

Abstract
Clamp loaders are multi-subunit complexes that use the energy derived from ATP binding and hydrolysis to assemble ring-shaped sliding clamps onto DNA. Sliding clamps in turn tether DNA polymerases to the templates being copied to increase the processivity of DNA synthesis. Here, the rate of clamp release during the clamp loading reaction was measured directly for the first time using a FRET-based assay in which the E. coli γ complex clamp loader (γ3δδ’χψ) was labeled with a fluorescent donor and the β-clamp was labeled with a nonfluorescent quencher. When a β·γ complex is added to DNA, there is a significant time lag before the clamp is released onto DNA. To establish what events take place during this time lag, the timing of clamp release was compared to the timing of DNA binding and ATP hydrolysis by measuring these reactions directly “side-by-side” in assays. DNA binding is relatively rapid and triggers hydrolysis of ATP. Both events occur prior to clamp release. Interestingly, the temporal correlation data and simple modeling studies indicate that the clamp loader releases DNA prior to the clamp and that DNA release may be coupled to clamp closing. Clamp release is relatively slow and likely to be the rate-limiting step in the overall clamp loading reaction cycle.
Clamp loaders load sliding clamps, which serve as processivity factors for DNA polymerases, onto DNA. Sliding clamps are ring-shaped complexes of crescent-shaped monomers that encircle duplex DNA and bind DNA polymerases to tether them to the template being copied. Binding to sliding clamps increases the processivity of DNA polymerases from tens to thousands of nucleotides. Clamp loaders are molecular machines that ultimately open the ring-shaped clamps and place the clamps around duplex DNA. Many structural and functional features of clamps and clamp loaders are conserved from bacteria to man, and the Escherichia coli sliding clamp and clamp loader have served as a useful model system for defining the mechanism of the clamp loading reaction (reviewed in (1-4)).
The E. coli clamp loader contains seven polypeptides, three copies of the dnaX gene product, and one copy each of the δ, δ’, χ and ψ subunits (5-7). The DnaX protein is present in two forms in the cell, a long form, τ, and a short form, γ. The short form is the result of a translational frameshift that truncates DnaX such that the γ subunit is about two-thirds the length of τ and of identical sequence except for the last amino acid residue (8-10). The clamp loader present in the DNA polymerase III holoenzyme most likely contains two copies of τ and one copy of γ. Clamp loaders containing any combination of γ and τ subunits are fully active in clamp loading (11). The additional C-terminal extension on τ mediates other protein·protein interactions at the replication fork that are important for efficient DNA replication (reviewed in (12, 13)). The form of the clamp loader used in these studies is the γ complex (γ3δδ’ψχ), which contains three copies of the γ subunit.
Clamp loaders use ATP to modulate their interactions with the clamp and DNA in order to catalyze the mechanical clamp loading reaction. Functional ATP binding sites are located in the DnaX subunits of the E. coli clamp loader (6, 14, 15). When the γ complex binds ATP, it promotes conformational changes that give the clamp loader a high affinity for the clamp and DNA (16-18), whereas ATP hydrolysis reduces the affinity for the clamp and DNA (18, 19). These ATP-induced changes in affinity help drive the clamp loading reaction, but other mechanisms are likely required to help promote an ordered series of interactions that results in productive clamp loading. For example, if the clamp loader were simply to bind and hydrolyze ATP on its own, it may be in the wrong state to productively interact with the clamp or DNA at the necessary point in the clamp loading reaction cycle. It is likely that interactions between the clamp loader and clamp, and between the clamp loader and DNA also promote conformational changes in the clamp loader that facilitate an ordered series of interactions. Prior to binding DNA, the ATP hydrolysis activity of the γ complex is relatively weak (14). Binding to primed template DNA specifically triggers hydrolysis of bound ATP (20), most likely by altering the conformation of the clamp loader to increase the efficiency of the reactions at the ATP active sites. Recent structural data indicates that this is likely to be the case. Interactions between clamp loader subunits and DNA induce a symmetrical arrangement of ATP sites that are poised for catalysis (21). Similarly, the clamp stimulates DNA-dependent ATP hydrolysis by the clamp loader (14, 22). In the presence of the clamp, three molecules of ATP are hydrolyzed rapidly when the clamp loader binds DNA whereas in the absence of the clamp only 2 molecules are hydrolyzed rapidly (23).
Our working model is that each interaction the clamp loader makes (with the clamp, DNA, ATP and ADP) promotes a change in the clamp loader that facilitates the next step in the clamp loading reaction to favor an ordered series of steps for an efficient clamp loading reaction. Thus, the interactions are dynamic in that each binding event alters the activity of the clamp loader. Given the dynamic nature of these interactions, it is necessary to define the temporal order of events in the clamp loading reaction as a first step in defining the mechanism for the clamp loading reaction.
MATERIALS AND METHODS
Buffers
Buffer Z contains 20 mM Tris·HCl pH 7.5 and 0.5 mM EDTA. Buffer A contains 20 mM Tris·HCl pH 7.5, 2 mM DTT, 0.5 mM EDTA, and 10% glycerol. Assay buffer contains 20 mM Tris·HCl pH 7.5, 50 mM sodium chloride, and 8 mM magnesium chloride.
DNA oligonucleotides
The DNA substrate used was a 30-nt primer annealed (as described (24)) to a 60-nt template. The sequence of the 60mer was 5′ TTC AGG TCA GAA GGG TTC TAT CTC TGT TGG CCA GAA TGT CCC TTT TAT TAC TGG TCG TGT3′ and the 30mer was complementary to the 30 nucleotides at the 3′ end. For anisotropy experiments, a C6-amino linker was incorporated at the 5′ end of the 60mer, and the amino group was covalently modified with mixed isomers of X-rhodamine-5-isothiocyanate and X-rhodamine-6-isothiocyante (Invitrogen) (25).
Protein expression
Coding sequences for the γ, δ, and δ’ subunits were inserted into separate pET-15B expression vectors (Novagen) between the NcoI and BamHI sites to express full-length proteins without epitope tags. Coding sequences for the χ and ψ subunits were inserted into the same pET-15b expression vector to create an artificial operon to express χ and ψ in the same cell. Co-expression of χ and ψ produces a soluble χψ complex (26) whereas expression of ψ alone gives insoluble protein that must be refolded (27).
All bacterial growth was performed in Luria Broth media supplemented with 100 μg/mL ampicillin. Competent E. coli BL21(DE3) cells were transformed with pET-15b constructs and plated. A 10-mL starter culture, inoculated with a single colony from a fresh transformation, was grown at 37 °C for 4 hours and stored overnight at 4 °C. Cells from the starter culture were harvested by centrifugation and resuspended in 2 mL of fresh media, and 500 μL of resuspended cells were used to inoculate a 500-mL expression culture. Expression cultures were grown at 37 °C to an O.D.600 of 0.6 - 0.7 (about 4 hrs) and protein expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Cells were incubated for 3 hours and harvested by centrifugation at 5000 x g for 30 min. Cell pellets were stored at -80 °C.
Protein purification
Proteins, β (28), γ (29), δ (30), δ’ (30), and χψ (26), were purified following published procedures with minor modifications. For ease of reference, these procedures are compiled in the Supplemental Methods. The γ complex (γ3δδ’χψ) was reconstituted from purified subunits as described previously (11). The concentration of γ complex containing AF488-labeled1 δ’ was determined by measuring the absorbance at 280 nm (ε280 = 220,050 M-1cm-1) in 6 M guandinium hydrochloride (31) and correcting for the contribution due to AF488 absorbance as described for AF488-labeled δ’.
AF488-labeled δ’
Purified δ’ (250 nmol) was dialyzed against a total volume of 1 L of 50 mM sodium phosphate buffer pH 7.0 for 5 hours and fresh 1 L of 50 mM sodium phosphate buffer pH 7.0 overnight. Dialyzed δ’ was pooled and incubated with 1.56 μmol Alexa Fluor 488 (AF488, Invitrogen) in the dark for 3.5 hours at 4 °C. Excess AF488 was removed from the labeled protein, δ’-AF488, by passing it through a desalting column (BioGel P6 DG) equilibrated with Buffer Z. The eluent was diluted with an equal volume of Buffer Z supplemented with 20% glycerol before loading it on a 1-mL HiTrap Q column equilibrated with Buffer A. The δ’-AF488 was eluted with a linear gradient of 0 to 600 mM sodium chloride, and dialyzed against Buffer A supplemented with 100 mM sodium chloride. The concentration of labeled protein was determined by measuring the absorbance at 280 nm (ε280 = 59, 960 M-1cm-1) for denatured protein (31) and correcting for the contribution from the AF488. The absorbance of AF488 at 280 nm is 0.11 times the value at 495 nm. The concentration of AF488 was determined by measuring the absorbance at 495 nm (ε495 = 71,000 M-1cm-1). Based on the relative concentrations of AF488 and protein, labeling efficiencies were in the range of 60 - 130%. The residue(s) modified by AF488 was not determined.
QSY9-labeled β
Purified β (65 nmol) was dialyzed against a total volume of 1 L of 50 mM sodium phosphate buffer pH 7.0 for 7 hours and fresh 1 L of 50 mM sodium phosphate buffer pH 7.0 overnight. Dialyzed β was pooled and incubated with 1.05 μmol QSY9 carboxylic acid, succimidyl ester (Invitrogen) for 4 hours in the dark at 4 °C. Excess QSY9 was removed from the labeled protein, β-QSY9, by passing it through a desalting column (BioGel P6 DG) equilibrated with Buffer Z. The eluent was loaded on a 1-mL HiTrap Q column equilibrated with Buffer A. The β-QSY9 was eluted with a linear gradient of 0 to 500 mM sodium chloride, and dialyzed against Buffer A minus DTT. The concentration of labeled protein was determined by measuring the absorbance at 280 nm (ε280 = 14,890 M-1cm-1) in 6.0 M guanidinium hydrochloride (31) and correcting for the contribution from the QSY9. The absorbance of QSY9 at 280 nm is 0.23 times the value at 562 nm. The concentration of QSY9 was determined by measuring the absorbance at 562 nm (ε495 = 85,000 M-1cm-1). Because the absorbance at 280 nm for β is relatively small, a modified Lowry Assay (Pierce) was used to verify the protein concentrations. Both techniques gave the same concentrations. Based on the relative concentrations of QSY9 and β, labeling efficiencies were in the range of 100 - 120%. The site(s) of modification by QSY9 was not determined.
Fluorescence emission spectra
Fluorescence emission spectra of AF488 were taken on an Edinburgh FS900 exciting at 490 nm and using a 3.6 nm bandpass on excitation and emission monochromators. Assay buffer (68 μL), γc-AF488 (4 μL), β-QSY9, unlabeled β, or protein dilution buffer (4 μL), and ATP (4 μL) were added sequentially to a Hellma microcuvette (105.251-QS) and an emission spectrum was recorded following each addition. The background signal for assay buffer was subtracted from each spectrum, and the intensity relative to the intensity of free γc-AF488 at 515 nm is plotted. Spectra were not corrected for the decrease in fluorescence due to dilution by sequential addition of reagents.
Equilibrium binding
For each concentration of β-QSY9, assay buffer and ATP (72 μL), γc-AF488 (4 μL), and β-QSY9 (4 μL) were added sequentially to the cuvette and an emission spectrum was recorded following each addition. Buffer background signals were subtracted from each spectrum, and the intensity at 515 nm relative to free γc-AF488 was calculated. Intensities relative to the intensity of γc-AF488 in the absence of β-QSY9 are plotted. The dissociation constant (Kd) was calculated by fitting the observed intensity data (Iobs) to equation 1 in which γc is the concentration of γc-AF488, β is the concentration of β-QSY9, Imax is the intensity of free γc-AF488 and Imin is the intensity of γc-AF488·β-QSY9. The intensity of γc-AF488·β-QSY9 (Imin) and Kd were fit as adjustable parameters.
| (1) |
Pre-steady-state kinetic assays
All kinetic measurements were made using an Applied Photophysics SX18MV stopped-flow at 20 °C. Experiments were done in single-mix mode in which equal volumes of two solutions were mixed immediately prior to entering the cuvette as indicated in the diagrams given in each figure. Final concentrations after mixing the two solutions are given in the Figure Legends.
DNA binding/RhX anisotropy measurements
Vertically and horizontally polarized emission were measured simultaneously when exciting RhX with vertically polarized light at 580 nm. Emission data were measured using a 610-nM cut-on filter at 2.5-ms intervals over a 5-second time period. Six to nine reactions were run, and polarized intensities were signal averaged. Anisotropy, robs, as a function of time was calculated from the polarized intensities using equation 2 where Ivv and Ivh are the vertically and horizontally polarized intensities, respectively, measured when exciting with vertically polarized light, and g is the g-factor. The g-factor corrects for differences in the efficiencies of the detectors and is the ratio of Ihv to Ihh where Ihv and Ihh are the intensities of vertically and horizontally polarized light, respectively, when exciting with horizontally polarized light. The experimentally determined g-factor was 0.996. Anisotropy time courses were smoothed over 5 data points.
| (2) |
ATP hydrolysis/MDCC intensity measurements
E. coli PBP was purified and labeled with N-(2-(1-maleimidyl)ethyl)-7-(diethylamino)coumarin-3-carboxamide (MDCC; Invitrogen) as described previously (23). MDCC was excited at 425 nm using 1.9 nm bandpass and emission was collected through a 455-nm cut-on filter. Four thousand data points were collected over a 5-second time period.
Clamp release/PY anisotropy measurements
Anisotropy measurements were made as described for DNA binding experiments. Polarized emission was measured when exciting PY with vertically polarized light at 327 nm. Emission data were measured using a 375-nM cut-on filter at 1.25-ms intervals over a 5-second time period. Polarized intensities from 24 runs were averaged and used to calculate anisotropy according to equation 2. The g-factor for these experiments was 0.94. Anisotropy time courses were smoothed over 3 points.
Clamp release/AF488 intensity measurements
AF488 was excited at 490 nm using 1.9 nm bandpass. Emission data were collected with a 515-nm cut-on filter at 1.25 ms intervals over a 5-second time period.
Kinetic modeling
Kinetic data were fit and simulated using DynaFit.
RESULTS
FRET-based assay to measure γ complex binding β
In previous studies (24, 32), a pyrene (PY) anisotropy-based assay was used to measure γ complex binding to a PY-labeled β clamp. The PY anisotropy assay is not ideal because PY fluorescence is too weak for performing experiments at the low nanomolar protein concentration range. Therefore, a more sensitive FRET-based assay, in which the γ complex was labeled with a fluorescent donor and the β-clamp was labeled with a nonfluorescent quencher, was developed to measure clamp loader·clamp interactions. Available structural data indicates that fluorophores covalently attached to the N-terminal amino group of the δ’ subunit and the N-terminal amino group of β will be within a suitable distance for FRET to occur in a β·γ complex. In addition, both amino groups are located on solvent-exposed surfaces that are not expected to be directly involved in protein·protein or protein·DNA interactions so that covalent modification of these groups would not be anticipated to negatively impact the functions of the proteins. The δ’ subunit of the γ complex was covalently labeled with a fluorescent donor, Alexa Fluor 488 (AF488), and β was covalently labeled with a nonfluorescent quencher, QSY9. Labeling reactions were performed at pH 7.0 to promote selective labeling of the N-terminal amino residues, which typically have pKa values about a unit lower than lysine residues. Donor-labeled γ complex was reconstituted from purified subunits and the AF488-labeled δ’ subunit. The β-clamp (β-QSY9) should contain two quenchers, one per monomer. Given the head-to-tail symmetry of the dimeric β ring, γc-AF488 binding β-QSY9 in either orientation will place a quencher near the donor on δ’ (Fig. 1).
Figure 1. Schematic diagram illustrating the locations of the fluorescent donor (D), AlexaFluor 488 (AF488), and nonfluorescent quencher (Q), QSY9.
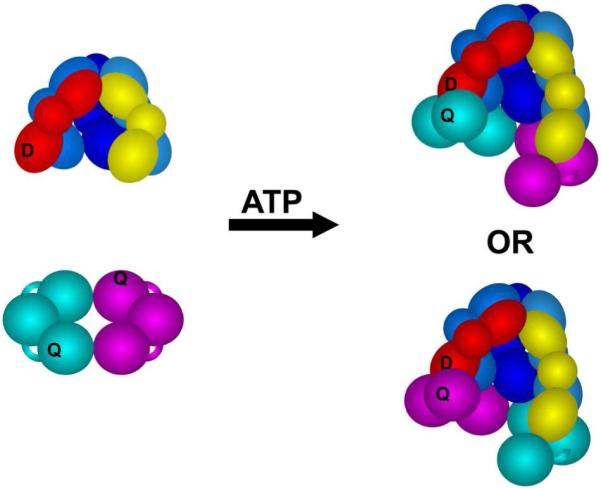
The five-subunit core, γ (shades of blue), δ (yellow), and δ’ (red), of the γ complex (γ3δδ’χψ) is shown in this diagram. Each subunit is composed of three globular domains represented by spheres and ovals. The χ and ψ subunits are omitted from the figure, but present in the complex used in all experiments. The δ’ subunit (red) of the γ complex was covalently labeled with AF488 (D) and the β clamp was covalently labeled with QSY9 (Q). The β-clamp is a dimer of identical subunits each containing three globular domains, and there are two quencher sites within each clamp, one quencher on the cyan subunit and a second on the magenta subunit. Given the C2-axis of symmetry through the center of the ring-shaped clamp, the donor on the δ’ subunit should be near a quencher when the γ complex binds β in either orientation.
Because the quencher is on one macromolecule and the donor on the other, FRET quenching of AF488 fluorescence should occur when γc-AF488 binds β-QSY9 and quenching should be relieved when γc-AF488 dissociates from β-QSY9. Emission spectra of AF488 were measured before and after the successive addition of β-QSY9 and ATP to γc-AF488, and fluorescence intensities relative to free γc-AF488 (light gray spectra) are shown in Fig. 2A. Addition of β-QSY9 to γc-AF488 (Fig. 2A, dark gray solid line) quenches AF488 fluorescence and addition of ATP further quenches fluorescence (Fig. 2A, black solid line) because γ complex binds β weakly in the absence of ATP and with higher affinity in the presence of ATP (17). A control experiment in which buffer was added in place of β-QSY9 (Fig. 2A, dotted lines) gave decreases in fluorescence due to dilution that were not as large as the quench due to β-QSY9 binding. A parallel experiment with unlabeled β (Fig. 2B) shows that unlabeled β (solid lines) gives the same decrease in AF488 fluorescence as dilution by buffer (dotted lines). These results demonstrate that the quench in AF488 fluorescence on addition of β-QSY9 to γc-AF488 is due to FRET rather than an environmental effect due to changes in protein·protein interactions.
Figure 2. FRET occurs when β-QSY9 binds γc-AF488 and results in a β-QSY9-concentration-dependent quench in AF488 fluorescence.

The emission spectrum of γc-AF488 (solid light gray lines) was recorded prior to the sequential addition of either A) β-QSY9 (solid dark gray line) or B) unlabeled β (solid dark gray line) followed by ATP (solid black lines). Controls were done in which protein storage buffer was added instead of β (dotted lines) to measure the decrease in fluorescence due to dilution by each addition. Final concentrations after addition of ATP were 20 nM γc-AF488, 200 nM β-QSY9 or unlabeled β, 0.5 mM ATP in assay buffer. In panel C, the quench in AF488 fluorescence was measured as a function of β-QSY9 concentration to determine the dissociation constant, Kd, for γc-AF488·β-QSY9. At each β-QSY concentration, spectra of AF488 were measured before and after β-QSY9 addition to γc-AF488 and the relative intensity of AF488 fluorescence after β-QSY9 addition is plotted. Assays contained 10 nM γc-AF488 and 0.5 mM ATP in assay buffer. Error bars show the standard deviation of three independent experiments and the solid line through the data represents the fit to equation 1 (Materials and Methods). The average Kd value calculated was 15 ± 6.1 nM. Assay buffer for spectral measurements contained 20 mM Tris·HCl pH 7.5, 50 mM NaCl, and 8 mM MgCl2.
A complete titration of γc-AF488 with β-QSY9 was done in the presence of ATP to determine the dissociation constant for the γc-AF488·β-QSY9 interaction. The relative intensity of AF488 at 515 nm is plotted as a function of β-QSY9 concentration in Fig. 2C. A Kd value of 15 ± 6.1 nM was calculated from the fits of three independent experiments at 10 nM γc-AF488 (averages are shown in Fig. 2C). This Kd value is 5-fold greater than the value of 3 nM reported previously (17). Perhaps one of the proteins is labeled at a site that affects protein interactions and gives rise to this small decrease in affinity.
Clamp loading catalyzed by γc-AF488
Given that binding of γc-AF488 to β-QSY9 was somewhat weaker than reported for the unlabeled proteins, clamp loading activity of γc-AF488 was measured to determine whether the presence of a fluorophore affected enzyme activity. Clamp loading was measured in experiments in which a pre-incubated solution of β, γ complex, and ATP was added to a solution of DNA and ATP. During the pre-incubation period, a β·γ complex forms that is poised to bind DNA and load clamps. Three independent assays were performed that report on 1) DNA binding (25, 33), 2) ATP hydrolysis (34, 35), and 3) clamp release (24, 32). DNA binding was measured in an anisotropy-based assay in which p/t-DNA was covalently labeled with X-rhodamine (RhX) on the 5′ template end. The anisotropy of RhX reports on the fraction of p/t-DNA molecules bound by protein and increases when the fraction of DNA bound increases. When a β·γ complex is added to p/t-DNA-RhX, the anisotropy initially increases as the β·γ complex binds DNA, but then decreases as the clamp loader deposits the clamp and dissociates from the clamp·DNA complex (20, 25, 33) (Fig. 3A). The time courses for loading unlabeled β clamps catalyzed by γc-AF488 (black trace) and unlabeled γ complex (gray trace) show the same rates of increases and decreases in RhX-anisotropy, and we conclude that labeling does not affect the kinetics of DNA binding/release. There is a small variation in amplitude that is most likely due to variation that occurs from experiment to experiment even with the same protein.
Figure 3. Clamp loading reactions catalyzed by labeled γc-AF488 and unlabeled γ complex.

The clamp loading activities of γc-AF488 and unlabeled γ complex were compared to determine whether the fluorescent label affected enzyme activity. Clamp loading reactions were initiated by adding a solution of γc-AF488 (black reaction traces) or unlabeled γ complex (gray reaction traces), β, and ATP to a solution of p/t-DNA and ATP. Clamp loading was measured in three separate assays: A) a DNA binding assay containing final concentrations of 100 nM γc-AF488 or unlabeled γc, 800 nM β, 100 nM p/t-DNA-RhX, 0.5 mM ATP, B) an ATP hydrolysis assay containing final concentrations of 100 nM γc-AF488 or unlabeled γc, 800 nM β, 100 nM p/t-DNA, 0.5 mM ATP, 3 μM MDCC-PBP, and C) a clamp release assay containing final concentrations 400 nM γc-AF488 or unlabeled γc, 400 nM β-PY, 420 nM p/t-DNA, 0.5 mM ATP, 8 μM unlabeled β.
Similarly, DNA-triggered ATP hydrolysis that occurs during the clamp loading reaction was measured for the AF488-labeled and unlabeled γ complex (Fig. 3B). Hydrolysis was measured as a function of time using E. coli phosphate binding protein (PBP) labeled with a coumarin dye (MDCC) to report on the release of inorganic phosphate (Pi) by ATP hydrolysis (35). When MDCC-PBP binds Pi, the fluorescence of MDCC increases. Binding of the β·γ complex to DNA triggers a rapid burst of ATP hydrolysis over about 0.1 s for both labeled and unlabeled clamp loaders (19, 34) (Fig. 3B). This burst is followed by a linear increase in MDCC fluorescence associated with a steady-state clamp loading reaction. The rates of ATP hydrolysis were the same for the AF488-labeled (black trace) and unlabeled (gray trace) clamp loaders. The greater burst amplitude for γc-AF488 compared to unlabeled γ complex is likely due to differences in active site concentrations between the two different enzyme preparations.
A second anisotropy assay was used to measure clamp loading from the perspective of the β-clamp, which was covalently labeled with PY (32). The anisotropy of PY decreases when the large clamp loader (250 kDa) releases the clamp (80 kDa) on the small p/t-DNA (30 kDa). This clamp loading reaction was limited to a single turnover by including an excess of unlabeled β-clamp in the solution of p/t-DNA and ATP. After releasing the labeled β, the γ complex has a higher probability of binding the unlabeled β, which is in excess. After addition of the β·γ complex to DNA, the anisotropy of PY decreased in the first 0.8 s of the reaction as the clamp loader released the clamp on DNA (Fig. 3C). The time courses for clamp release were the same for both γc-AF488 (black trace) and unlabeled γ complex (gray trace). Taken together, these three experiments show that the clamp loading activity of the labeled γ complex, γc-AF488, is the same as unlabeled γ complex, and therefore, is not negatively affected by covalent attachment of a fluorophore.
Passive dissociation of the clamp from the clamp loader versus active release of the clamp onto DNA
The main goal of this work is to correlate the timing of clamp release with timing of ATP hydrolysis and DNA binding and release. However, our clamp release assays simply report on the dissociation of a clamp loader·clamp complex and this can occur by two mechanisms. The clamp can dissociate from the clamp loader via 1) a passive mechanism that is a function of equilibrium binding and 2) an active mechanism that results from loading the clamp on DNA. To determine whether we could distinguish between these two reactions in kinetics assays, two experiments were done. First, the rate of passive dissociation was measured by adding a solution of γc-AF488, β-QSY9, and ATP to a solution of ATP and excess unlabeled β (Fig. 4A). When the quencher-labeled-β dissociates from the γ complex, the γ complex is “trapped” by the excess unlabeled β. In this passive dissociation reaction, the relative intensity of AF488 increased slowly as β-QSY9 dissociated from γc-AF488 and the FRET quench was relieved. Overall, this reaction was slow taking about 100 s to reach completion, however, two phases were present and the data is best fit by a double exponential (Fig. 4A, solid black curve) yielding observed rate constants of 0.34 s-1 and 0.040 s-1.
Figure 4. Passive dissociation of the clamp from the clamp loader versus active clamp loading onto DNA.

Dissociation of the clamp from the clamp loader was measured in two different experiments by adding a solution of γc-AF488, β-QSY9, and ATP to a solution of A) excess unlabeled β and ATP or B) p/t-DNA, excess unlabeled β, and ATP. Reactions in panel B contain 0 nM (gray trace), 25 nM (blue trace), 50 nM (purple trace), 100 nM (red trace), 200 nM (green trace), or 400 nM (cyan trace) p/t-DNA. Solid black lines through the gray traces containing no p/t-DNA in panels A and B are exponential fits of the data. Final concentrations were 20 nM γc-AF488, 400 nM β-QSY9, 8 μM unlabeled β, and 0.5 mM ATP in assay buffer containing 20 mM Tris·HCl pH 7.5, 50 mM NaCl, 8 mM MgCl2, and 4% glycerol.
The second experiment measuring active loading of β was the same as the first except that p/t-DNA was included in the solution of unlabeled β and ATP (Fig. 4B). The concentration of p/t-DNA was varied from 25 - 400 nM to determine how the rate of DNA binding influenced the rate of clamp release. The rate of clamp release increased with the concentration of DNA and begins to approach a maximal rate at the highest concentration. The DNA-concentration-dependence of the rates is consistent with an active mechanism in which DNA binding triggers a subsequent clamp release reaction. Overall, the active clamp release reaction is much faster than the passive dissociation reaction, nearing completion after about 3 seconds at DNA concentrations greater than or equal to the concentration of γ complex. The shape of time courses for active clamp loading reactions, containing DNA at concentrations equal to (100 nM) or greater than the β·γ complex concentration, also differs from that of the passive dissociation reaction. In the passive dissociation reaction, AF488 intensity increases as an exponential function whereas in the active clamp release reactions, there is a substantial time lag of about 50 ms (Fig. 4B inset) before the fluorescence increases giving rise to a sigmoidal-shaped increase. The sigmoidal shape indicates that some kinetic steps that do not affect the fluorescence signal take place prior to clamp release. These “silent” events are likely the clamp loader·clamp complex binding DNA and hydrolysis of some or all of the ATP bound to the γ complex.
Although the DNA-dependent clamp release reaction is about 30 times faster than the DNA-independent passive release reaction, a reaction containing no p/t-DNA performed at the same time and under the same conditions as the DNA-triggered clamp release reactions showed that a small but significant amount of DNA-independent dissociation occurs on the time scale of the active clamp loading reaction. A fit of this DNA-independent reaction to a single exponential yielded an observed rate constant of 0.41 s-1, which matches the rate constant for the rapid phase of the passive dissociation reaction measured on a longer time scale in Fig. 4A. This DNA-independent dissociation reaction appears to compete with active release and to contribute reaction kinetics measured at substoichiometric DNA concentrations of 25 and 50 nM. The 25 nM DNA time course does not contain a lag but instead the fluorescence increases linearly over about the first 300 ms of the reaction (Fig. 4B inset). In assays with substoichiometric concentrations of DNA, only a fraction of clamp loader·clamp complexes can bind DNA during the first turnover and the remaining complexes are free to dissociate via the passive mechanism. In contrast, when enough DNA is present to bind all the clamp loader·clamp complexes, a distinct lag of about 50 ms in the fluorescence increase is present (Fig. 4B inset). In these reactions, DNA binding is likely to be rapid relative to passive dissociation so that the passive dissociation reaction does not effectively compete. It is not clear why two phases are present in the passive dissociation reaction. It is possible that some or all of the proteins are labeled at a site that adversely affects binding and gives rise to a rapid dissociation rate (0.3 - 0.4 s-1). This rapid dissociation rate may explain the higher Kd value measured for labeled proteins as compared to unlabeled proteins. Because the DNA-independent dissociation reaction makes a smaller contribution to clamp release kinetics at DNA concentrations greater than or equal to 100 nM, remaining clamp release experiments were done at these concentrations.
Temporal correlation of DNA binding, ATP hydrolysis, and clamp release
In order to dissect the temporal order of events in the clamp loading reaction, the kinetics of DNA binding, ATP hydrolysis, and clamp release were measured directly under identical reaction conditions. In each case, clamp-loading reactions were initiated by the addition of a solution of β, γ complex, and ATP to a solution of p/t-DNA and ATP (Fig. 5A). The concentration of p/t-DNA was varied, 100, 200, and 400 nM, to evaluate the dependence of reaction kinetics on DNA binding rates. Concentrations of γ complex, β, and ATP were held constant at 100 nM, 500 nM, and 0.5 mM, respectively. DNA binding and subsequent release of the clamp on DNA were measured in the RhX-anisotropy assay using RhX-labeled p/t-DNA. Hydrolysis of ATP was measured using the MDCC-PBP assay in which MDCC-PBP, at a final concentration of 3 μM, was included in the syringe with p/t-DNA. The clamp release reaction measured by FRET differed from the DNA binding and ATPase assays in that inclusion of excess unlabeled β in the syringe with p/t-DNA limited clamp release reactions to a single turnover whereas the other reactions included both pre-steady-state and steady-state phases.
Figure 5. Temporal correlation of DNA binding, ATP hydrolysis, and release of the clamp on DNA.

A) RhX anisotropy, MDCC intensity, and AF488 intensity were measured as a function of time to follow DNA binding/release (red traces), ATP hydrolysis (blue traces), and clamp release (black traces), respectively, during the course of clamp loading reactions. For each reaction, a solution of β, γ complex, and ATP was added to a solution of p/t-DNA and ATP. Final concentrations were 100 nM γ complex, 500 nM β, 0.5 mM ATP, and B) 100 nM, C) 200 nM, or D) 400 nM p/t-DNA. Proteins were unlabeled in DNA binding/release experiments, the clamp, clamp loader, and DNA were unlabeled in ATP hydrolysis experiments, and DNA was not labeled in clamp release experiments.
Time courses for DNA binding, ATP hydrolysis, and clamp release are shown on the same graphs by plotting RhX anisotropy (DNA binding, red traces) on the right-hand y-axis and the relative intensities of MDCC (ATPase, blue traces) and AF488 (clamp release, black traces) on the left-hand ordinate. Data for reactions containing 100 nM DNA are plotted in Fig. 5B. As shown previously, the anisotropy of RhX increases rapidly on addition of the clamp loader·clamp complex to DNA and then decreases when the clamp loader releases DNA. The intensity of MDCC fluorescence increases rapidly over about 0.1s, levels off, and then increases again linearly. The increase in anisotropy occurs before the increase in MDCC intensity, and the anisotropy reaches a peak value around the same time, about 70 ms, as the midpoint of the initial rapid increase in MDCC fluorescence. This timing is consistent with DNA release following hydrolysis of ATP (34), and previous work has shown that all three molecules of ATP bound to the clamp loader are hydrolyzed in this initial rapid phase (23, 24).
A comparison of all three assays in Fig. 5B shows that the increase in AF488 fluorescence due to clamp release occurs after the increase in RhX anisotropy and the increase in MDCC intensity. There is a substantial lag before the intensity of AF488 fluorescence increases such that the intensity of AF488 begins to increase rapidly when the RhX anisotropy has reached its peak and begins to decrease. The rate of decrease in RhX anisotropy is slower than the actual DNA dissociation rate because the observed anisotropy is the sum of binding events that increase anisotropy and dissociation events that decrease anisotropy. This combined with the observation that AF488 intensity increases more slowly than RhX anisotropy decreases strongly suggests that DNA is released before the clamp. When the concentration of DNA is increased to 200 nM (Fig. 5B) and 400 nM (Fig. 5C), the rate of DNA binding increases but the relative timing of the fluorescence changes associated with DNA binding/release, ATP hydrolysis, and clamp release remain the same. The peak in the anisotropy time course shifts to shorter times with increasing DNA concentrations because DNA binding is faster. The amplitude of the anisotropy peak decreases because a smaller fraction of RhX-labeled DNA is bound due to an excess of p/t-DNA over the clamp loader·clamp complex. Together, these DNA binding/release, ATP hydrolysis and clamp release data support a model in which the β·γ complex binds DNA, DNA binding triggers ATP hydrolysis, and DNA and the clamp are released sequentially.
Kinetic modeling of clamp loading reactions
Kinetic modeling studies were done with two main goals in mind. The first goal was to determine whether kinetic data for clamp release were consistent with the temporal order of events in the clamp loading reaction that have been defined by previous studies of ATP hydrolysis and DNA binding/release. And the second goal was to determine whether the kinetic data supported a model in which the DNA was released before the clamp. Previous studies have shown that: 1)ATP binding supports formation of a β·γ complex but that ATP hydrolysis is not efficient in this complex (16-18), 2) β·γ complex binding to primed template DNA triggers ATP hydrolysis (34), 3) the β·γ complex hydrolyzes all 3 molecules of ATP after binding DNA2 (23, 24), 4) DNA is released after hydrolysis of ATP (34). ATP hydrolysis and clamp release data were fit to a very simple temporal model based on these results and illustrated in the schematic diagram in Fig. 6A. In this model, the β·γ complex binds DNA, γ complex hydrolyzes three molecules of ATP sequentially at the same rate for each, and then DNA and the clamp are released. DNA dissociates from the clamp immediately on release. An assumption made in this model is that the clamp and DNA are released at the same time. ATP hydrolysis and clamp release data were fit to this model independently to determine whether both data sets yielded similar rate constants. The first turnover (about 0 - 0.2 s) of ATP hydrolysis time courses, for experiments containing 25 - 400 nM DNA, were fit to the model using DynaFit (36) to yield the rate constants shown in black text in Fig. 6A and calculated fits (black curves in Fig. 6B).3 Clamp release time courses, for experiments done at 100 - 400 nM DNA in Fig. 4B, were fit to the same model to yield the rate constants shown in blue text in Fig. 6A and calculated fits (blue curves) in Fig. 6C. The rate constant for DNA binding obtained from the fits to clamp release data is about 3 times slower than from the ATP hydrolysis data whereas the hydrolysis rate constants are about twice as fast. Notably, the rate constant obtained for clamp and DNA release is about 10 times slower for the fit to the clamp release reaction than for the fit to the DNA release reaction. The anisotropy experiments measuring DNA binding were not fit because we do not have a direct measure of the anisotropy values for the transiently bound DNA species (the ternary β·γ·DNA complexes with bound nucleotide). Instead, the rate constants obtained from the fits to ATP hydrolysis and clamp release data were used to calculate the fraction of DNA bound as a function of time in Fig. 6D, black and blue traces, respectively, for a reaction containing 100 nM β·γ complex and 100 nM DNA. Given that the anisotropy for bound DNA is greater than for free DNA, the observed anisotropy should change in the same way, increase on binding and decrease on release, as the fraction of DNA bound. The overlays of the simulated time courses show that the rate constants obtained from the ATP hydrolysis kinetics more accurately reflect the timing of DNA binding and release. Importantly, the rate of DNA dissociation is much to slow when the rate constant obtained from the clamp release fits is used.
Figure 6. Kinetic modeling of clamp loading reactions with simultaneous release of the clamp and DNA.

A) Simple kinetic model used to fit and simulate data. Rate constants given in black text are from fits of ATP hydrolysis data and rate constants in blue text are from fits to clamp release data. B) The first 0.2 s of ATP hydrolysis reactions (the first turnover) were fit to the model illustrated in panel A using DynaFit (36). Calculated fits are shown in black. C) The first 1.2 s of clamp release time courses were fit to the same model and calculated fits are shown in blue. Reaction time courses in panels A and B were color-coded to the DNA concentrations as follows: 25 nM (blue), 50 nM (purple), 100 nM (red), 200 nM (green), and 400 nM (cyan). In experiments, β is present in excess (500 nM) over γ complex (100 nM) to ensure that all the γ complex was bound. The concentration of β·γ complex used in modeling was 100 nM. D) The fraction of DNA bound as a function of time was calculated using rate constants derived from fitting ATP hydrolysis kinetics (black curve) and clamp release kinetics (blue curve) for reactions containing 100 nM β·γ complex and 100 nM DNA. These simulated time courses were overlayed on the anisotropy data for DNA binding/release done at the same concentrations.
Based on these results, the simple model shown in Fig. 6A was modified to include a two-step dissociation reaction in which DNA is released prior to β and DNA slips out of the clamp loader·clamp complex (Fig. 7A). In addition, the possibility that DNA dissociation was coupled to clamp closing was also tested by fitting the molar response (effective quantum yield) for this intermediate clamp loader·clamp complex. Rate constants for DNA binding, ATP hydrolysis, and DNA release were fixed at values obtained from the fits to the ATP hydrolysis kinetics, and the rate constant for β release and molar responses for the closed β·γ complex and free γ complex were fit as adjustable parameters. Results of these fits (black curves) are shown with the clamp release data in Fig. 7B. The calculated rate constant for β dissociation was 3 s-1. The molar response obtained for the putative closed β·γ complex was 10.4 which is in between the initial value of 10 for the open β·γ complex and 12.5 for free γ complex. Time courses for DNA binding/release, ATP hydrolysis, and clamp release were calculated using these rate constants for a reaction containing 100 nM DNA and 100 nM β·γ complex to compare with the temporal correlation kinetics shown in Fig. 5B. Four steps were added to generate the steady-state phases of the DNA binding and ATP hydrolysis reactions (see Supplemental Methods): 1) dissociation of ADP, 2) binding ATP, 3) an ATP-induced conformational change, and 4) binding β. These time courses in Fig. 7C calculated using the simple model have many features in common with the data in Fig. 5B.
Figure 7. Kinetic model for clamp loading with sequential release of DNA and the clamp.

A) Diagram illustrating a simple kinetic model with sequential release of DNA and β in which clamp closing is coupled to DNA release. B) The kinetics of clamp release (Fig. 4B) measured at 100 nM (red), 200 nM (green) and 400 nM (cyan) were fit to the model illustrated in panel A using DynaFit (36). Rate constants in black type were fixed and the rate constant for clamp release (blue type) as well as the molar responses for the closed clamp loader·clamp complex and the free clamp loader were fit as adjustable parameters. Calculated fits are solid black lines through the data. C) The model in panel A was used to simulate the fraction of DNA bound (red trace), relative intensity of MDCC (blue trace) reporting on ATP hydrolysis, and relative intensity of AF488 (black trace) reporting on clamp release as a function of time. This model was modified to include steady-state phases for the DNA binding and ATP hydrolysis reactions (Supplementary Methods). Simulations were done for the reaction conditions used experimentally in Fig. 5B which contained 100 nM γ complex, 500 nM β, 100 nM p/t-DNA and 0.5 mM ATP.
Although a main goal of kinetic modeling was to determine whether these results were consistent with the temporal order of events in the clamp loading reaction as described in previous work, other models that differed in the timing of hydrolysis of individual molecules of ATP were tested. The kinetic data could not be adequately described by models in which the three molecules of ATP were hydrolyzed in different phases of the reaction, for example, two molecules hydrolyzed before DNA release and the third molecule hydrolyzed after DNA release. The data could be described equally as well by a model in which all three molecules of ATP were hydrolyzed simultaneously in the same step before DNA and clamp release rather than one-at-a-time or sequentially as shown in Figs. 6 and 7. Therefore, based on these data we cannot distinguish between sequential and simultaneous hydrolysis so this remains to be defined. However, both sequential and simultaneous models for ATP hydrolysis give rise to the same temporal order of events; the β·γ complex binds DNA, hydrolyzes 3 molecules of ATP, releases DNA, and finally releases the clamp.
DISCUSSION
Temporal order of events in the clamp loading reaction
Dynamic changes in protein·protein and protein·DNA interactions must occur for the clamp loader to bind and open the clamp, place the clamp around DNA, and then release the clamp·DNA complex. Affinity modulation is achieved, at least in part, by ATP binding and hydrolysis that promote conformational changes in the clamp loader that alter binding interactions with the clamp and DNA. But ATP binding and hydrolysis alone are unlikely to provide a mechanism for ordered affinity modulation required for an efficient clamp loading reactions. For example, if the clamp loader were simply to oscillate between binding and hydrolyzing ATP, it may be in the wrong state at a given point in the clamp loading reaction cycle to productively interact with the clamp and DNA. Interactions with the clamp and DNA are likely to provide an additional level of regulation. For example, a β·γ complex does not hydrolyze ATP efficiently in the absence of DNA suggesting that DNA binding promotes a conformational change that increases the ATPase activity of the γ complex. This suggestion is borne out by recent structural studies showing that DNA binding to the clamp loader aligns the ATP sites in the appropriate geometry for catalysis (21). Our working model is that each interaction the clamp loader makes promotes a change in the clamp loader that facilitates the next step in the clamp loading reaction to favor a defined temporal order of events that lead to an efficient clamp loading reaction.
As a first step in testing this model for affinity modulation, we are working to define the temporal order of events in the clamp loading reaction. Our approach is to use fluorescence-based assays to monitor individual species in real time during the course of a clamp loading reaction. The clamp loading reaction can be divided into two stages: 1) formation of a ternary clamp loader·clamp·DNA complex and 2) decay of the ternary complex to produce a clamp·DNA complex. The temporal order of events in the second stage is the focus of this work. In previous studies, DNA binding/release and ATP hydrolysis assays have been established (25, 34). Here, a FRET-based assay was developed and used to monitor the fate of an E. coli clamp loader·clamp complex, and to directly measure when the clamp is released onto DNA. To simplify reaction kinetics for initial studies, the clamp loading reaction is initiated from a defined state by adding a pre-formed clamp loader·clamp complex to DNA rather than mixing all three macromolecules and ATP together simultaneously. This mixing scheme allows for rapid formation of the ternary complex so that kinetics of the decay of the ternary complex are not complicated by differences in kinetics for different pathways of ternary complex formation.
A direct comparison of time courses for DNA binding/release, ATP hydrolysis, and clamp release (Fig. 5) established the temporal order of these events; the clamp loader·clamp complex rapidly binds DNA, hydrolyzes ATP, releases DNA, and then the clamp. Earlier work had shown that primed DNA templates containing single-stranded/double-stranded DNA junctions with 5′-single-stranded overhangs preferentially trigger ATP hydrolysis (20, 33), and that the clamp loader hydrolyzes all bound molecules of ATP in the same global step (23, 24, 34). That is not to say that all three molecules of ATP are hydrolyzed simultaneously, hydrolysis of individual molecules may occur sequentially in a series of reactions triggered by DNA binding as illustrated in the models in Figs. 6A and 7A, like a line of dominos falling when the first is pushed. A novel, and perhaps unexpected, finding from this work is that following ATP hydrolysis, the γ complex releases DNA before the clamp. Previous studies had shown that ATP hydrolysis reduces the affinity of the γ complex for both DNA and β (18, 19), but this work shows that they are not released simultaneously.
The ability to measure clamp release directly in real time using the FRET assay allowed us to address the question of whether γ complex loaded some clamps without hydrolysis of ATP. An earlier study suggested that the clamp loader associated with the DNA polymerase III holoenzyme could load a clamp on the leading strand without hydrolyzing ATP, but required ATP hydrolysis to load a second clamp on the lagging strand (37). However, in our FRET assay, two phases of clamp release, one preceding and one following ATP hydrolysis, were not observed. The γ complex only released β after hydrolyzing ATP. One possible reason for the difference in experimental results is that the clamp loader associated with the holoenzyme contains protein elements not present in the γ complex clamp loader used in these studies. While both clamp loaders contain three copies of the dnaX gene product, the holoenzyme contains two copies of τ and one copy of γ whereas the γ complex contains three copies of γ. Although both clamp loaders are fully active in loading clamps (11), the additional C-terminal extension present in τ or its interactions with the α subunit of the polymerase could alter the ATP requirements for clamp loading and impart this asymmetry. This FRET assay could easily be adapted to studies with the holoenzyme, and it will be interesting to see if the two clamp loaders differ in their ATP requirements for clamp loading.
Kinetic mechanism of clamp loading
The kinetic mechanism of the clamp loading reaction is complicated. There are many discrete steps required to describe a linear pathway for assembling clamps on DNA that include: 1) the clamp loader binding three molecules of ATP (38), 2) ATP-induced conformational changes that give the clamp loader a high affinity for the clamp and DNA (17, 18), 3) clamp loader·clamp binding, 4) clamp opening (16, 18), 5) DNA binding, 6) hydrolysis of three molecules of ATP (23, 24), 7) DNA and clamp release, and 8) release of ADP. This list does not include conformational changes in the clamp loader that are likely to be promoted by interactions with DNA and the clamp, and by ATP hydrolysis, or the possibility that individual molecules of ATP are bound or hydrolyzed in separate steps to promote different steps in the reaction. This complexity is further increased by the possibility of alternate pathways for clamp loading. For example, two parallel pathways could exist, one in which the clamp loader binds the clamp first and a second in which the clamp loader binds DNA first (39, 40). Side reactions could also occur, for example, intermediate complexes could dissociate without completing a productive clamp loading cycle. Given this complexity, a complete kinetic mechanism for clamp loading cannot be defined by a single set of experiments as described here. That being said, simple models can be used to show that the kinetics of DNA binding, ATP hydrolysis, and DNA and clamp release are consistent with the temporal order of events proposed.
Time courses for DNA binding and release, ATP hydrolysis, and clamp release were simulated (Fig. 7C) by a very simple model that is illustrated in Fig. 7A, and this simple model recapitulates many of the features present in the clamp loading data. The overall shapes of the simulated time courses are similar to the observed data as is the timing of the changes in each time course. DNA binding is rapid. There is a short lag in time before MDCC fluorescence increases (ATP hydrolysis) and a longer lag in time before AF488 fluorescence increases (clamp release). The peak in DNA binding occurs at the same point in time as the midpoint of the increase in MDCC fluorescence (ATP hydrolysis) and as AF488 fluorescence (clamp release) begins to increase. The unusual features of ATP hydrolysis and DNA binding/release time courses are also present in the simulated data. These traces do not resemble typical pre-steady-state time courses in which a rapid pre-steady-state phase transitions directly into a steady-state phase. For example, if the ATPase reaction followed typical “burst” kinetics, it would consist of a rapid exponential increase in MDCC fluorescence that transitioned directly into a linear increase in fluorescence. Instead, MDCC fluorescence levels out prior to a faster linear increase. Similarly, in the time course for DNA binding/release, the anisotropy of RhX dips below the steady-state level and this dip occurs in the same time frame as the transition in MDCC fluorescence from the lag to linear phases. These unusual features likely result from initiating clamp loading reactions with a pre-formed clamp loader·ATP·clamp complex which bypasses several kinetic steps in the first turnover that are required to reform this complex in subsequent turnovers. These steps include binding three molecules of ATP, a relatively slow ATP-induced conformational change in the clamp loader (23), and clamp binding/opening. In addition, the clamp loader must release three molecules of ADP prior to binding ATP and this may also include conformational change steps. This series of kinetic steps, that are invisible in terms of the signals being measured, RhX anisotropy and MDCC intensity, generate the lag before the steady-state reaction is established.
Several interesting features of the clamp loading reaction were uncovered by directly measuring kinetics of clamp release and by comparing these data to the timing of DNA binding/release and ATP hydrolysis. Visual inspection of DNA and clamp release time courses suggested that DNA was released prior to the clamp and this interpretation is supported by our simple modeling studies. Kinetics of ATP hydrolysis, particularly those at substoichiometric DNA concentrations where DNA must be released to trigger additional rounds of ATP hydrolysis, indicated that DNA release must be relatively rapid (Fig. 6B). This is rapid rate of DNA release is consistent with anisotropy data measuring DNA binding/release (Fig. 6D) as well as previous studies (34). In contrast, modeling clamp release data suggested that β release was about 10 times slower than DNA release (Fig. 6C). This is the first evidence that DNA and the clamp may be released sequentially rather than simultaneously. Further, we hypothesize that clamp closing may be coupled to DNA release. Modeling studies suggested that in the FRET-based clamp release assay, rapid clamp closing coupled to DNA release gave a partial increase in fluorescence and slower clamp release accounted for the rest of the increase to give rise to the biphasic increase in fluorescence that is observed. In vitro, the short p/t-DNA (30/60mer) could slip out of the closed clamp loader·clamp complex to account for the relatively rapid rate of DNA release. In vivo, clamp closing would limit unproductive clamp loading events by preventing the longer chromosomal DNA from dissociating from the clamp loader·clamp complex. In addition, DNA is not likely to rapidly slide through the closed clamp loader·clamp complex in vivo due to the interaction between the χ subunit of the clamp loader and single-stranded DNA binding protein (41, 42). The observation that clamp release is relatively slow is also interesting when considering the complete clamp loading reaction cycle. Steady-state rates of ATP hydrolysis of 2 - 3 s-1 have been reported for the clamp loading reaction cycle on synthetic p/t-DNA substrates such as ours (22, 34). Together with the slow rate of clamp release of about 3 s-1 calculated from our FRET assays, this means that clamp release is likely to be the rate-limiting step in the clamp loading reaction cycle.
Conclusions
The rate of clamp release onto DNA was measured directly for the first time using a FRET-based assay in which the γ complex clamp loader was labeled with a fluorescent donor and the β-clamp was labeled with a nonfluorescent quencher. Together with real-time DNA binding and ATP hydrolysis assays, these experiments have established a temporal order of events that occur when a β·γ complex binds primed template DNA. DNA binding triggers the clamp loader to hydrolyze ATP, which in turn facilitates the release of DNA and then the clamp. The combination of these real time assays will be invaluable for defining additional steps in the clamp loading reaction cycle.
Supplementary Material
Footnotes
- AF488
- Alexa Fluor 488
- γc
- γ complex
- γc-AF488
- γ complex covalently labeled on the δ’ subunit with AF488
- β-QSY9
- β covalently labeled with QSY9
- p/t-DNA
- primed template DNA
- QSY9
- a nonfluorescent quencher available from Invitrogen
In the absence of β, the γ complex hydrolyzes ATP in two phases following DNA binding. Two molecules are hydrolyzed relatively rapidly before DNA release, and the third is hydrolyzed slowly after DNA release (23).
The ATP hydrolysis time courses for 100 - 400 nM DNA are also shown in Fig. 5. The reactions containing 25 and 50 nM DNA were part of the same experiment but only shown in Fig. 6B.
This work was supported by NIH grant GM055596 to L.B.B. and NIH grant GM038839 to M.OD.
REFERENCES
- 1.Bloom LB. Loading clamps for DNA replication and repair. DNA Repair (Amst) 2009;8:570–578. doi: 10.1016/j.dnarep.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell M, Kuriyan J. Clamp loaders and replication initiation. Curr. Opin. Struct. Biol. 2006;16:35–41. doi: 10.1016/j.sbi.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Trakselis MA, Alley SC, Abel-Santos E, Benkovic SJ. Creating a dynamic picture of the sliding clamp during T4 DNA polymerase holoenzyme assembly by using fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. (U.S.A) 2001;98:8368–8375. doi: 10.1073/pnas.111006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeruzalmi D, O’Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader gamma (γ) complex of E. coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 6.Maki S, Kornberg K. DNA polymerase III holoenzyme of Escherichia coli. II. A novel complex including the γ subunit essential for processive synthesis. J. Biol. Chem. 1988;263:6555–6560. [PubMed] [Google Scholar]
- 7.Pritchard AE, Dallman HG, Glover BP, McHenry CS. A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of δδ’ with DnaX4 forms DnaX3δδ’. EMBO J. 2000;19:6536–6545. doi: 10.1093/emboj/19.23.6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blinkowa AL, Walker JR. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res. 1990;18:1725–1729. doi: 10.1093/nar/18.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flower AM, McHenry CS. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc. Natl. Acad. Sci. (USA) 1990;87:3713–3717. doi: 10.1073/pnas.87.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsuchihashi Z, Kornberg A. Translational frameshifting generates the γ subunit of DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. (USA) 1990;87:2516–2520. doi: 10.1073/pnas.87.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onrust R, Finkelstein J, Naktinis V, Turner J, Fang L, O’Donnell M. Assembly of a chromosomal replication machine: Two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. I. Organization of the clamp loader. J. Biol. Chem. 1995;270:13348–13357. doi: 10.1074/jbc.270.22.13348. [DOI] [PubMed] [Google Scholar]
- 12.McHenry CS. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol Microbiol. 2003;49:1157–1165. doi: 10.1046/j.1365-2958.2003.03645.x. [DOI] [PubMed] [Google Scholar]
- 13.Pomerantz RT, O’Donnell M. Replisome mechanics: insights into a twin DNA polymerase machine. Trends Microbiol. 2007;15:156–164. doi: 10.1016/j.tim.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Onrust R, Stukenberg PT, O’Donnell M. Analysis of the ATPase subassembly which initiates processive DNA synthesis by DNA polymerase III holoenzyme. J. Biol. Chem. 1991;266:21681–21686. [PubMed] [Google Scholar]
- 15.Tsuchihashi Z, Kornberg A. ATP interactions of the τ and γ subunits of DNA polymerase III holoenzyme of Escherichia coli. J. Biol. Chem. 1989;264:17790–17795. [PubMed] [Google Scholar]
- 16.Hingorani MM, O’Donnell M. ATP binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 1998;273:24550–24563. doi: 10.1074/jbc.273.38.24550. [DOI] [PubMed] [Google Scholar]
- 17.Naktinis V, Onrust R, Fang F, O’Donnell M. Assembly of a chromosomal replication machine: Two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem. 1995;270:13358–13365. [PubMed] [Google Scholar]
- 18.Turner J, Hingorani MM, Kelman Z, O’Donnell M. The internal workings of a DNA polymerase clamp-loading machine. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertram JG, Bloom LB, Turner J, O’Donnell M, Beechem JM, Goodman MF. Pre-steady state analysis of the assembly of wild type and mutant circular clamps of Escherichia coli DNA polymerase III onto DNA. J. Biol. Chem. 1998;273:24564–24574. doi: 10.1074/jbc.273.38.24564. [DOI] [PubMed] [Google Scholar]
- 20.Ason B, Handayani R, Williams CR, Bertram JG, Hingorani MM, O’Donnell M, Goodman MF, Bloom LB. Mechanism of loading the Escherichia coli DNA polymerase III beta sliding clamp on DNA. Bona fide primer/templates preferentially trigger the gamma complex to hydrolyze ATP and load the clamp. J. Biol. Chem. 2003;278:10033–10040. doi: 10.1074/jbc.M211741200. [DOI] [PubMed] [Google Scholar]
- 21.Simonetta KR, Kazmirski SL, Goedken ER, Cantor AJ, Kelch BA, McNally R, Seyedin SN, Makino DL, O’Donnell M, Kuriyan J. The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell. 2009;137:659–671. doi: 10.1016/j.cell.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hingorani MM, Bloom LB, Goodman MF, O’Donnell M. Division of labor-sequential ATP hydrolysis drives assembly of a DNA polymerase sliding clamp around DNA. EMBO J. 1999;18:5131–5144. doi: 10.1093/emboj/18.18.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams CR, Snyder AK, Kuzmic P, O’Donnell M, Bloom LB. Mechanism of Loading the Escherichia coli DNA Polymerase III Sliding Clamp: I. Two distinct activities for individual ATP sites in the γ complex. J. Biol. Chem. 2004;279:4376–4385. doi: 10.1074/jbc.M310429200. [DOI] [PubMed] [Google Scholar]
- 24.Anderson SG, Williams CR, O’Donnell M, Bloom LB. A function for the psi subunit in loading the Escherichia coli DNA polymerase sliding clamp. J. Biol. Chem. 2007;282:7035–7045. doi: 10.1074/jbc.M610136200. [DOI] [PubMed] [Google Scholar]
- 25.Bloom LB, Turner J, Kelman Z, Beechem JM, O’Donnell M, Goodman MF. Dynamics of loading the β sliding clamp of DNA polymerase III onto DNA. J. Biol. Chem. 1996;271:30699–30708. doi: 10.1074/jbc.271.48.30699. [DOI] [PubMed] [Google Scholar]
- 26.Olson MW, Dallmann HG, McHenry CS. DnaX of Escherichia coli DNA polymerase III holoenzyme. The χψ complex functions by increasing the affinity of τ and γ for δ·δ’ to a physiologically relevant range. J. Biol. Chem. 1995;270:29570–29577. [PubMed] [Google Scholar]
- 27.Xiao H, Crombie R, Dong Z, Onrust R, O’Donnell M. DNA polymerase III accessory proteins. III. holC and holD encoding χ and ψ. J. Biol. Chem. 1993;268:11773–11778. [PubMed] [Google Scholar]
- 28.Johanson KO, Haynes TE, McHenry CS. Chemical characterization and purification of the beta subunit of the DNA polymerase III holoenzyme from an overproducing strain. J. Biol. Chem. 1986;261:11460–11465. [PubMed] [Google Scholar]
- 29.Maki S, Kornberg A. DNA polymerase III holoenzyme of Escherichia coli. I. Purification and distinctive functions of subunits tau and gamma, the dnaZX gene products. J. Biol. Chem. 1988;263:6547–6554. [PubMed] [Google Scholar]
- 30.Dong Z, Onrust R, Skangalis M, O’Donnell M. DNA Polymerase III Accessory Proteins. I. holA and holB encoding delta and delta’. J. Biol. Chem. 1993;268:11758–11765. [PubMed] [Google Scholar]
- 31.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 32.Snyder AK, Williams CR, Johnson A, O’Donnell M, Bloom LB. Mechanism of loading the Escherichia coli DNA polymerase III sliding clamp: II. Uncoupling the β and DNA binding activites of the γ complex. J. Biol. Chem. 2004;279:4386–4393. doi: 10.1074/jbc.M310430200. [DOI] [PubMed] [Google Scholar]
- 33.Ason B, Bertram JG, Hingorani MM, Beechem JM, O’Donnell M, Goodman MF, Bloom LB. A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends: DNA triggers a change in binding specificity of the γ complex clamp loader. J. Biol. Chem. 2000;275:3006–3015. doi: 10.1074/jbc.275.4.3006. [DOI] [PubMed] [Google Scholar]
- 34.Bertram JG, Bloom LB, Hingorani MM, Beechem JM, O’Donnell M, Goodman MF. Molecular mechanism and energetics of clamp assembly in Escherichia coli. The role of ATP hydrolysis when γ complex loads β on DNA. J. Biol. Chem. 2000;275:28413–28420. doi: 10.1074/jbc.M910441199. [DOI] [PubMed] [Google Scholar]
- 35.Brune M, Hunter JL, Corrie JET, Webb MR. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994;33:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 36.Kuzmic P. Program DynaFit for the analysis of enzyme kinetic data: application to HIV protease. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 37.Glover BP, McHenry CS. The DNA polymerase III holoenzyme: an asymmetric dimeric replicative complex with leading and lagging strand polymerases. Cell. 2001;105:925–934. doi: 10.1016/s0092-8674(01)00400-7. [DOI] [PubMed] [Google Scholar]
- 38.Johnson A, O’Donnell M. Ordered ATP hydrolysis in the gamma complex clamp loader AAA+ machine. J. Biol. Chem. 2003;278:14406–14413. doi: 10.1074/jbc.M212708200. [DOI] [PubMed] [Google Scholar]
- 39.Smiley RD, Zhuang Z, Benkovic SJ, Hammes GG. Single-molecule investigation of the T4 bacteriophage DNA polymerase holoenzyme: multiple pathways of holoenzyme formation. Biochemistry. 2006;45:7990–7997. doi: 10.1021/bi0603322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhuang Z, Berdis AJ, Benkovic SJ. An alternative clamp loading pathway via the T4 clamp loader gp44/62-DNA complex. Biochemistry. 2006;45:7976–7989. doi: 10.1021/bi0601205. [DOI] [PubMed] [Google Scholar]
- 41.Glover BP, McHenry CS. The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem. 1998;273:23476–23484. doi: 10.1074/jbc.273.36.23476. [DOI] [PubMed] [Google Scholar]
- 42.Kelman Z, Yuzhakov A, Andjelkovic J, O’Donnell M. Devoted to the lagging strand-the subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J. 1998;17:2436–2449. doi: 10.1093/emboj/17.8.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.