

Abstract
Experimental animals’ seizures are often defined arbitrarily based on duration, which may lead to misjudgement of the syndrome and failure to develop a cure. We employed a functional definition of seizures based on the clinical practice of observing epileptiform electrocorticography and simultaneous ictal behaviour, and examined post-traumatic epilepsy induced in rats by rostral parasagittal fluid percussion injury and epilepsy patients evaluated with invasive monitoring. We showed previously that rostral parasagittal fluid percussion injury induces different types of chronic recurrent spontaneous partial seizures that worsen in frequency and duration over the months post injury. However, a remarkable feature of rostral parasagittal fluid percussion injury is the occurrence, in the early months post injury, of brief (<2 s) focal, recurrent and spontaneous epileptiform electrocorticography events (EEEs) that are never observed in sham-injured animals and have electrographic appearance similar to the onset of obvious chronic recurrent spontaneous partial seizures. Simultaneous epidural-electrocorticography and scalp-electroencephalography recordings in the rat demonstrated that these short EEEs are undetectable by scalp electrocorticography. Behavioural analysis performed blinded to the electrocorticography revealed that (i) brief EEEs lasting 0.8–2 s occur simultaneously with behavioural arrest; and (ii) while behavioural arrest is part of the rat's behavioural repertoire, the probability of behavioural arrest is greatly elevated during EEEs. Moreover, spectral analysis showed that EEEs lasting 0.8–2 s occurring during periods of active behaviour with dominant theta activity are immediately followed by loss of such theta activity. We thus conclude that EEEs lasting 0.8–2 s are ictal in the rat. We demonstrate that the assessment of the time course of fluid percussion injury-induced epileptogenesis is dramatically biased by the definition of seizure employed, with common duration-based arbitrary definitions resulting in artificially prolonged latencies for epileptogenesis. Finally, we present four human examples of electrocorticography capturing short (<2 s), stereotyped, neocortically generated EEEs that occurred in the same ictal sites as obvious complex partial seizures, were electrographically similar to rat EEEs and were not noted during scalp electroencephalography. When occurring in the motor cortex, these short EEEs were accompanied by ictal behaviour detectable with simultaneous surface electromyography. These data demonstrate that short (<2 s) focal recurrent spontaneous EEEs are seizures in both rats and humans, that they are undetectable by scalp electroencephalography, and that they are typically associated with subtle and easily missed behavioural correlates. These findings define the earliest identifiable markers of progressive post-traumatic epilepsy in the rat, with implications for mechanistic and prophylactic studies, and should prompt a re-evaluation of the concept of post-traumatic silent period in both animals and humans.
Keywords: traumatic brain injury; epileptogenesis; biomarkers; drug screening, prophylaxis
Introduction
A clear understanding of the mechanisms of acquired epileptogenesis after brain insult and the development of prophylactic treatments requires accurate and sensitive identification of chronic spontaneous recurrent seizures (i.e. epilepsy) in experimental animals. However, both behavioural and electrophysiological correlates of experimental partial epilepsy are poorly defined, making detection and evaluation of chronic recurrent spontaneous partial seizures (CRSPSs) in experimental animals challenging for several reasons. First, even in humans, detection of CRSPSs is often difficult. The observation of brief stereotyped recurrent abnormal behaviour in patients at risk of developing epilepsy is considered a tell-tale sign of the condition (Beghi et al., 2005; Fisher et al., 2005). However, complex partial seizures—the seizure type for which novel treatments are most needed (Juul-Jensen, 1986; Mattson et al., 1985; 1996; Semah et al., 1998)—often present subtly with staring, memory lapses, muscle twitching or impairment in normal function that may go unrecognized by both patients and physicians (Blum et al., 1996; Tatum et al., 2001), and can be virtually impossible to detect in animals. Indeed, before diagnosis is confirmed by scalp encephalography (EEG), human complex partial seizures are often first identified through either the patients’ altered state of consciousness or their self-reported internal subjective experience, information not obtainable from animals. Secondly, studying genuine epilepsy mandates observation of spontaneous seizures, a daunting task in a freely behaving animal in which the unpredictable onset of rare subtle ictal behaviours must be distinguished from the normal behavioural repertoire of the animals. Finally, we have a very poor understanding of the features and variability of behavioural phenotypes of rodent partial seizures.
Because of these difficulties with behavioural analysis, electrophysiological markers of epilepsy are a promising alternative, but are also currently poorly defined. It has been common practice to define spontaneous epileptiform electrocorticography (ECoG)/EEG events that last longer than an arbitrarily set duration as ‘seizures’. Many laboratories choose a 5-s cut-off, while others employ longer durations as deemed more practical for the specific problem at hand. These arbitrary definitions, while practical, inevitably affect our understanding of an epilepsy syndrome and its mechanisms, which hampers the development of genuine prophylactic treatments. While a process that prolongs seizure duration beyond a set threshold will appear epileptogenic, one that shortens seizures below that threshold will appear anti-epileptogenic. This would lead to wrong estimates of the time course of epileptogenesis, of the overall incidence of epilepsy and, ultimately, to missed opportunity for treatment. Drugs that affect mechanisms of seizure spread, maintenance and termination probably have therapeutic value, as shortening seizures may reduce their cognitive, behavioural and pathological consequences for patients. However, the rational identification of a true cure for epilepsy will require a clear understanding of each type of mechanism and the development of strategies to target them jointly or independently.
Thus, the next step in preclinical mechanistic and prophylactic studies requires better definition of genuine seizures and epilepsy, and the identification of their electrophysiological markers arising after a clinically relevant experimental insult to the brain. These studies have thus far been hampered by the lack of animal models of acquired epileptogenesis in which (i) the initiating insult is temporally limited and well distinct from ensuing epileptogenic processes, thus facilitating following the onset and evolution of CRSPSs and their behavioural and electrical correlates; and (ii) the aetiology closely replicates the human condition, leading to confidence that the ensuing CRSPSs are mechanistically similar to the corresponding ones in humans. The discovery of rostral parasagittal fluid percussion injury (rpFPI) induced post-traumatic epilepsy (PTE) in the rat, in which CRSPSs develop peri-lesionally following an injury mechanically identical to human contusive closed head injury (D'Ambrosio et al., 2004, 2005; D'Ambrosio and Perucca, 2004), now provides such an opportunity.
In this study, we hypothesized that (i) short (<2 s) perilesional spontaneous recurrent EEEs include ictal events that should be considered as part of the PTE syndrome; and (ii) definitions of seizures that are based on arbitrary durations, rather than on functional features, bias the assessment of acquired epileptogenesis. The data demonstrate that the earliest ictal, clinically relevant manifestation of progressive PTE in head injured rats consists of short (>0.8 s) CRSPSs, and we hypothesize that human PTE may first manifest with similar short partial seizures and may therefore be under-recognized. A preliminary report of this study was previously reported (D'Ambrosio et al., 2007).
Materials and Methods
This study relied upon 31 outbred male Sprague-Dawley rats (Charles Rivers, Hollister, CA) evaluated with video-ECoG and scalp-EEG, and four epilepsy patients evaluated with video-ECoG and electromyography (EMG). All animal experimental procedures were approved by the University of Washington Institutional Animal Care and Use Committee. The human examples were provided with the patient's consent and in accordance with the guidelines for reporting clinical data by the Institutional Review Boards of University of Washington and University of Texas Southwestern.
Human ECoG/skin-electrode recordings
Patients previously diagnosed with medically refractory neocortical epilepsy underwent craniotomy for implantation of standard subdural electrodes (Ad-Tech, Racine, WI) using both 8 × 8 grid arrays and 6- to 8-electrode strips (contact spacing 1 cm) to localize the seizure onset zone. Preoperative evaluation included MRI and video-EEG telemetry. Anti-epileptic medications were suspended during the assessment period. In three patients cutaneous EMG electrodes were placed, as part of the clinical evaluation, either on the right forearm (Patient 1), laterally to the right eye (Patient 3), or on the left pectoral muscle (Patient 4). Data from Patients 1, 3 and 4 were acquired using the XLTEK telemetry system (Oakville, Ontario, Canada) either at 2 KHz with low-pass filter at 500 Hz (Patients 1 and 4), or at 512 Hz with low-pass filter at 200 Hz (Patient 3). High-pass filtering was 0.1 Hz in all three cases. Data from Patient 2 were acquired with the Stellate telemetry system (Montreal, Quebec, Canada) at 1 KHz, with no additional filters applied.
Rostral parasagittal fluid percussion injury in the rat
Rostral parasagittal fluid percussion injury (rpFPI) was performed as previously described (D’Ambrosio et al., 2005). Rats (33–35 days post-natal) were anaesthetized with halothane, intubated and mounted on a stereotaxic frame. A burr hole of 3 mm in diameter was drilled 2 mm posterior to bregma and 3 mm lateral to midline, on the right convexity. Maximal care was taken to avoid any damage to the underlying dura during drilling to ensure reproducible fluid percussion injury (FPI). A 8–10 ms pressure pulse of 3.25–3.5 atm was delivered through the FPI device (Scientific Instruments, University of Washington). After a 10 s pause in breathing upon injury, the animal was re-connected to the ventilator. Sham-injured animals underwent the same procedure but the pressure pulse was generated with the stopcock of the FPI device closed. The mortality rate from acute post-traumatic complications was ∼11%.
Video-ECoG/EEG in the rat
One week after rpFPI, guiding craniotomies ∼ф = 0.75 mm were drilled and epidural electrodes (stainless-steel screws of ф = 1 mm) were implanted in them. Two ECoG montages were employed in different groups of animals. Montage A (Fig. 2G) consisted of five epidural electrodes: one was placed midline in the frontal bone and used as reference, while two electrodes per parietal bone were placed at coordinates bregma 0 and –6.5 mm, 4 mm from the midline. Montage B (Fig. 2A) consisted of a denser array of 12 epidural electrodes, in which four electrodes were placed peri-lesionally (electrodes 3, 4, 6, 7) to provide improved coverage of the perilesional epileptiform ECoG activity. All electrodes were connected through insulated wire to a gold-plated pin in a plastic pedestal, and the entire assembly was cemented onto the skull with dental acrylic. Headsets were occasionally lost; when possible they were re-implanted, otherwise animals were included in the study only until the last day of recording. The electrodes’ headset, video-ECoG/EEG acquisition, amplification and storage were as previously described (D'Ambrosio et al., 2005). Briefly, electrical brain activity was amplified (×5000–10 000) and filtered (0.3 Hz high-pass, 100 Hz low-pass) using a Neurodata 12 or a M15 amplifier (Grass Instruments, Quincy, Mass.), acquired at 512 Hz per channel, stored and analysed on Pentium-based computers equipped with SciWorks with Experimenter V3 software (Datawave Technologies Inc., Longmont, CO) and DT3010 acquisition boards (DataTranslation Inc., Marlboro, MA). Montage A was used for 8 h video-ECoG recordings performed weekly from 2 to 28 weeks post-injury. Montage B was used for 24 h video-ECoG recordings performed weekly from 2 to 10 weeks post-injury. For paired scalp-EEG/epidural-ECoG, montage A was complemented with two removable cup scalp electrodes (ϕ = 6 mm, Grass Technologies) glued to the rat's skin with collodion before each recording and removed afterwards. Epidural electrodes 1–4 were referenced to epidural electrode 5, while the parietal scalp electrode was referenced to the occipital one. All data were analysed off-line.
Figure 2.

Electroclinical syndrome of PTE induced by rpFPI in the rat. The spectrum of focal epileptiform ECoG activities observed by 12-electrode video-ECoG recordings (A–F), and the three types of CRSPSs observed by 5-electrode video-ECoG recordings (I–L), are shown. (A) Schematic of the location of the 12 epidural electrodes (filled circles) and of the injury site (hollow circle) in respect to the rat skull for events in A–F. Representative examples of a perilesional double spike (A), a spike complex followed by higher frequency oscillation (B), and a triple spike (C), all associated with no overt behavioural change of the animal. Filled arrows indicate the first spike of the event. Representative examples of longer multi-spike EEEs associated with movement arrest of the animal, ranging from short events resembling grade 1 seizures and evident in electrodes 3, 4 and 7 (D), to a grade 1 seizure evident in all perilesional electrodes 3, 4, 6 and 7 (E) to a grade 2 seizure with perilesional onset and subsequent spread to all the electrodes (F). Scales in A apply to A–F. (G) Schematic of the locations of the five epidural ECoG electrodes (filled circles) and of the injury site (hollow circle) in respect to the rat skull for data shown in H–L. (H) Summary results of the ECoG analysis of sham-injured and FPI animals performed blinded to the treatment, behaviour and time after surgery. All EEEs observed segregated to FPI animals. The numbers above the columns represent counts. (I) ECoG recording acquired 2 weeks post-injury shows a typical electrical grade 1 seizure associated with behavioural arrest. (J) ECoG recording acquired at 28 weeks post-injury shows a typical grade 2 seizure associated with behavioural arrest. This was first detected at the perilesional frontal-parietal cortex and then propagated to multiple channels. (K) Representative ECoG recording of a grade 3 seizure recorded 27 weeks post-injury, with onset detected simultaneously on multiple channels. This event was associated with behavioural arrest. (L) The epileptic syndrome as defined by the relationship between electrical seizure types and their behavioural correlate as ranked by the behavioural scale. Note that all seizures that are longer than 2 s are also coincident to a detectable ictal behavioural change (behavioural score >0). Throughout the panels, numbers next to each ECoG trace indicate the electrodes by which the trace is recorded, and its reference.
Quality control of surgical procedures
As in our previous work (D’Ambrosio et al., 2004, 2005), precautions were taken to ensure that the ECoG events we examined were induced by rpFPI and not by the neuropathology associated with substandard drilling or epidural electrode implantation. Compression heating associated with drilling can cause cortical damage. Thus, the skull and drill bit were cooled during drilling with room-temperature sterile saline, and we took care never to deform the skull or the dura with the drill bit. Suboptimal electrode implantations resulted in exclusion from the study. Glial fibrillary acidic protein (GFAP) immunostaining was also routinely performed, as previously described, and examined at bregma 0 and –6.5 mm in most rpFPI and sham-injured animals after the completion of recordings. Cases of electrode implantation damage, as indicated by ∼0.5–1.5 mm diameter neocortical damage and focus of GFAP positive astroglial reactivity right under the electrodes (Fig. 1), were excluded from the study (∼5% of implanted animals).
Figure 1.

Quality control of chronic implantation of epidural electrodes. Pathological examination of the ECoG electrode implantation sites is routinely performed in sham-injured and rpFPI animals in order to study genuine epilepsy induced by closed head injury and insure against epileptiform ECoG activity due to substandard implantation of epidural electrodes. Coronal sections from two rpFPI animals obtained at coordinates bregma 0 mm demonstrate acceptable (A) versus unacceptable (B) chronic ECoG electrode implantation. (A) A typical animal that we include in our studies must not present any focal cortical abnormalities or focal GFAP positive immunostaining right underneath the position of implanted epidural electrodes. Pathology was studied 1 month after electrode implantation. GFAP positive immunostaining underlying electrode 1 (A1) and electrode 4 (A2) presents no focal damage. (B) A typical animal that we discard from our studies as technical failure presented with a focal deformity of the neocortex of ∼0.5–1.5 mm in diameter, associated with increased focal glial reactivity. GFAP positive immunostaining underlying electrode 1 is severe (B1), while the one underlying electrode 4 is milder but noticeable (B2). Pathology was studied 2 weeks after electrode implantation. These kinds of electrode-induced pathology are sometimes associated with focal epileptiform ECoG. Note both animals present the characteristic rpFPI-induced band of enhanced GFAP positive immunostaining in layer 6—white matter in the hemisphere ipsilateral to the injury (black arrows). Dotted boxes in A and B represents areas of the coronal section magnified in the corresponding underlying panels. White arrows indicate electrode damage. Scale bars represent 1 mm.
Identification of seizures, epileptiform ECoG events, measurements and definitions
ECoG events of interest accepted for analysis had amplitudes exceeding twice the previous 2 s baseline and an epileptiform appearance defined by the presence of one or more spikes lasting 150–250 ms each. The duration of EEEs was determined as follows: (i) to be considered part of an EEE, spikes had to be visible on the perilesional electrode in both referential and average montages; and (ii) the first and last spike of the EEE had to be members of a sequence of spikes with no more than a 200 ms interruption. In this study, we use the terms ‘ictal event’ and ‘seizure’ interchangeably. We consider epilepsy as the disorder of chronic recurrent spontaneous seizures (Beghi et al., 2005; Fisher et al., 2005). Stereotyped behavioural changes coinciding with epileptiform ECoG activity are taken as evidence that EEEs are clinical seizures, while not all seizures may present with overt behavioural change (subclinical seizures). Behavioural changes in the absence of simultaneous epileptiform ECoG are not considered seizures. As all rpFPI animals were at risk of developing PTE, we diagnosed epilepsy upon their first proven ictal event (Beghi et al., 2005; Fisher et al., 2005).
Spectral analysis for the seizure test
We compared the power spectra of the ECoG immediately following epileptiform versus randomly chosen events to determine whether the occurrence of epileptiform ECoG activity was temporally associated with changes in background ECoG activity and cognitive/behavioural state of the rats. Random ECoG event segments were (i) picked by a software randomizer (Research Randomizer; http://www.randomizer.org); (ii) selected from the same data files containing epileptiform events; and (iii) set to last the average duration of the EEEs. To ensure the cognitive/behavioural state of the animals at the time of the ECoG event was well defined, all evaluated events, epileptiform or random, were accepted for analysis only if they occurred after at least 1 min of active exploratory or grooming behaviour of the animal with dominant theta activity. For each event, the power spectrum of the ECoG was obtained by fast Fourier transformation (MATLAB R2007b, The Mathworks Inc., Natick, MA) during the 2 s immediately following it. Spectral power was computed at 0.5 Hz intervals in the range of 0.5–12.5 Hz. We computed the summed power Ps in the theta (6–8 Hz) and the delta (1–4 Hz) bands, and their ratio RΘ/δ, as follows:
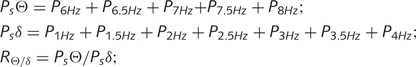 |
These data were exported to SPSS (Statistical package for the Social Sciences) 14.0 for statistical analysis.
Behavioural analysis
The association of ECoG events with behavioural changes was evaluated off-line as previously described for rpFPI-induced PTE (D’Ambrosio et al., 2005): 0 = no detected behavioural change (subclinical); 1 = freeze-like pause in behaviour without impaired posture; 2 = freeze-like pause in behaviour without impaired posture and accompanied by facial automatisms (twitching of vibrissae, sniffing, eye blinking or jaw automatisms); 3 = head nodding; 4 = body myoclonus; 5 = behavioural arrest with loss of posture or atonia; 6 = behavioural arrest with loss of posture followed by motor manifestations (facial automatisms or contralateral limb dystonia); 7 = tonic–clonic convulsion; and 8 = partial status epilepticus.
Behavioural changes were assessed by comparing the behavioural pattern of the animal during the time window defined by the onset and termination of the epileptiform ECoG discharge with the preceding behaviour. Cases when animals’ behavioural changes could not be appreciated because of an unfavourable position with respect to the camera, or because they were already motionless at the time of the ECoG event, were considered unrankable and excluded from analysis. As internal controls, random ECoG events were picked by a software randomizer (Research Randomizer) from within the same data files as epileptiform ECoG events. To ensure they represented true negatives, random events within 5 s of any identified epileptiform activity or recording artefact were excluded from analysis. To characterize the behaviour associated with long (≥2 s) epileptiform ECoG events (Figs 3 and 4), we employed random ECoG events lasting 4 s, that matched the average duration of focal EEEs at 2–4 weeks post-injury. To determine whether short (<2 s) EEEs (Figs 5 and 6) were seizures, we employed random ECoG events lasting 2 s and behavioural analysis blinded to the ECoG. A blinder (J.S.F or D.R.V) randomly chose 18 video-ECoG recordings, each lasting 8 h, from 15 different injured rats. Enough random ECoG events were identified so that when added to the short epileptiform ECoG events to evaluate, each video-recording contained 50 events to be ranked by blinded raters. This approach was used to (i) maintain the blind; and (ii) examine more random ECoG events than EEEs available to determine the sensitivity of the test better. Three blind raters (A.H.S., T.S., R.D.) independently assessed the behavioural association of all ECoG events while blinded to the ECoG of the animal. After independent scoring, the raters consulted to agree on the final grading of each event while still being blinded.
Figure 3.

Behavioural changes are temporally specifically associated with rpFPI-induced CRSPSs lasting at least 2 s. Graphs show the proportion of epileptiform and random ECoG events recorded between 2 and 28 weeks post-injury that were associated with a behavioural change. Data were analysed by time post-injury (A–C) and by electrical seizure type (D–F). Numbers over each column represent counts. In each panel, statistical comparison of the population distribution of behavioural changes associated with epileptiform versus random ECoG segments were performed by Fisher's Exact test (P < 0.001 in all cases). Note all rankable epileptiform ECoG events lasting at least 2 s are associated with behavioural change and are therefore ictal.
Figure 4.

Loss of theta rhythm following rpFPI-induced CRSPSs lasting longer than 2 s. Changes in the power spectrum of the rat ECoG immediately after either CRSPSs lasting >2 s or ECoG events randomly chosen within the same files, are examined by fast Fourier transformation when animals are engaged in active behaviour with dominant theta activity. Note the theta activity, with average peak power at 7.5 Hz, after random ECoG events, indicating the animals persisted in the active behaviour, while CRSPS are followed by loss of power in the theta band and an increase in power in the delta band, indicating they are associated with change in behaviour/cognitive state of the animal. Summed power in delta (1–4 Hz) or theta (6–8 Hz) bands in the post-ictal versus post-random ECoG were compared using the Mann–Whitney test.
Figure 5.

Electroclinical evidence that short (<2 s) recurrent spontaneous focal epileptiform ECoG discharges induced by rpFPI are ictal. Two different blinded analyses of behaviour concurrent with focal epileptiform ECoG discharges lasting between 0.8 and 2 s are shown. (A and B) Analysis of behavioural arrests occurring during animal's ongoing behaviour, performed blind to the ECoG; (C and D) Analysis of behavioural changes occurring during either epileptiform or randomly selected ECoG events, performed blind to the ECoG. (A) Schematic of the temporal sequence of behavioural arrests and EEE in 1 min video-ECoG data files examined blind to the ECoG to assemble the cross-correlogram shown in B. Upper arrow: sequence of behavioural arrests (vertical ticks) observed over the time examined. Lower arrow: occurrence of the EEE (vertical tick) at random time within the file. (B) Cross-correlogram of behavioural arrests versus EEEs reported as counts of behavioural arrests versus the time before or after the EEE represented at time 0. The time scale is in bins of 1 second. Note the greatly elevated incidence of behavioural arrest during EEEs (bin 0). (C) Association of epileptiform or randomly chosen ECoG events to a behavioural change. Of the rankable recurrent spontaneous focal epileptiform ECoG events observed, 88.2% were associated with a behavioural change, a freeze-like pause in behaviour in all cases. Random ECoG events were associated with no behavioural change in 86.6% of the cases. Numbers above each column represent counts. The statistical comparison between the behavioural distribution of epileptiform versus random ECoG was computed by Fisher's Exact test (P < 0.001). (D) Population distribution of the focal epileptiform ECoG events plotted as count and percentage versus their duration on the ECoG. Grey columns: events with evident ictal behavioural change. Black columns: events associated with no detectable behavioural change, corresponding to either interictal or subclinical seizures. Hollow circles: percentage of short EEEs with a behavioural change. Notably, there is no relationship between the fraction of EEEs without behavioural change and their duration on the ECoG. (E) Example of a focal epileptiform ECoG event recorded at week 2 post-injury, lasting about 1.3 s and coincident with evident ictal behaviour (movement arrest). (F) Example of a focal epileptiform ECoG event recorded at week 2 post-injury and lasting about 1 s with no evidence of ictal behaviour. The numbers next to each ECoG trace in E–F indicate the electrodes by which the trace is recorded, and its reference.
Figure 6.

Spectral evidence that short (<2 s) recurrent spontaneous focal epileptiform ECoG discharges are ictal in the rat. Changes in the power spectrum of the rat ECoG immediately after either recurrent spontaneous focal EEEs lasting between 0.8 and 2 s, or random ECoG events lasting 2 s chosen from the same files. All events occurred in animals displaying dominant theta activity and engaged in active behaviour. Note the theta dominance after random ECoG events, indicating the animals persisted in its active behaviour and cognitive state, while short EEEs are immediately followed by loss of power in the theta band and increase in power in the delta band, indicating a change in the behavioural/cognitive state of the animal, and demonstrating that these EEEs are ictal events. Summed power in delta (1–4 Hz) or theta (6–8 Hz) bands in the post-ictal versus post-random ECoG were compared using the Mann–Whitney test.
Results
A total of 1196 h of video-ECoG recordings, obtained 2–28 weeks post-rpFPI, were examined in 31 animals operated on in our laboratory from 2003 to 2008. An initial analysis of rpFPI-induced PTE in 23 of these animals has previously been published (D’Ambrosio et al., 2005). We now further examine the properties of rpFPI-induced chronic recurrent spontaneous partial seizures and present novel 12-channel grid recordings in the rat, simultaneous epidural ECoG/scalp EEG recordings in the rat, and simultaneous subdural ECoG/EMG in the human.
Varying epileptiform ECoG activity induced by rpFPI
A five-electrode epidural ECoG montage (Fig. 2G) previously allowed us to observe the temporal evolution after injury of three different types of ictal events (D’Ambrosio et al., 2004, 2005). Electrical grade 1 and grade 2 seizures were first detected at the perilesional electrode (Fig. 2I and J), and were hypothesized to originate from the perilesional neocortex, while grade 3 seizures appeared bilateral at their cortical onset (Fig. 2K) and were hypothesized to originate from a subcortical focus. At weeks 2–4 post-injury these EEEs lasted an average of ∼5 s, presented with behavioural arrest and thus represented CRSPSs (D'Ambrosio et al., 2004, 2005). Those previous analyses were not performed blind to the injury status and were based on our clinical and experimental experience with epilepsy. We now present a blind analysis of the rat ECoG (Fig. 2H). Eighteen different 8 h long ECoG files were randomly selected from five different FPI rats and six different sham-injured rats, all recorded 2–5 weeks after surgery. ECoG files were scored manually while blind to the treatment (FPI versus sham), the behaviour of the animal and the time after surgery at which the files were collected. All of the 350 EEEs detected segregated to FPI animals. No EEEs were found in sham-injured animals, thus demonstrating that EEEs are a pathological feature of FPI. These 350 EEEs ranged in duration from 0.4 s (perilesional double spike) to 40.5 s (grade 2 seizure) and demonstrated a remarkable feature of rpFPI-induced epilepsy: the occurrence in the early weeks post-injury of epileptiform activity covering a wide range of durations.
To describe the spatial and temporal evolution of these EEEs better, we obtained 12-channel epidural grid-ECoG recordings (Fig. 2A–F) between 2 and 10 weeks post-injury in 3 rpFPI animals for a total of 206 h of recording. These provided a higher spatial resolution of EEEs than previously available. We observed perilesional single and double spikes (Fig. 2A), each spike lasting ∼150–250 ms, that were never associated with overt behavioural change. Two weeks post-injury, these events occurred with an average frequency of six events per hour. We also observed transitional EEEs consisting of perilesional spike complexes followed by higher frequency oscillations (Fig. 2B), that lasted 400–1000 ms, and triple spikes lasting 500–600 ms (Fig. 2C) that, combined, had a frequency of 0.8 events per hour 2 weeks post-injury. We further observed perilesional quadruple spikes, longer multi-spike EEEs associated with movement arrest (Fig. 2D and E) that were electrographically similar to grade 1 seizures, and transitioned to obvious grade 2 seizures, that spread contralaterally, and were also associated with behavioural arrest (Fig. 2F). While an exhaustive characterization of the properties of these transitional EEEs was beyond our scope, we wanted to determine whether short (<2 s) EEEs could include ictal events. To this end, we first identified functional properties of longer CRSPSs and then determined whether shorter EEEs shared these properties.
Analysis of the behavioural correlate of EEEs longer than 2 s in the rat
In order to determine whether all EEEs lasting ≥2 s are seizures, or whether non-ictal events can be distinguished among them, we defined the epileptic syndrome induced by rpFPI based on the five-electrode ECoG montage (n = 16 animals). This was described by a scatter plot relating each electrical type of EEEs lasting ≥2 s with its associated behavioural change (Fig. 2L). We found that all EEEs lasting ≥2 s were associated with a detectable change in behaviour which typically consisted of a movement arrest with or without facial automatisms, or a stereotyped loss of posture in which the animal crouched down and was motionless at the bottom of the cage, with or without motor manifestations such as facial automatisms or contralateral limb dystonia. We also observed rarer cases of myoclonus associated with all types of electrical seizures and of partial status epilepticus associated with electrical grade 3 type seizures. No tonic–clonic convulsions were observed. Grades 1 and 2 EEEs lasting ≥2 s were mostly associated with a simple behavioural arrest that became more easily detectable the longer the EEE lasted, while grade 3 EEEs most commonly were associated with crouching.
We then examined whether EEEs lasting ≥2 s were specifically temporally associated with stereotyped changes in behaviour, which is one of the criteria used to diagnose clinical seizures in humans, or whether the behavioural changes observed during EEEs were due to random coincidence with normal behavioural repertoire of the animal (Fig. 3). As a control, we used random ECoG events selected within the same data files as the EEEs. These random ECoG events were matched in number to each type of EEE in each file, and were evaluated in the exact same manner. Data were compiled by ECoG event type (grades 1, 2, 3 and random) and by time post-injury (Weeks 2–3, 14–15 and 27–28). We found that all rankable EEEs lasting ≥2 s were associated with a detectable behavioural change, regardless of their type and the time post-injury. All 139 EEEs that were rankable at 2–3 weeks post-injury (Fig. 3A) were associated with a behavioural change. Conversely, 96% of the random ECoG events were not associated with any detectable behavioural change. Similar findings were obtained at Weeks 14–15 (Fig. 3B) and 27–28 post-injury (Fig. 3C); and also when data at each of the three time points post-injury were analysed by seizure type (Fig. 3D–F). For all groups, the comparison of behavioural change occurrence with random versus epileptiform ECoG events was significant (all P-values <0.001; Fisher's exact test). We conclude that the behavioural changes we observe during EEEs lasting ≥2 s are not due to random association with normal behaviour but represent ictal behaviour that defines the behavioural phenotype of the post-traumatic epileptic syndrome after rpFPI.
Analysis of ECoG power spectrum after EEEs longer than 2 s in the rat
To further characterize the epileptic syndrome induced by rpFPI we studied the consequence of electrical seizures lasting >2 s on the background ECoG state of the animals assessed by the post-ictal power spectrum (Fig. 4). For this analysis we used only EEEs that occurred during at least 1 min of active exploratory or grooming behaviour of the animal with dominant theta activity, indicating the animal was involved in spatial orientation and learning. Of all these seizure events, we found nine that satisfied these conditions, including five grade 1, three grade 2 and one grade 3 seizure, which were pooled for group analysis (n = 5 animals). As controls, we used an equal number of 4 s long ECoG events selected at random times within the same data files of the seizures, and occurring during at least 1 min of active behaviour of the animal with dominant theta activity. We found that random ECoG events were followed by ECoG that maintained the dominant theta activity, with an average dominant peak at 7.5 Hz. Their summed power in theta was 59 000 ± 11 000 μV2s, and in delta was 30 000 ± 4000 μV2s, consistent with the theta activity typical of active/exploratory behaviour of the animal. Conversely, seizures were followed by a significantly slower ECoG that displayed dominant delta activity, with an average dominant peak at 1.5 Hz. Their summed power in theta was only 25 000 ± 4000 μV2s (P = 0.02; Mann–Whitney), and that in delta 69 000 ± 8000 μV2s (P = 0.002; Mann–Whitney). This phenomenon was further investigated by examining the ratio of the summed power in the theta band and that in the delta band. RΘ/δ was 2.1 ± 0.4 following random ECoG events, and was significantly decreased to 0.4 ± 0.1 after EEEs (P = 0.001; Mann–Whitney). We interpret these findings as further evidence that EEEs that last >2 s are indeed ictal events because, when they occur in animals engaged in active behaviour with theta dominance, they are immediately followed by ECoG slowing with disruption of that theta activity.
Analysis of the temporal correlation between behavioural arrests and short EEEs
The most common ictal behavioural correlate of rpFPI-induced seizures in the early weeks post-injury is a freeze-like behavioural arrest of the animal (D'Ambrosio et al. 2004, 2005). Behavioural pauses, however, also occur as part of the normal rat behaviour. Thus, we tested whether there is a specific temporal association between the occurrence of EEEs and behavioural arrests, or whether normal behavioural arrests occur frequently enough to coincide with EEEs by chance (Fig. 5A and B). To this end, we computed the cross-correlogram of the timing of the animals’ behavioural arrests identified on video by raters who were blinded to the ECoG, and the times of EEEs identified by raters who were blinded to the video. The blinder selected fourteen 1 min long clips of video-ECoG from 10 different animals at 2–5 weeks post-injury. These videoclips were chosen to include only one EEE lasting 0.8–2 s, located at a random time after the file's beginning. The 1 min long videos were then examined by a rater blinded to both the ECoG and the time of occurrence of the EEE, who marked each time the animal appeared to present a freeze-like behavioural arrest. After all 14 files were scored, the blinder created the 14 rasters of animals’ pauses and computed the time difference between the onset of each behavioural arrest and the one of the EEE (Fig. 5A). The cross-correlogram between behavioural arrests and EEEs was then obtained by setting time bins of 1 s, and representing each observed behavioural arrest as one bin count at the time difference between the behavioural arrest and the EEE and aligning the time zero of the cross correlogram with the occurrence of the EEEs (Fig. 5B). The cross-correlogram shows that, while some behavioural arrests occurred randomly within the time interval as expected for normal behaviour of the animals (1.9 events per minute on average), all EEEs occurred simultaneously with a behavioural arrest (14 out of 14). Thus, we observed an average of 2.9 behavioural arrests per minute, all sources included, corresponding to a probability of occurrence in a bin of 0.0483. Therefore, the simultaneous occurrence of EEEs with a behavioural arrest is statistically highly significant (P << 0.001; probability of 14/14 events occurring in the same time bin), and cannot be explained with chance association of randomly occurring normal behavioural arrests. We thus conclude that the occurrence of short seizures manifests with both the EEE and a simultaneous behavioural arrest, which is therefore a form of ictal behaviour.
Analysis of behavioural correlate of EEEs shorter than 2 s in the rat
We further investigated whether perilesional, chronic, recurrent and spontaneous EEEs shorter than 2 s were specifically associated with ictal behaviour by examining the behavioural correlate of EEEs while blinded to the ECoG. To this end we performed a visual analysis of the behavioural changes occurring during either epileptiform or randomly selected ECoG events, performed blind to the ECoG (Fig. 5C and D). In a pilot study, we estimated that EEEs shorter than 0.8 s were too fast for reliable visual examination. Thus we examined EEEs lasting between 0.8 and 2 s. First we randomly chose 18 different 8 h long video-ECoG recordings obtained from 15 different rpFPI animals at 2–14 weeks post-injury. As internal controls, we chose 2 s long normal ECoG epochs (true negatives) at random times within the same recordings. The behaviour of the animal coinciding with all ECoG events was scored as per our behavioural scale by raters who were blinded to the ECoG. Of the 34 rankable EEEs lasting 0.8–2 s that were identified (Fig. 5C), 88.2% were associated with a behavioural change (a freeze-like movement arrest in all cases) while 11.8% were not. Conversely, the 127 rankable random ECoG events identified presented the opposite association, with 86.6% having no behavioural change, and only 13.4% having a behavioural change (P < 0.001, Fisher's Exact test), which also consisted of a movement arrest in all cases. The sensitivity of the blinded video analysis in detecting a behavioural pause coincident with EEEs, as defined by the true-negative random ECoG events, was therefore 86.6%. Consistent with the pilot study, we found no relationship between the duration of the EEE and its association with behavioural change (Fig. 5D). We interpret these findings as evidence that EEEs lasting 0.8–2 s represent seizures.
Analysis of ECoG power spectrum after EEEs shorter than 2 s in the rat
We then studied the consequences of these short focal recurrent and spontaneous EEEs on the background ECoG activity of the animal (Fig. 6). For this analysis we used only short EEEs that (i) lasted 0.8–2 s; and (ii) occurred during at least 1 min of active behaviour of the animal with dominant theta activity. Of the ECoG events identified in our database, 10 satisfied these conditions. As controls, we used an equal number of 2 s long ECoG events randomly chosen within the same data files and occurring during at least 1 min of active behaviour of the animal with dominant theta activity. For each epileptiform and random ECoG event we obtained the power spectrum of the 2 s immediately following the event. We found that random ECoG events were followed by dominant theta oscillations, with average dominant peak at 6.5 Hz. Their summed power in the theta band was 51 000 ± 6000 μV2s, and that in delta was 44 000 ± 5000 μV2s, consistently with the active or exploratory behaviour of the animal. Conversely, EEEs were followed by a significantly slower ECoG. Their summed power in the theta band was only 25 000 ± 4000 μV2s (P = 0.003; Mann–Whitney), and that in delta 77 000 ± 10 000 μV2s (P = 0.043; Mann–Whitney). This was further investigated by examining the ratio of the summed power in theta over that in delta. RΘ/δ was 1.30 ± 0.2 following random ECoG events, and was significantly decreased to 0.3 ± 0.1 after EEEs (P = 0.0004; Mann–Whitney). We interpret these findings as further evidence that EEEs lasting 0.8–2 s are ictal events because, when they occur in the animal during periods of active behaviour with dominant theta activity, they are immediately followed by loss of theta activity and the interruption of the active behavioural state.
Simultaneous epidural ECoG/scalp EEG in the rat
Scalp EEG is commonly employed for diagnosis of human epilepsy and estimates of human PTE incidence rely heavily on it. To put rpFPI-induced PTE in the rat into context with scalp EEG we performed simultaneous epidural ECoG and scalp EEG recordings (Fig. 7). To this end, five animals underwent rpFPI and received the standard five-electrode montage for epidural ECoG. Two scalp electrodes (Fig. 7A) were added in parietal (ipsilateral to the injury site) and occipital positions. Animals were then monitored from 4 through 10 weeks post-injury for a total of 22 h of recordings. We found that grade 1 ECoG seizures, regardless of their duration, were never detected by the scalp EEG (n = 94; Fig. 7B, C). Conversely, grades 2 and 3 seizures became visible on the scalp only when and if they spread to all four epidural electrodes 1–4 (n = 20 out of 52; Fig. 7D). Seizures detected by only two or three epidural ECoG electrodes were not detected on the scalp.
Figure 7.

Simultaneous scalp-EEG and epidural ECoG recordings in the rat reveal focal seizures undetected by scalp EEG. (A) Schematics of the montage employed: five epidural electrodes (1 through 5) and two scalp electrodes placed parieto-temporally (pt) and occipitally (o). The light grey oval indicates the FPI craniotomy. Number 4 is the perilesional epidural electrode. (B–C) Two examples of short grade 1 seizures clearly detected by the ECoG, but undetected by scalp-EEG. (D) A grade 2 seizure first detected by ECoG electrode 4 and later spreading to all epidural electrodes was detected by scalp EEG during the spreading. The dotted box in D shows the electrographic seizure simultaneously detected by epidural electrode 4 and scalp electrode pt at higher magnification. All seizures shown were associated with behavioural arrest of the animal. The numbers next to each ECoG trace indicate the electrodes by which the trace is recorded, and its reference.
Brief focal seizures in humans
We present four clinical cases with brief (<2 s) seizures observed during intracranial recordings (Fig. 8; Table 1). During evaluation for the neurosurgical treatment of their pharmacoresistant epilepsy all four patients manifested typical complex partial seizures lasting ∼1 min. However, invasive monitoring revealed the occurrence of brief seizures in the same ictal sites. The details are as follows:
Figure 8.

Short recurrent spontaneous focal seizures in the human. Representative examples of short seizures lasting 1–2 s are presented from four pharmacoresistant patients evaluated for resective surgery. For aetiology and clinical history of patients see Table 1 (Patients 1– 4 correspond to panels A–D, respectively). In all patients longer complex partial seizures with secondary generalization were recorded originating from the same electrodes, and with identical electrographic onset patterns, as observed in the corresponding short seizures presented. In Patients 1 and 2, who presented with ictal sites in the frontal lobe (A and B), the brief electrographic seizures were not detected clinically, while longer and spreading complex partial seizures were. In Patients 3 and 4, who presented with ictal sites in the sensori-motor and motor cortex (C and D), EMG revealed the occurrence of simultaneous ictal behaviour during the short seizures. Note the skin electrode traces present the evidence of eye blinking (C) and muscle contraction (D) during the EEEs (vertical dotted lines). Lateral X-ray images of patient's skulls after electrode placement are shown for all patients (A1, B1, C1 and D1). ECoG sites and labels are shown in the schematic diagrams (A2, B2, C2 and D2–3). ECoG is presented with referential montage in A, C and D, and bipolar montage in B. The voltage scales refer to ECoG electrodes only and not EMG. EMG electrodes were placed on the face lateral to the right eye in Patient 3 (C), and on the surface of the skin overlying the left pectoralis muscle in Patient 4 (D). Asterisk marks one of the cardiac signals detected by the skin electrode. In the schematic diagrams, grey circles show electrodes not involved with seizure onset. Crossed-out circles show inactive electrodes not displayed. Red circles show electrodes proximal to seizure onset. Yellow circles show areas of direct electrographic seizure spread. The labels on schematics match the labels on the corticographic tracings: g: grid; sA: strip A; sB: strip B; sC: strip C; sD: strip D. Numbers within the grids and next to the strips define their orientation. The recording montage covered most of the left (Patients 1, 2 and 3; panels A, B and C) or right (Patient 4; panel D) lateral perisylvian neocortex.
Table 1.
Clinical features of pharmacoresistant epilepsy patients presenting short neocortical seizures
| Patient number | Aetiology of epilepsy | Clinical features and ictal semiology | MRI Findings | ECoG localization |
|---|---|---|---|---|
| 1 | Traumatic brain injury to left frontal lobe at age 6 years. | Epilepsy diagnosed at the age of 6 years, a few weeks after injury and refractory by age 18. At time of evaluation complex partial seizures included aphasia and fluttering of eyes. Secondary generalized seizures presented with right body hemiclonic movements. | Left frontal encephalomalacia | Left inferior frontal. |
| 2 | Traumatic brain injury at age 2 years. | Epilepsy diagnosed at age 23. At time of evaluation complex partial seizures presented with head version to the right and right body hemiclonic movements. | Bilateral frontal but left greater than right frontal encephalomalacia | Lateral superior frontal. |
| 3 | Cryptogenic aetiology. Patient had life-long developmental delay and learning disability | Epilepsy diagnosed at age 16. At time of evaluation patient suffered from as many as 100 complex partial with or without secondary generalization per month. Seizures had an initial clinical manifestation of right sided eye-blinking or eyelid twitching. The patient's family had noted a ‘pre-seizure’ warning of episodic right eye blinking (‘twitching’). | Non-specific, with only a left anterior frontal venous angioma on MRI. | Inferior-parietal/lateral temporal. |
| 4 | Perinatal infarction to right middle cerebral artery territory. | Diagnosis of epilepsy was at infancy. At time of evaluation, the patient was experiencing two dozen complex partial seizures and two secondarily generalized tonic–clonic seizures per month. Seizures lasted >1 min and were manifested by movements on the left side including left hand twitching and posturing. | Encephalomalacia in right parietal, occipital and posterior temporal areas consistent with prior infarction. | Superior parietal. |
Patient 1 (Fig. 8A) is a 19-year-old man who suffered severe traumatic brain injury at six years of age from a sporting accident. The first recognized convulsive seizure occurred a few weeks later, and others followed every few months. Complex partial seizures consisting of fluttering of eyes, confusion and speech impairments also occurred frequently but were not initially recognized. They were later noted to occur several times per week and were found to be refractory to several anti-epileptic drugs. Invasive monitoring confirmed the ictal sites to be in left inferior frontal region. The patient was seizure-free after surgery. Patient 2 (Fig. 8B) is a 31-year-old woman who sustained severe closed head injury, from a motor-vehicle accident, at age 2. Seizures were diagnosed at age 23. Typical complex partial seizures started with a period of staring and unresponsiveness, with head turning towards the right, which could be followed by body hemiclonic movements and tonic–clonic convulsions. Seizures were refractory to several anti-epileptic drugs. Invasive monitoring confirmed the onset of the typical complex partial seizures to be in the left lateral superior frontal region. The patient was seizure-free after surgery. Both Patients 1 and 2 presented short electrographic seizures originating in the same ictal site as the longer complex partial seizures, which were distant from motor areas. These brief seizures were not reported by patients, nor were they associated with any detectable external behaviour, but were electrographically identical to the onset of the patient's longer complex partial seizures. Patient 3 (Fig. 8C) was a 22-year-old man, with long standing developmental delay who suffered from as many as 100 complex partial or secondarily generalized tonic–clonic seizures per month. Scalp video-EEG monitoring demonstrated typical focal seizures lasting >1 min and with onset suspected to be in the left lateral frontoparietal neocortex. During ECoG monitoring, several of the patient's typical complex partial and secondarily generalized seizures were captured, but it also became clear that the patient suffered many shorter seizures manifested only by brief runs of right eye blinking (‘twitching’) that were often unnoticed by the patient but recognized by family members as the earliest signs of seizures. Additional surface skin EMG electrodes were placed peri-orbitally to the right eye to capture the earliest clinical manifestations of seizures. Eye-twitching episodes, lasting <5 s, each correlated with brief discharges limited to a few contacts over the left sensori-motor cortex. The ECoG changes appeared to precede EMG changes by ∼200 ms. Patient 4 (Fig. 8D) was a 47-year-old woman, with a history of perinatal infarction in the right middle cerebral artery territory, and subsequent cerebral palsy with mild left spastic hemiparesis. The patient experienced two dozen complex partial seizures and two secondarily generalized tonic–clonic seizures per month. The seizures lasted >1 min and were manifested by movements on the left side including left hand twitching and posturing. Scalp video-EEG monitoring captured multiple focal simple and complex partial seizures suspected to begin in the right lateral hemispheric neocortex. Invasive ECoG monitoring was carried out with additional skin surface electrodes placed over left forearm, upper arm, face and leg areas to identify the earliest clinical manifestations of the seizures. Monitoring showed typical complex partial seizures with onset in the contralateral primary motor cortex. The patient also had brief simple partial motor seizures with cortical ECoG signals lasting <2 s with associated EMG activity from the contralateral pectoralis muscle. Both Patients 3 and 4 presented short electrographic seizures originating in the same ictal site, within sensory-motor areas, as the longer complex partial seizures. These brief seizures were similar in ECoG appearance to the initial portion of longer complex partial seizures, and were accompanied by evident ictal behaviour consisting of twitching of the corresponding contralateral muscles as confirmed by simultaneous skin-EMG recordings.
Impact of seizure definition on seizure frequency and incidence of epilepsy in the rat
Video-ECoG recordings performed in 10 rpFPI animals, from 2 to 28 weeks post-injury, were examined to assess the impact of seizure definition on the observed seizure frequency. As expected, seizure frequency was progressively lower as seizures were defined by progressively longer EEEs (Fig. 9A). Because seizure duration increases over time post-injury (D'Ambrosio et al., 2005), and because all animals present with short EEEs coexisting with longer ones, the greater biases were observed at weeks 2–4 post-injury, and for seizures defined as EEEs ≥5 s. We also examined how the incidence of PTE is affected by different definitions of epilepsy in the rat (Fig. 9B). We plotted the cumulative probability of PTE after rpFPI by the following definitions: (i) one EEE lasting ≥0.8 s; (ii) one EEE lasting ≥5 s; (iii) one EEE lasting ≥15 s; and (iv) one tonic–clonic convulsion. In addition, we estimated the cumulative probability of PTE as it would be detected by a longitudinal scalp EEG study performed from week 2–28 post-injury. This was simulated by applying to the longitudinal ECoG data obtained from 2 to 28 weeks post-injury, the rule, obtained during the simultaneous ECoG/EEG recordings, that only ECoG seizures spreading to all four epidural ECoG electrodes could be detected by scalp EEG (Fig. 7). For our analysis, we considered that seizures would be detected by scalp EEG if they were observed simultaneously on all four ECoG electrodes for either 1 or 5 s. We found that both the incidence of PTE and the time course of epileptogenesis greatly depended upon the definition of seizure employed. After injury, if tonic–clonic convulsions were required for diagnosis, no rpFPI animals could be considered epileptic at any time point. If at least one episode of 5 s long epileptiform activity was required on the scalp EEG, then the overall probability of epilepsy was ≅80% by 20 weeks post-injury, with a half time for epileptogenesis of ∼17 weeks. However, if at least one 0.8 s long epileptiform ECoG event needed to be observed, then the overall probability of epilepsy was 100% by week 5 post-injury, with a half time for epileptogenesis of ∼1 week.
Figure 9.

Seizure frequency and time course of epileptogenesis after rpFPI depend on the definition of epilepsy and on the diagnostic technique employed. (A) Seizure frequency (counts/h) is reported for 10 rpFPI animals over time post-injury. Data are presented by different definitions of electrographic seizures as lasting at least 0.8, 2, 5 or 15 s on the ECoG. Data are presented as mean ± SEM. Note the dramatically lower seizure frequency as seizures are arbitrarily defined as being longer than 5 or 15 s. (B) The cumulative probability of PTE is plotted over time after rpFPI as defined by six different criteria. Animals were defined as epileptic after: one ECoG seizure lasting ≥0.8 s (filled circles); one ECoG seizure lasting ≥5 s (filled triangles); one ECoG seizure lasting ≥15 s (filled diamonds); one simulated scalp-EEG seizure lasting ≥1 s (hollow squares); one simulated scalp-EEG seizure lasting ≥5 s (hollow stars); one tonic–clonic convulsion (hollow diamonds). Note that by week 5 post-injury (vertical dotted line) all rpFPI animals were epileptic if the condition was defined by the most stringent definition available, while all other definitions underestimated the fraction of epileptic animals and overestimated the duration of the silent period. Fitting lines are hand-drawn.
Discussion
To our knowledge this is the first attempt to put duration-based identification of seizures on a solid empirical basis using functional criteria. We applied the clinical practice of functionally evaluating seizures in humans to experimental seizures in animals, and considered EEEs—a pathological feature of FPI that is absent in sham-injured animals (Fig. 2H)—as proven seizures if we found evidence that hypersynchronous ECoG occurred simultaneously with behavioural changes. We demonstrate that rpFPI in the rat induces CRSPSs lasting <2 s, and that similar short CRSPSs occur in humans. Regardless of the electrical type, all EEEs lasting ≥2 s are associated with stereotyped behavioural changes (Fig. 2L and 3). The temporal progression of both electrical and behavioural seizures from 2 to 28 weeks post-injury have previously been described (D'Ambrosio et al., 2004, 2005). We now demonstrate that these changes are not just randomly coincident normal behaviour but are specifically linked to the epileptiform ECoG, as demonstrated by the paired behavioural analysis of random ECoG events within the same files (Fig. 3). In fact, while all rankable EEEs lasting ≥2 s were coincident with a behavioural change, the vast majority of random EEEs were not. We further found that all the EEEs lasting ≥2 s that occur during active behaviour of the animal with theta dominance were simultaneous with behavioural arrest and were followed by a loss of this theta activity (Fig. 4). Thus, we conclude that all chronic, recurrent and spontaneous EEEs that last ≥2 s are seizures. Notably, theta activity in the rat ECoG is thought to represent an active cognitive and behavioural phase in which the animal is engaged in sensorimotor integration, spatial orientation and learning, and during which the hippocampus sustains a theta rhythm (Winson, 1978; Buzsáki, 2005; Kay, 2005; McNaughton et al., 2006). The fact that these theta rhythms are absent immediately after seizures suggests that the cognitive and behavioural states of the animal are affected by EEEs.
EEEs lasting between 0.8 and 2 s presented with the same functional properties as longer seizures. These short EEEs coincided with behavioural arrests (Fig. 5), and when they occurred during active behaviour of the animal with theta dominance, they coincided with a behavioural arrest and were immediately followed by loss of theta activity (Fig. 6). These EEEs are a pathological feature of the FPI brain (Fig. 2H), occur chronically, spontaneously and recurrently, and are associated with behavioural changes. Thus, they fit the clinical definition of seizures and must therefore be considered as such, despite their duration. The cross-correlogram of the temporal sequence of animals’ behavioural arrests versus EEEs (Fig. 5A and B) demonstrates that short seizures manifest with both an EEE and a simultaneous behavioural arrest, which is, therefore, a form of ictal behaviour. Notably, the behavioural arrest is the most common ictal behaviour in the early months after rpFPI (Fig. 2L). Because behavioural pauses are part of normal behaviour, this may explain why FPI-induced epilepsy was not recognized until ECoG was employed to aid behavioural observation (D'Ambrosio et al., 2004). The analysis of the animals’ behaviour during short epileptiform versus randomly selected ECoG events (Fig. 5C) further demonstrates that short EEEs are associated with behavioural changes (a pause in behaviour in all cases) and are therefore clinical seizures. Two independent sources of uncertainty affect this blind behavioural seizure test of epileptiform ECoG. The first, U1, is due to chance association of interictal events or subclinical seizures with a behavioural arrest that is a normal component of the animal's behaviour, and results in an overestimate of clinical seizures. Our blinded behavioural analysis test returned 110 true negatives correctly identified as such, and 17 false positives (a pause in behaviour not associated with an EEE). Thus this test had a specificity of 86.6%, and we expect that 13.4% of short EEEs may be misclassified as clinical seizures because of a chance association to behavioural arrest. Thus, U1 = 0.134. The second source of uncertainty, U2, is due to limitations in visual examination of behaviour because, as the duration of the ECoG event to evaluate gets shorter, the behavioural arrest associated with it also gets shorter, blends into the normal behaviour of the animal and becomes progressively more difficult to distinguish by visual examination. This phenomenon results in the underestimate of clinical seizures. U2 was negligible in the EEE-duration interval studied, as demonstrated by the duration-independence of the proportion of EEEs with behavioural change (Fig. 5D). Thus, we can estimate the overall uncertainty that EEEs lasting 0.8–2 s are clinical seizures to be U1 = 13.4%. Because we observe EEEs of similar duration with or without behavioural changes (Fig. 5D), it is likely that all EEEs lasting 0.8–2 s are seizures, and at least 86.6% of them are expected to be clinical seizures. Considering that rpFPI animals present a wide range of durations of EEEs, ranging from frequent perilesional single spikes lasting 150–250 ms and associated with no overt behavioural changes that may be considered interictal by definition, to perilesional epileptiform activity consisting of a few sequential spikes (Figs 2A–C and 5F), to trains of perilesional spikes lasting 0.8–2 s (Figs 2D–E, 5E and 7B–C), that we found to satisfy the definition of ictal just as prolonged seizures do (Fig. 2I–K), we then conclude that the transition from interictal to ictal activity occurs in the rpFPI animal for EEEs lasting between 0.2 and 0.8 s. This is a duration range in which the functional tests of seizures used in the present work cannot be reliably applied. Intrinsic limitations in techniques available (including the identification of very fast or subtle ictal behaviour, or changes in internal subjective behaviour), and in the definition of the duration of seizures as detected by ECoG, as well as the prohibitively time consuming count of these sub-second EEEs in the absence of sensitive and specific automatic spike-detection software, all prevent a finer definition of this transition. However, for the purpose of testing prophylactic treatments, a finer definition of this transition, while desirable, is not necessary because the frequency of proven ictal events (∼2/h) already gives adequate power to run preclinical studies with limited group size. Future work may reveal whether EEEs lasting <0.8 s are useful pathological markers to consider during prophylactic studies and/or to predict later onset of PTE. Notably, the existence of a continuous spectrum of perilesional epileptiform activity, ranging from single spikes, to multiple-spike complexes, to short seizures, to spreading seizures (Fig. 2) also indicates that we are observing epileptiform activity right where it is generated, and provides additional evidence that the perilesional neocortex is not simply receiving epileptiform activity from other areas but develops into an epileptic focus as previously hypothesized (D'Ambrosio et al., 2004, 2005).
While seizures lasting <5–10 s are generally considered in studies of idiopathic epilepsy in animals (Coenen and Van Luijtelaar, 2003; Shaw, 2004; Magil et al., 2005), they have thus far been mostly neglected in animal acquired epilepsy research, in which the predominant practice consists of defining as a seizure epileptiform activities longer than an arbitrarily set duration. There are several practical reasons for this: (i) to avoid defining the transition from interical to ictal events; (ii) to disregard the variable presence of age-dependent short (<10 s) idiophatic seizures in experimental rodents so that they are not confounded with acquired ones (D'Ambrosio et al., 2005); (iii) to disregard seizures induced by cortical damage from sub-optimal recording electrode implantation (see ‘Materials and methods’ section); and (iv) to simplify the implementation of algorithm-based automated and visual identification of seizures to obtain fewer false positives. However, this practical approach misses opportunities to develop better treatments, by biasing toward mechanism and treatments that affect seizure evolution, rather than their genesis. Indeed, by changing the definition of epileptic seizures in the rat we were able to demonstrate dramatic differences in the estimated incidence of PTE and duration of the silent period during which epileptogenesis is hypothesized to occur (Fig. 9B). Notably, all rpFPI animals had developed epilepsy by week 5 post-injury when we used a definition of seizure as ECoG discharge lasting at least 0.8 s, while epilepsy was diagnosed in progressively fewer animals when it was defined as the onset of progressively longer ECoG or scalp-EEG events.
Acquired partial seizures lasting <5 s on the ECoG are of strong experimental and clinical interest because they have several properties that render them very promising targets for mechanistic, translational and clinical studies of epileptogenesis. First, these short seizures in the rat are clinically relevant since similar acquired focal seizures also occur in humans (Fig. 8). We were able to identify four pharmacoresistant patients who presented with short chronic recurrent spontaneous focal EEEs with electrographic appearance similar to short grade 1 seizures in the rpFPI rat. Similar to what we observed for the neocortical epileptic focus induced by rpFPI (Fig. 2), all short seizures in humans occurred in the same ictal sites as longer complex partial seizures, indicating that the neocortical epileptic focus generates a range of epileptic activities in the human as it does in the rat. In two of these patients (Fig. 8C and D), these short seizures occurred in motor cortex areas and were associated with a mild stereotyped ictal behaviour (eye blinking or muscle twitching) that, while subtle and easily missed, could be reliably detected by EMG. When similar EEEs occurred distally from the motor cortex (Fig. 8A and B), as expected, they were not associated with detectable behavioural output. We believe that, depending on the neocortical location of the focus, brief EEEs would probably go unnoticed because of the lack of apparent ictal behaviour. We conclude that short seizures observed in head-injured rats are not a feature of the small rodent brain, but one of genuine epileptic foci. Second, these short partial seizures in the rat, while readily detected by ECoG, are mostly invisible on scalp EEG (Fig. 7), just as most short ECoG seizures are in humans (Cukiert et al., 2001; Binnie, 2003). By performing simultaneous epidural ECoG and scalp EEG in rpFPI-injured animals we were able to determine that no grade 1 and only ∼38% of grades 2 and 3 seizures were detected by scalp EEG. The onset of ECoG seizures could never be appreciated by the scalp EEG, which only picked up the activity when it had spread to a sufficiently large volume of cortex. Yet, the experimental conditions greatly favoured the detection of short seizures by scalp EEG in the rat compared to the human. Indeed, the rat has a significantly thinner skull and scalp, and the montage employed placed the parietal scalp electrode much closer (∼0.5 cm) to the neocortical focus in the rat than what typically occurs with a standard 10–20 electrode montage in humans (∼2 cm if in a gyrus; ∼5 cm if in a sulcus). Third, rpFPI-induced epilepsy is progressive in both the number of seizures and their duration (D’Ambrosio et al., 2004, 2005), and while short EEEs may occur at any time point after rpFPI, they are present in 100% of the animals investigated, most commonly 2–4 weeks post-injury (Fig. 9A). Thus, these are the earliest and most common markers of PTE that can be identified after head injury in the rat. Therefore, short EEEs constitute a powerful preclinical endpoint for a number of crucial studies. They are the ideal markers to guide the screening for prophylactic treatments of PTE, as all animals present them by week 5 post-rpFPI (Fig. 9). There is no need to wait longer periods post-injury for more severe electrical or behavioural manifestation of PTE to occur in the FPI rat to screen for antiepileptogenic compounds, which will allow for faster and more powered preclinical screening. Drugs that are effective for short seizures could then be tested for their effect at later times post-injury on longer or subcortical seizures, allowing characterization of the effect of drugs on different pro-epileptic mechanisms and anatomical foci. Also, the identification of the earliest frank partial seizures arising after an insult will help address the longstanding question of whether and how ‘seizures beget seizures’ and cause brain damage (Sutula et al., 1988, 2003; Cavazos et al., 1994; Sutula and Pitkanen, 2001; Holmes, 2002).
Finally, it should be noted that the vast majority of the short partial seizures we observed in both rats and humans are difficult to detect by simple behavioural observation or by scalp EEG. Short seizures in PTE patients (Fig. 8) may or may not be associated with obvious clinical signs depending on the brain location of the focus. Short seizures in the FPI rat are associated with easily missed behavioural arrest in the early weeks post-injury before evolving to more behaviourally severe seizures (D’Ambrosio et al., 2004, 2005). Thus, our data lends support to the hypothesis (Sloviter, 2008; Dichter, 2009) that mild CRSPSs may occur in patients significantly earlier than when epilepsy is first diagnosed, and that they may be unrecognized until they have progressed in frequency and severity to become a burden to the patient and to be detectable at scalp EEG. Future prospective studies will be necessary to address this hypothesis.
Funding
CURE Epilepsy foundation [5154001.01 to R.D.]; the National Institutes of Health [NS053928 to R.D. and T32NS07144].
Glossary
Abbreviations
- CRSPSs
chronic recurrent spontaneous partial seizures
- ECoG
electrocorticography
- EEE
epileptiform ECoG events
- EEG
electroencephalograpy
- EMG
electromyography
- EEG
electroencephalograpy
- GFAP
Glial fibrillary acidic protein
- PTE
post-traumatic epilepsy
- rpFPI
rostral parasagittal fluid percussion injury
References
- Beghi E, Berg A, Carpio A, Forsgren L, Hesdorffer DC, Hauser WA, et al. Comment on epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–2. doi: 10.1111/j.1528-1167.2005.00273_1.x. [DOI] [PubMed] [Google Scholar]
- Binnie CD. EEG, Paediatric neurophysiology, special techniques and applications. Vol. 2. Amsterdam: Elsevier; 2003. Clinical neurophysiology. [Google Scholar]
- Blum DE, Eskola J, Bortz JJ, Fisher RS. Patient awareness of seizures. Neurology. 1996;47:260–4. doi: 10.1212/wnl.47.1.260. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta rhythm of navigation: link between path integration and landmark navigation, episodic and semantic memory. Hippocampus. 2005;15:827–40. doi: 10.1002/hipo.20113. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14(5 Pt 2):3106–21. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen AM, Van Luijtelaar EL. Genetic animal models for absence epilepsy: a review of the WAG/Rij strain of rats. [Review] Behav Genet. 2003;33:635–55. doi: 10.1023/a:1026179013847. [DOI] [PubMed] [Google Scholar]
- Cukiert A, Buratini JA, Machado E, Sousa A, Vieira JO, Argentoni M, et al. Results of surgery in patients with refractory extratemporal epilepsy with normal or nonlocalizing magnetic resonance findings investigated with subdural grids. Epilepsia. 2001;42:889–94. doi: 10.1046/j.1528-1157.2001.00201.x. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Perucca E. Epilepsy after head injury. Curr Opin Neurol. 2004;17:731–5. doi: 10.1097/00019052-200412000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle D, Miller JW. Posttraumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127(Pt 2):304–14. doi: 10.1093/brain/awh038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R, Fender JS, Fairbanks JP, Simon E, Born DE, Doyle D, et al. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain. 2005;128(Pt 1):174–88. doi: 10.1093/brain/awh337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R, Hakimian S, Drane DL, Ojemann J, Miller JW, Sheerin AH, et al. What is an epileptic seizure? Insights from chronic recurrent partial seizures induced by fluid percussion injury in the rat. Proceedings of the American Epilepsy Society Meeting, Philadelphia, PA. Epilepsia. 2007;48(Suppl 6):294–5. [Google Scholar]
- Dichter MA. Posttraumatic epilepsy: the challenge of translating discoveries in the laboratory to pathways to a cure. Epilepsia. 2009;50(Suppl 2):41–5. doi: 10.1111/j.1528-1167.2008.02009.x. [DOI] [PubMed] [Google Scholar]
- Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–2. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- Holmes GL. Seizure-induced neuronal injury: animal data. Neurology. 2002;59(9 Suppl 5):S3–6. doi: 10.1212/wnl.59.9_suppl_5.s3. [DOI] [PubMed] [Google Scholar]
- Juul-Jensen P. Epidemiology of intractable epilepsy. In: Schmidt D, Morselli P, editors. Intractable epilepsy. New York: Raven Press; 1986. pp. 5–11. [Google Scholar]
- Kay LM. Theta oscillations and sensorimotor performance. Proc Natl Acad Sci USA. 2005;102:3863–8. doi: 10.1073/pnas.0407920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill PJ, Sharott A, Harnack D, Kupsch A, Meissner W, Brown P. Coherent spike-wave oscillations in the cortex and subthalamic nucleus of the freely moving rat. Neuroscience. 2005;132:659–64. doi: 10.1016/j.neuroscience.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Mattson RH, Cramer JA, Collins JF, Smith DB, Delgado-Escueta AV, Browne TR, et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic-clonic seizures. N Engl J Med. 1985;313:145–51. doi: 10.1056/NEJM198507183130303. [DOI] [PubMed] [Google Scholar]
- Mattson RH, Cramer JA, Collins JF. Prognosis for total control of complex partial and secondarily generalized tonic clonic seizures. Department of Veterans Affairs Epilepsy Cooperative Studies No. 118 and No. 264 Group. Neurology. 1996;47:68–76. doi: 10.1212/wnl.47.1.68. [DOI] [PubMed] [Google Scholar]
- McNaughton N, Ruan M, Woodnorth MA. Restoring theta-like rhythmicity in rats restores initial learning in the Morris water maze. Hippocampus. 2006;16:1102–10. doi: 10.1002/hipo.20235. [DOI] [PubMed] [Google Scholar]
- Semah F, Picot MC, Adam C, Broglin D, Arzimanoglou A, Bazin B, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology. 1998;51:1256–62. doi: 10.1212/wnl.51.5.1256. [DOI] [PubMed] [Google Scholar]
- Shaw FZ. Is spontaneous high-voltage rhythmic spike discharge in Long Evans rats an absence-like seizure activity? J Neurophysiol. 2004;91:63–77. doi: 10.1152/jn.00487.2003. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the “latent period” and other concepts. Epilepsia. 2008;49(Suppl 9):85–92. doi: 10.1111/j.1528-1167.2008.01931.x. [DOI] [PubMed] [Google Scholar]
- Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–50. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Hagen J, Pitkanen A. Do epileptic seizures damage the brain? Curr Opin Neurol. 2003;16:189–95. doi: 10.1097/01.wco.0000063770.15877.bc. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Pitkanen A. More evidence for seizure-induced neuron loss: is hippocampal sclerosis both cause and effect of epilepsy? Neurology. 2001;57:169–70. doi: 10.1212/wnl.57.2.169. [DOI] [PubMed] [Google Scholar]
- Tatum WO, 4th, Winters L, Gieron M, Passaro EA, Benbadis S, Ferreira J, et al. Outpatient seizure identification: results of 502 patients using computer-assisted ambulatory EEG. J Clin Neurophysiol. 2001;18:14–19. doi: 10.1097/00004691-200101000-00004. [DOI] [PubMed] [Google Scholar]
- Winson J. Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science. 1978;201:160–3. doi: 10.1126/science.663646. [DOI] [PubMed] [Google Scholar]