigens can replace wild-type DnaJ in vivo (23). Likewise, chimeras of T antigen containing the J-domain from DnaJ or a eukaryotic Hsp40 support SV40 DNA replication (24). Nevertheless, the role of the T antigen J-domain in viral DNA replication has not been elucidated.
C-terminal domains of DnaJ-family proteins may confer specificity for substrates and/or function. The C-terminal domains are less well-conserved than the J-domain (25). Most, but not all, DnaJ homologs contain a domain that binds two Zn2+ atoms, a feature shared with polyomavirus small t antigens (26). Genetic and biochemical evidence indicate that both the Zn2+-binding domain and the remaining C-terminal portion of the protein are involved in binding substrates (27, 28).
NMR perturbation mapping has been used previously to identify several protein–protein binding sites including those for histidine-containing phosphocarrier protein (Hpr) on the N-terminal domain of enzyme I (29), CD48 on CD2 (30), Ras on Raf-1 (31), and plastocyanin on cytochrome c (32). We have used two-dimensional 1H-15N correlation NMR perturbation mapping to identify the DnaK-binding site on the J-domain of DnaJ, DnaJ2–75. Resonance line broadening and chemical shift perturbations in the 1H-15N correlation spectrum of [15N]DnaJ2–75 were monitored during titration with unlabeled DnaK or a truncated DnaK molecule containing only the ATPase domain (DnaK2–388). In the 1H-15N correlation spectrum, each amide group reports changes in the chemical environment caused by side-chain rearrangements, exclusion of solvent at the interface, or changes in electrostatic potential on complex formation (32). Our results show that the J-domain forms a 1:1 complex with both the MgADP- and MgATP-bound forms of the DnaK ATPase domain. Mapping the most severe NMR perturbations to the DnaJ2–75 sequence reveals that the DnaK-binding site is localized to helix II and the adjacent loops, including the conserved HPD tripeptide. Perturbation of two buried residues suggests that DnaJ2–75 undergoes a conformational change on binding to DnaK.
MATERIALS AND METHODS
Expression and Purification of Proteins.
Expression and purification of DnaJ2–75 and DnaK were as described by Karzai and McMacken (17). For [15N]DnaJ2–75, NapIV/pRLM233 cells were grown at 30°C in minimal medium (33) containing [15N]NH4Cl as the sole nitrogen source supplemented with thiamine. DnaK from the MonoQ column (peak I), which contains 0.6–0.8 mol bound ADP per mol of enzyme (ref. 34 and R. Russell and R. McMacken, personal communication) was pooled and frozen in ethanol/dry ice and stored at −80°C. Before NMR, DnaK was incubated with one equivalent of MgATP at room temperature for 30 min, and excess nucleotide was removed by ultrafiltration in a centricon-30 concentrator. DnaK2–388 was prepared in an identical manner.
NMR.
Heteronuclear NMR experiments were performed with a 2-mM sample of uniformly 15N-labeled DnaJ2–75 on a Bruker AMX500 (Louisiana State University, Baton Rouge) or a GE Omega PSG 500 spectrometer. Two-dimensional 1H-15N correlation spectra were collected by using several pulse sequences: heteronuclear single-multiple quantum coherence (HSMQC) (35), heteronuclear single quantum coherence (HSQC) (36), or heteronuclear multiple quantum coherence with spin-echo water suppression (HMQC-JR) (37). For sequential assignments, HMQC-nuclear Overhauser effect spectroscopy (100 and 200 ms mixing times) and HMQC-total correlation spectroscopy (30 and 60 ms mixing times) (36, 38) experiments were recorded. Typically, 256 increments of 1,024 complex data points were collected for each experiment. Spectral widths of 8.0 kHz and 2.5 kHz were used in 1H and 15N, respectively.
All data were processed by using Felix 95.0 (Molecular Simulations). For the majority of the resonances, assignment of the backbone 15N resonances was straightforward by using the HMQC-nuclear Overhauser effect spectroscopy and HMQC-total correlation spectroscopy spectra. The cross peaks corresponding to A2, K3, and D40 were not observed at all at 25°C and pH 6.8 independently of the way of dealing with the water signal. The cross peak for G39 could be seen only at 25°C and pH 6.8 by using HMQC with spin-echo water suppression.
Titration of DnaJ2–75 with Wild-Type, Full-Length DnaK.
The initial [15N]DnaJ2–75 sample contained 50 mM Mops⋅KOH, pH 6.8, 10 mM potassium acetate, 90% H2O/10% D2O, trace trimethylsilyl propionate, and 0.05% NaN3 in a total volume of 0.6 ml, and the initial [15N]DnaJ2–75 concentration was 0.3 mM. The stock DnaK(MgADP) concentration was 3.5 mM in 50 mM Mops⋅KOH, pH 6.8, 10 mM potassium acetate. At the end of the DnaK(MgADP) titration, ATP and magnesium chloride were added to a final concentration of 7 mM. Two-dimensional 1H-15N HSMQC NMR experiments were performed after each addition of DnaK(MgADP) and after the addition of MgATP. NMR spectroscopy was carried out as described above except that the spectral width in the 1H dimension was 7.017 kHz. A total of 256 complex t1 increments of 1,024 complex t2 points were recorded in each experiment with 128 scans per increment. HSMQC gave distorted cross peaks at low DnaJ2–75 concentrations, whereas HMQC gave symmetrical cross peaks. HSMQC was used because it gave better resolution in crowded regions of the spectrum.
Titration of DnaJ2–75 with DnaK2–388.
The titration of [15N]DnaJ2–75 with DnaK2–388 was carried out as with wild-type DnaK except for the following changes: the sample buffer contained 50 mM Mops⋅NaOH, pH 6.8 and the stock DnaK2–388 concentration was 4.2 mM in 50 mM Mops⋅NaOH, pH 6.8. At the end of the DnaK2–388 titration, ATP and magnesium chloride were added to a final concentration of 5 mM. Two-dimensional 1H-15N HMQC NMR experiments were performed after each addition of DnaK2–388 and after the addition of MgATP. NMR spectroscopy was carried out as described above.
The fraction of bound ligand, α, was determined at each point in the titrations by the equation
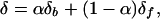 |
1 |
where δ is the chemical shift, and the subscripts b and f refer to the bound and free ligands, respectively (39). From the fraction of DnaK(MgADP) or DnaK2–388(MgADP) bound, dissociation constants were calculated by linear regression of a Scatchard analysis assuming a 1:1 complex.
RESULTS
The HSMQC spectrum of [15N]DnaJ2–75 was obtained first in the absence of DnaK and then in the presence of increasing ratios of DnaK(MgADP) to DnaJ2–75: 0.0:1.0, 0.32:1.0, 0.64:1.0, 0.81:1.0, 0.96:1.0, 1.12:1.0, 1.61:1.0, 2.09:1.0, and 2.57:1.0. As the DnaK(MgADP):DnaJ2–75 ratio increased to 0.96:1.0, most cross peaks decreased to between 15 and 40 percent of their original intensity (Fig. 1A and B). 1H-15N cross peaks broadened, and some also shifted. Cross peaks corresponding to Y6 and D35 broadened beyond detection at 0.96:1.0 DnaK/DnaJ2–75.
Figure 1.

Portions of three two-dimensional 1H-15N HSMQC spectra of [15N]DnaJ2–75 are shown. (A) 0.0:1.0 DnaK(MgADP) to DnaJ2–75 molar ratio. (B) 0.96:1.0 DnaK(MgADP) to DnaJ2–75 molar ratio. (C) 2.57:1.0 DnaK to DnaJ2–75 plus MgATP. B and C have been plotted at levels adjusted to compensate for changes in sample volume. All cross peaks are in the region plotted except that for D35, which is displaced downfield. The boxed region corresponds to the expanded view in Fig. 2B.
On addition of MgATP, the DnaK-bound MgADP is expected to exchange with MgATP in a reaction associated with discharge of protein substrates from DnaK (11). When MgATP was added to the sample, all of the previously broadened cross peaks increased in intensity (Fig. 1C) to approximately 50–60% of their original intensity. Thus, MgATP partially reversed the line broadening caused by DnaK(MgADP) addition. DnaK exists in oligomeric forms as isolated by standard procedures (40, 41). Oligomeric DnaK(MgADP) is converted to the monomeric form in the presence of MgATP (41). Dissociation of DnaK(MgADP) oligomers into DnaK(MgATP) monomers is expected to decrease the rotational correlation time of the complex with DnaJ2–75 and therefore reduce line broadening.
Residues of DnaJ2–75 that interact with DnaK were identified by line broadening and chemical shift changes. Broadening was analyzed by plotting the change in peak intensity at the lowest nonzero DnaK(MgADP) concentration divided by the change in peak intensity at the 0.96:1.0 DnaK(MgADP)/DnaJ2–75 ratio (30). Residues for which the linewidth was most sensitive include Y6, S13, I21, A24, R27, H33, D35, Y54, and T58 (Fig. 2A). The chemical shifts of each cross peak before and after DnaK(MgADP) addition were used to calculate the length of the vector change in hertz from the original position of the cross peak in the reference spectrum to the position in the sample with a 0.96:1.0 DnaK(MgADP)/DnaJ2–75 molar ratio (30). Residues S13, E20, I21, R22, A24, K26, R27, L28, M30, Y32, and T58 had changes greater than 10 Hz (data not shown). All residues that exhibited severe broadening except Y6 and Y54 also exhibited large chemical shift changes.
Figure 2.

(A, Upper) Fractional change in peak intensity at 0.32:1.0 DnaK(MgADP) to DnaJ2–75 plotted against DnaJ2–75 residue number. The horizontal dashed line is the cutoff for residues among the 10% most sensitive to line broadening by DnaK(MgADP) and for mapping onto the DnaJ2–75 structure (Fig. 3). Residues that broadened beyond detection at 0.96:1 DnaK(MgADP)/DnaJ2–75 are shown as black bars of uniform height. The position of proline 34 is indicated by ∗. G39 and D40 for which the NH groups exchange rapidly with solvent are indicated by x. Residues 9, 49, 51, 52, 53, 55, 61, 72, and 73 could not be monitored quantitatively because of spectral overlap, and their positions are indicated by o. The positions of helices I–IV of DnaJ2–75 are indicated by bars at the top. (A, Lower) Combined 1H/15N DnaK(MgATP)-induced chemical shift changes measured as the length of the resultant vector in hertz plotted against DnaJ2–75 residue number. The horizontal dashed line at 10 Hz is the cutoff for residues considered to be most perturbed and for mapping onto the structure of DnaJ2–75. (B) Superposition of a selected region of two-dimensional 1H-15N HSMQC spectra of [15N]DnaJ2–75 alone (light contours) and in the presence of DnaK(MgATP) (heavy contours). The spectrum of DnaJ2–75 with DnaK(MgATP) was plotted at a level that compensates for dilution of the sample. Residues I21, A24, and L28 were most perturbed in this region of the spectrum whereas residues A43, A45, I50 and A65 changed little. (C, Upper) Fractional change in peak intensity at 0.32:1.0 DnaK2–388(MgADP) to DnaJ2–75 plotted against DnaJ2–75 residue number. The horizontal dashed line is the cutoff for residues most sensitive to line broadening by DnaK2–388(MgADP). Residues 9, 49, 50, 51, 52, 53, 55, 60, 61, 72, and 73 could not be monitored quantitatively because of spectral overlap, and their positions are indicated by o. Other symbols are the same as that in A. (C, Lower) Combined 1H/15N DnaK2–388(MgATP)-induced chemical shift changes measured as the length of the resultant vector in hertz plotted against DnaJ2–75 residue number. The horizontal dashed line at 10 Hz is the cutoff for residues considered to be most perturbed. (D) Titration of [15N]DnaJ2–75 with DnaK(MgADP) and DnaK2–388(MgADP). Proton chemical shift changes are plotted as a function of DnaK(MgADP)/DnaJ2–75 ratio (open symbols) or DnaK2–388:DnaJ2–75 ratio (closed symbols).
When MgATP was added, signal broadening was reversed, but chemical shift changes persisted (Fig. 2B). Because of reduced line broadening, the chemical shifts of more cross peaks can be analyzed. The set of residues perturbed after addition of MgATP includes all residues perturbed by DnaK(MgADP). Residues V12, S13, A16, R19, E20, I21, R22, A24, Y25, K26, R27, L28, M30, Y32, H33, D35, and T58 had changes greater than 10 Hz in the presence of DnaK(MgATP) (Fig. 2 A and B). The magnitude of chemical shift changes after addition of MgATP was comparable to that in the presence of DnaK(MgADP). Thus DnaJ2–75 interacts with both MgADP and MgATP forms of DnaK. A 31P NMR spectrum taken at the end of the HSMQC experiment indicated that all of the MgATP had been converted to MgADP (data not shown). Thus, the HSMQC spectrum with ATP is an average spectrum of DnaJ2–75 in the presence of MgATP and MgADP forms of DnaK.
The titration of [15N]DnaJ2–75 was repeated with a truncated DnaK molecule, DnaK2–388, that contains only the ATPase domain (R. McMacken, personal communication). NMR signals for DnaJ2–75 decreased to between 25 and 50 percent of their original intensity at the ratio of 0.98:1.0 DnaK2–388 to DnaJ2–75. Reduced line broadening by DnaK2–388 compared with that by full-length DnaK is consistent with a requirement for an intact peptide-binding domain for oligomerization of DnaK (42).
1H-15N cross peaks of [15N]DnaJ2–75 broadened and shifted in the presence of DnaK2–388. Residues for which linewidths were most sensitive to the addition of DnaK2–388 are Y6, K14, T15, E20, I21, Y25, K26, L28, M30, K31, Y54, and T58 (Fig. 2C). Residues for which chemical shifts changed greater than 10 Hz at 0.98:1.0 DnaK2–388 to DnaJ2–75 molar ratio are S13, T15, E17, R19, E20, I21, R22, A24, K26, R27, L28, M30, K31, and Y32 (Fig. 2C). Because these residues are nearly identical to the residues affected with DnaK(MgATP), the binding site for the J-domain is contained in the ATPase domain of DnaK.
On addition of MgATP to the DnaK2–388/DnaJ2–75 complex, cross peaks increased in intensity by only about 5%. Because DnaK2–388 is not expected to form oligomers to the same extent as full-length DnaK, the increase in intensity of DnaJ2–75 cross peaks could be caused by a decrease in affinity of DnaK2–388 for DnaJ2–75 in the presence of ATP. However, because DnaJ2–75, DnaK2–388, and ATP concentrations are substantially greater than the Kd measured for DnaJ2–75 binding to DnaK2–388, the amount of J-domain-DnaK2–388 complex formed in this sample is not very sensitive to changes in affinity.
Titration of [15N]DnaJ2–75 with DnaK2–388 was performed in the absence of potassium ions, thus inhibiting the ATPase (12, 43). After the final HMQC experiment, 31P NMR showed that not all of the MgATP had been converted to MgADP (data not shown). Nevertheless, the pattern of chemical shift perturbation in the presence of MgATP was nearly identical to that in the presence of MgADP (data not shown).
To determine whether the DnaK-binding site on the J-domain of DnaJ was saturated during the titrations, the change in amide proton chemical shift for three [15N]DnaJ2–75 residues was plotted as a function of the DnaK(MgADP)/DnaJ2–75 or DnaK2–388(MgADP)/DnaJ2–75 molar ratio (Fig. 2D). Chemical shift perturbation reached saturation at about 1.5:1.0 DnaK(MgADP)/DnaJ2–75 and 1.32:1.0 DnaK2–388(MgADP)/DnaJ2–75, respectively, suggesting that DnaJ2–75 formed a 1:1 complex with DnaK(MgADP) and with DnaK2–388(MgADP). The fraction of DnaK bound at each point in the titrations was calculated for three amide protons (A24, M30, and Y32). Bound and free DnaK concentrations were calculated from the fraction bound (Eq. 1). Scatchard analysis yielded a dissociation constant of 10 × 10−6 ± 30 × 10−6 M for DnaK(MgADP) and 5 × 10−6 ± 10 × 10−6 for DnaK2–388.
DISCUSSION
The interaction of the J-domain with both ADP- and ATP-bound forms of the DnaK ATPase domain provides a mechanism for DnaJ to tether DnaK to substrates during cycles of ATP-dependent binding and release by the DnaK peptide-binding domain. Others have shown that DnaJ targets substrates for DnaK binding (44, 45) and that DnaJ stabilizes DnaK binding in the presence of ATP (8). NMR chemical shift perturbation data indicate that the J-domain-DnaK interaction is not substantially affected by the exchange of ADP for ATP, although J-domain affinity for DnaK(MgATP) may be slightly lower than for DnaK(MgADP). Because the DnaK peptide-binding domain is thought to mediate oligomerization of Hsp70 proteins, the continued J-domain-DnaK interaction during the ATP-induced dissociation of DnaK oligomers may represent one component of the interaction between DnaJ and DnaK that is maintained during cycles of substrate binding and release. It seems likely that one of the principal roles of DnaJ is to facilitate delivery of substrates to the DnaK peptide-binding domain, and the resulting increase in local concentration of substrate could be a mechanism for stimulation of the DnaK ATPase.
Mapping of perturbed residues onto the three-dimensional structure of DnaJ2–76 (20, 46) shows that the DnaK-binding site is localized to the outer surface of helix II (Fig. 3), which is consistent with the speculation of Szyperski et al. (20), and includes the tripeptide HPD as anticipated by the genetic (4, 16, 18, 48) and NMR studies (20, 46, 49). The minimal sequence necessary for the interaction of DnaJ-like proteins with Hsp70 proteins could be residues 1–35, including helix II and the tripeptide HPD. Alignment of J-domain sequences from DnaJ homologs and T antigens shows that the N-terminal portion up to and including the HPD cluster is more conserved than the portion following the HPD (25). In a study with yeast YDJ1 (an Hsp40) and SSA1 (an Hsp70), a peptide containing the YDJ1 sequence corresponding to residues 21–40 competes with YDJ1 for binding to SSA1 (50). A peptide corresponding to residues 21–40 with the H34Q mutation did not compete as well as the wild-type peptide.
Figure 3.

Two views of a space-filling depiction of DnaJ2–76 identifying surface-exposed residues perturbed by the interaction with DnaK(MgATP) and DnaK(MgADP). Residues that have greater than 10 Hz shift (defined in Fig. 2A, legend) in the presence of DnaK(MgATP) are colored blue. Residues that are severely broadened in the presence of DnaK(MgADP) but whose chemical shift is not affected in the presence of DnaK(MgATP) are colored red. In the view at right, the molecule has been rotated 180° about the vertical axis relative to the view at left. The figure was prepared by using the program molscript (47) with frame 1 of Protein Data Bank file 1XBL.
DnaJ2–75 binding to DnaK affects residues involved in packing the core of the J-domain. Resonances for Y6 and Y54 are substantially broadened in the presence of DnaK(MgADP) but do not exhibit a significant chemical shift change. These residues are remote from the site of DnaK interaction on the outer face of helix II. Perturbation of these residues may be caused by a conformational change in the molecule on formation of the complex with DnaK(MgADP). In the structure of DnaJ2–75, helix II forms an antiparallel coiled coil with helix III, and helix I packs against one face of the helix II–helix III pair (46, 51). Our results suggest that mutations (52, 53) in helix III could disrupt the packing of helices and interfere with the conformational change. This model is consistent with the proposal that the flexible loop between helices II and III becomes ordered on binding to DnaK (46).
Seven of 17 residues in DnaJ2–75 that were perturbed by DnaK(MgATP) have charged functional groups. Therefore, the DnaJ2–75-DnaK(MgATP) binding mode seems to be dominated by electrostatic interactions. Helix II contains five charged residues, and the polarity of the charge is always conserved (20). The net positively charged helix II probably interacts with a negatively charged surface of DnaK. A plot of electrostatic potential for DnaJ2–76 reveals a prominent region of positive potential at the outer surface of helix II and largely negative potential elsewhere (Fig. 4), whereas a plot of the DnaK ATPase domain reveals negative potential covering most of its surface (Fig. 5). Ziegelhoffer et al. (54) showed that at low ionic strength, YDJ1 stimulates the ATPase activity of SSA1, but at higher ionic strength, stimulation was greatly reduced. These data indicate that the interaction between Hsp40s and Hsp70s has an important electrostatic component. Because ionic strength is likely to deviate from the cellular average near both charged surfaces and hydrophobic surfaces, the J-domain-DnaK interaction could facilitate selective binding and dissociation of substrates near these surfaces.
Figure 4.

(Left) Contours of electrostatic surface potential in and around DnaJ2–76. Blue and red contours indicate potentials of 2kT and −2kT, respectively. The potentials were calculated with the program delphi 95.0 (Molecular Simulations) by using default values for solvent and protein dielectric constants. Helix II contains a large positive surface potential that could interact with an area of negative surface potential on DnaK2–388. (Right) Ribbon diagram of DnaJ2–76 in the same orientation as the molecule to the left with the helices I–IV labeled. The figure was prepared by using frame 1 of Protein Data Bank file 1XBL.
Figure 5.

(Left) Contours of potentials in and around DnaK3–383. Contours indicate potentials as specified in the Fig. 4 legend. The potentials were calculated as in Fig. 4 by using Protein Data Bank file 1DKG after removal of all atoms associated with GrpE. (Right) Ribbon diagram of DnaK3–383 in the same orientation as the molecule to the left.
Our results clearly indicate that at least one function of the highly conserved J-domain is to bind the ATPase domain of Hsp70 protein. Thus, viral products containing J-domain-like sequences probably carry out the same function, except directing chaperone power to viral-specific products or redirecting host components to fates that promote the viral life cycle.
Acknowledgments
We thank R. McMacken for plasmids and procedures for preparation of DnaJ2–75, DnaK, and DnaK2–388, J. Karam for providing NapIV cells, L. Gierasch for helpful discussion, J. Rizo-Rey for critical reading of the manuscript and helpful discussion, and N. K. Steede for technical assistance. We gratefully acknowledge the Tulane University Molecular and Cellular Biology Program, the Tulane Cancer Center, and the National Science Foundation (MCB-9512711) for support of this project.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: HSMQC, heteronuclear single-multiple quantum coherence; HMQC, heteronuclear multiple quantum coherence.
References
- 1.Saito H, Uchida H. Mol Gen Genet. 1977;164:1–8. doi: 10.1007/BF00267592. [DOI] [PubMed] [Google Scholar]
- 2.Zylicz M, LeBowitz J H, McMacken R, Georgopoulos C. Proc Natl Acad Sci USA. 1983;80:6431–6435. doi: 10.1073/pnas.80.21.6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wickner S, Hoskins J, McKenney K. Proc Natl Acad Sci USA. 1991;88:7903–7907. doi: 10.1073/pnas.88.18.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wild J, Altman E, Yura T, Gross C A. Genes Dev. 1992;6:1165–1172. doi: 10.1101/gad.6.7.1165. [DOI] [PubMed] [Google Scholar]
- 5.Liberek K, Georgopoulos C. Proc Natl Acad Sci USA. 1993;90:11019–11023. doi: 10.1073/pnas.90.23.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schroder H, Langer T, Hartl F U, Bukau B. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skowyra D, Georgopoulos C, Zylicz M. Cell. 1990;62:939–944. doi: 10.1016/0092-8674(90)90268-j. [DOI] [PubMed] [Google Scholar]
- 8.Langer T, Lu C, Echols H, Flanagan J, Hayer M K, Hartl F U. Nature (London) 1992;356:683–689. doi: 10.1038/356683a0. [DOI] [PubMed] [Google Scholar]
- 9.Hendrick J P, Langer T, Davis T A, Hartl F U, Wiedmann M. Proc Natl Acad Sci USA. 1993;90:10216–10220. doi: 10.1073/pnas.90.21.10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szabo A, Langer T, Schroder H, Flanagan J, Bukau B, Hartl F U. Proc Natl Acad Sci USA. 1994;91:10345–10349. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liberek K, Skowyra D, Zylicz M, Johnson C, Georgopoulos C. J Biol Chem. 1991;266:14491–14496. [PubMed] [Google Scholar]
- 12.Palleros D R, Reid K L, Shi L, Welch W J, Fink A L. Nature (London) 1993;365:664–666. doi: 10.1038/365664a0. [DOI] [PubMed] [Google Scholar]
- 13.Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Proc Natl Acad Sci USA. 1991;88:2874–2878. doi: 10.1073/pnas.88.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmid D, Baici A, Gehring H, Christen P. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 15.Wawrzynow A, Banecki B, Wall D, Liberek K, Georgopoulos C, Zylicz M. J Biol Chem. 1995;270:19307–19311. doi: 10.1074/jbc.270.33.19307. [DOI] [PubMed] [Google Scholar]
- 16.Wall D, Zylicz M, Georgopoulos C. J Biol Chem. 1994;269:5446–5451. [PubMed] [Google Scholar]
- 17.Karzai A W, McMacken R. J Biol Chem. 1996;271:11236–11246. doi: 10.1074/jbc.271.19.11236. [DOI] [PubMed] [Google Scholar]
- 18.Feldheim D, Rothblatt J, Schekman R. Mol Cell Biol. 1992;12:3288–3296. doi: 10.1128/mcb.12.7.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wall D, Zylicz M, Georgopoulos C. J Biol Chem. 1995;270:2139–2144. doi: 10.1074/jbc.270.5.2139. [DOI] [PubMed] [Google Scholar]
- 20.Szyperski T, Pellecchia M, Wall D, Georgopoulos C, Wuthrich K. Proc Natl Acad Sci USA. 1994;91:11343–11347. doi: 10.1073/pnas.91.24.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawai E T, Butel J S. J Virol. 1989;63:3961–3973. doi: 10.1128/jvi.63.9.3961-3973.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawai E T, Rasmussen G, Butel J S. Virus Res. 1994;31:367–378. doi: 10.1016/0168-1702(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 23.Kelley W L, Georgopoulos C. Proc Natl Acad Sci USA. 1997;94:3679–3684. doi: 10.1073/pnas.94.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell K S, Mullane K P, Aksoy I A, Stubdal H, Zalvide J, Pipas J M, Silver P A, Roberts T M, Schaffhausen B S, DeCaprio J A. Genes Dev. 1997;11:1098–1110. doi: 10.1101/gad.11.9.1098. [DOI] [PubMed] [Google Scholar]
- 25.Kelley W L, Landry S J. Trends Biochem Sci. 1994;19:277–278. doi: 10.1016/0968-0004(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 26.Bork P, Sander C, Valencia A, Bukau B. Trends Biochem Sci. 1992;17:129. doi: 10.1016/0968-0004(92)90319-5. [DOI] [PubMed] [Google Scholar]
- 27.Szabo A, Korszun R, Hartl F-U, Flanagan J. EMBO J. 1996;15:408–417. [PMC free article] [PubMed] [Google Scholar]
- 28.Banecki B, Liberek K, Wall D, Wawrzynow A, Georgopoulos C, Bertoli E, Tanfani F, Zylicz M. J Biol Chem. 1996;271:14840–14848. doi: 10.1074/jbc.271.25.14840. [DOI] [PubMed] [Google Scholar]
- 29.Garrett D S, Seok Y J, Peterkofsky A, Clore G M, Gronenborn A M. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 30.McAlister M S B, Mott H R, van der Merwe P A, Campbell I D, Davis S J, Driscoll P C. Biochemistry. 1996;35:5982–5991. doi: 10.1021/bi952756u. [DOI] [PubMed] [Google Scholar]
- 31.Emerson S D, Madison V S, Palermo R E, Waugh D S, Scheffler J E, Tsao K-L, Kiefer S E, Liu S P, Fry D C. Biochemistry. 1995;34:6911–6918. doi: 10.1021/bi00021a001. [DOI] [PubMed] [Google Scholar]
- 32.Ubbink M, Bendall D S. Biochemistry. 1997;36:6326–6335. doi: 10.1021/bi963199u. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 34.Gao B C, Emoto Y, Greene L, Eisenberg E. J Biol Chem. 1993;268:8507–8513. [PubMed] [Google Scholar]
- 35.Zuiderweg E R P. J Magn Reson. 1990;86:346–357. [Google Scholar]
- 36.Norwood T J, Boyd J, Soffe N, Campbell J D. J Magn Reson. 1990;87:488–501. [Google Scholar]
- 37.Sklenar V, Bax A. J Magn Reson. 1987;74:469–479. [Google Scholar]
- 38.Gronenborn A M, Bax A, Wingfield P T, Clore G M. FEBS Lett. 1989;243:93–98. doi: 10.1016/0014-5793(89)81224-4. [DOI] [PubMed] [Google Scholar]
- 39.Klotz I M. In: Protein Function: A Practical Approach. 1st Ed. Creighton T E, editor. New York: IRL; 1990. pp. 25–54. [Google Scholar]
- 40.Schonfeld H J, Schmidt D, Schroder H, Bukau B. J Biol Chem. 1995;270:2183–2189. doi: 10.1074/jbc.270.5.2183. [DOI] [PubMed] [Google Scholar]
- 41.Kim D H, Lee Y J, Corry P M. J Cell Phys. 1992;153:353–361. doi: 10.1002/jcp.1041530215. [DOI] [PubMed] [Google Scholar]
- 42.Benaroudj N, Fouchaq B, Ladjimi M M. J Biol Chem. 1997;272:8744–8751. doi: 10.1074/jbc.272.13.8744. [DOI] [PubMed] [Google Scholar]
- 43.Feifel B, Sandmeier E, Schonfeld H J, Christen P. Eur J Biochem. 1996;237:318–321. doi: 10.1111/j.1432-1033.1996.0318n.x. [DOI] [PubMed] [Google Scholar]
- 44.Gamer J, Bujard H, Bukau B. Cell. 1992;69:833–842. doi: 10.1016/0092-8674(92)90294-m. [DOI] [PubMed] [Google Scholar]
- 45.Liberek K, Wall D, Georgopoulos C. Proc Natl Acad Sci USA. 1995;92:6224–6228. doi: 10.1073/pnas.92.14.6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pellecchia M, Szyperski T, Wall D, Georgopoulos C, Wuthrich K. J Mol Biol. 1996;260:236–250. doi: 10.1006/jmbi.1996.0395. [DOI] [PubMed] [Google Scholar]
- 47.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 48.Scidmore M A, Okamura H H, Rose M D. Mol Biol Cell. 1993;4:1145–1159. doi: 10.1091/mbc.4.11.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill R B, Flanagan J M, Prestegard J H. Biochemistry. 1995;34:5587–5596. doi: 10.1021/bi00016a033. [DOI] [PubMed] [Google Scholar]
- 50.Tsai J, Douglas M G. J Biol Chem. 1996;271:9347–9354. doi: 10.1074/jbc.271.16.9347. [DOI] [PubMed] [Google Scholar]
- 51.Qian Y Q, Patel D, Hartl F U, McColl D J. J Mol Biol. 1996;260:224–235. doi: 10.1006/jmbi.1996.0394. [DOI] [PubMed] [Google Scholar]
- 52.Nelson M K, Kurihara T, Silver P A. Genetics. 1993;134:159–173. doi: 10.1093/genetics/134.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlenstedt G, Harris S, Risse B, Lill R, Silver P A. J Cell Biol. 1995;129:979–988. doi: 10.1083/jcb.129.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziegelhoffer T, Lopezbuesa P, Craig E A. J Biol Chem. 1995;270:10412–10419. doi: 10.1074/jbc.270.18.10412. [DOI] [PubMed] [Google Scholar]