

Abstract
Cyclin‐dependent kinase (CDK) 1 and the murine double minute 2 homolog (MDM2)–p53 interaction are potential therapeutic targets in cancer, and their inhibition has been reported to be more proapoptotic in malignant cells compared to normal cells. We investigated the effect of CDK1 inhibition on p53 signaling after simultaneous dual blockade using the CDK1 inhibitor RO‐3306 and the MDM2 inhibitor Nutlin‐3 in AML. Treatment of growing AML cells with RO‐3306 induced G2/M‐phase cell cycle arrest and apoptosis in a dose‐ and time‐dependent manner. We found that RO‐3306 acts cooperatively with Nutlin‐3 to induce mitochondrial apoptosis in a cell cycle‐independent fashion. RO‐3306 downregulated expression of the antiapoptotic proteins Bcl‐2 and survivin and blocked p53‐mediated induction of p21 and MDM2. CDK1 siRNA experiments showed that reduced CDK1 expression affects p53‐induced p21 transactivation. We suggest that RO‐3306 actively enhances downstream p53 signaling to promote apoptosis and that a combination strategy aimed at both inhibiting CDK1 and activating p53 signaling is potentially effective in AML, where TP53 mutations are rare and downstream p53 signaling is intact. (Cancer Sci 2009; 100: 1128–1136)
The different phases of the cell cycle are precisely controlled by the sequential activation of cyclin‐dependent kinases (CDK).( 1 ) In mammalian cells, the principal events of the cell cycle are driven by CDK1, CDK2, and CDK4 in association with various cyclin regulatory subunits. In contrast to CDK2 and CDK4, which are dispensable for cell cycle progression, CDK1 is a non‐redundant CDK with an essential role in mitosis.( 2 ) The effects of CDK2 inhibition have been evaluated in several studies, which found that selective CDK2 inhibition in multiple cancer cell lines has minimal antiproliferative effects.( 3 , 4 , 5 ) Because of the high degree of primary structure homology (86%) between the ATP‐binding domains of CDK1 and CDK2, most ATP‐competitive CDK2 inhibitors inhibit CDK1.( 3 ) It has also been reported that CDK1 inhibition downregulates survivin, an inhibitor of apoptosis protein, and induces apoptosis.( 6 , 7 , 8 ) These findings together suggest that CDK1 may be a better target for limiting cancer growth. Recently, a selective small‐molecule inhibitor of CDK1, RO‐3306, has been identified.( 9 ) RO‐3306 inhibits CDK1/cyclin B1 activity with a K i of 35 nM, a nearly 10‐fold increase in selectivity relative to CDK2/cyclin E and greater than 50‐fold increase in selectivity relative to CDK4/cyclin D. Interestingly, the selective CDK1 inhibition reversibly arrests human cells at the G2/M border of the cell cycle in normal human cells while inducing apoptosis in tumor cells, suggesting that selective CDK1 inhibitors may have utility in cancer therapy.
The most frequently inactivated protein in human cancer is p53; more than 50% of all solid tumors carry p53 mutations that abrogate its DNA binding and transactivation activity. Although TP53 mutations are rare in AML, it has been suggested that inactivation of wild‐type p53 frequently occurs through binding to its principal cellular regulator murine double minute 2 homolog (MDM2).( 10 ) MDM2 is a p53‐specific E3 ubiquitin ligase, and it mediates the ubiquitin‐dependent degradation of p53. MDM2 has been found to be overexpressed in approximately 50% of AML cases, a process that can actively enhance tumorigenic potential and resistance to apoptosis.
As many signaling pathway components are frequently affected in AML, synergistic targeted therapies that inhibit multiple targets are required.( 11 , 12 , 13 , 14 ) Here, we report the apoptotic effect of a potential targeted therapy, the simultaneous inhibition of CDK1 by RO‐3306 and activation of p53 signaling by the MDM2 inhibitor Nutlin‐3 in AML.( 9 , 15 ) Nutlin‐3 increases cellular p53 levels, a critical determinant of p53‐dependent apoptosis, and it efficiently induces p53‐mediated apoptosis in AML cells harboring wild‐type p53.( 16 ) The p53‐mediated apoptosis pathway has been shown to be well preserved in the model AML cell lines OCI‐AML‐3 and MOLM‐13.( 16 , 17 ) We found that RO‐3306 enhanced Nutlin‐induced p53‐mediated Bax conformational changes and apoptosis in AML cells independently of cell cycle phases. RO‐3306 cooperated with Nutlin‐3 in reducing levels of the antiapoptotic proteins Bcl‐2 and survivin. RO‐3306 treatment also blocked p53‐mediated induction of MDM2 and antiapoptotic p21. Our findings suggest that RO‐3306 actively enhances downstream p53 signaling toward mitochondrial apoptosis and that a combination strategy aimed at inhibiting CDK1 and activating p53 signaling could potentially be effective in AML, where TP53 mutations are rare and downstream p53 signaling is intact.
Materials and Methods
Reagents. The CDK1 inhibitor RO‐3306 and the selective small‐molecule antagonist of MDM2, Nutlin‐3, were purchased from Axxora (San Diego, CA, USA), dissolved in DMSO, and kept frozen at –20°C. In some experiments, cells were cultured with 70 µM cycloheximide, 0.2 µM MG132, or 50 µM Z‐VAD‐FMK (Axxora), which were added to the cells 1 h before drug administration. The final DMSO concentration in the medium did not exceed 0.1% (v/v). At this concentration, DMSO had no effect on cell growth or viability of AML cells used in this study for up to 72 h.
Antibodies. The following antibodies were used: rabbit polyclonal anti‐p53 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); mouse monoclonal anti‐MDM2 (Santa Cruz Biotechnology); mouse monoclonal anti‐p21 (EMD Biosciences, San Diego, CA, USA); rabbit polyclonal antisurvivin (R&D Systems, Minneapolis, MN, USA); mouse monoclonal anti‐Bcl‐2 (Dako Cytomation, Carpinteria, CA, USA); rabbit polyclonal anti‐Bcl‐XL (BD Biosciences, San Jose, CA, USA); rabbit polyclonal anti‐Bax (Cell Signaling Technology, Beverly, MA, USA); rabbit polyclonal anti‐Puma (Cell Signaling Technology); rabbit polyclonal anti‐Mcl‐1 (BD Biosciences); rabbit polyclonal anti‐CDK1 (Cell Signaling Technology); rabbit polyclonal antiphospho‐RNA polymerase II (Ser2) (Bethyl Laboratories, Montgomery, TX, USA); rabbit polyclonal antiphospho‐RNA polymerase II (Ser5) (Bethyl Laboratories); mouse monoclonal anti‐RNA polymerase II (Covance, Emeryville, CA, USA); and mouse monoclonal anti‐β‐actin (Sigma, St Louis, MO, USA).
Cell lines, primary samples, and cultures. Three human AML cell lines, OCI‐AML‐3, MOLM‐13 and HL‐60, were cultured in RPMI‐1640 medium containing 10% heat‐inactivated fetal calf serum (FCS). OCI‐AML‐3 and MOLM‐13 cells have wild‐type p53. In HL‐60, p53 is disabled by a large deletion of the TP53 gene. Stable p53 knockdown OCI‐AML‐3 cells were generated as described previously.( 17 ) HCT116 cells were cultured in DMEM containing 10% FCS. Heparinized bone marrow samples with more than 70% leukemia cells were obtained from AML patients after informed consent, according to institutional guidelines following the Declaration of Helsinki. Mononuclear cells were purified by Ficoll‐Hypaque (Sigma) density‐gradient centrifugation. AML cell lines were harvested in log‐phase growth, seeded at a density of 2 × 105 cell/mL, and exposed to RO‐3306 and/or Nutlin‐3. Primary AML mononuclear cells seeded at 5 × 105 cell/mL in RPMI‐1640 medium supplemented with 10% FCS were also exposed to RO‐3306 and/or Nutlin‐3. In combination experiments of Nutlin‐3 and RO‐3306, AML cell lines were treated with RO‐3306 at 0, 1, 2, 3, or 5 µM. The concentration ratio of RO‐3306 to Nutlin‐3 was 1 : 2 in OCI‐AML‐3 and 1 : 1 in MOLM‐13 and HL‐60 cells. The two agents were added simultaneously to cells and cultured for 18 h. In experiments involving a combination of RO‐3306 and doxorubicin, the two agents were added simultaneously to cells and cultured for 24 h. Cells were treated with doxorubicin at 0, 100, 250, or 500 nM and the concentration ratio of doxorubicin to RO‐3306 was 1 : 10. Primary AML cells were treated for 24 h with Nutlin‐3 at 0, 1, 2, or 4 µM in the absence or presence of 5 µM RO‐3306.
Transfection of CDK1 siRNA. HCT116 cells were transfected with siRNA oligonucleotides in six‐well plates using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). To evaluate the transfection efficiency, cells were transfected with the BLOCK‐iT Fluorescent Oligo (Invitrogen). Efficiency of transfection was estimated to be approximately 60%, with approximately 90% cell viability. Cells were transfected with scramble LO GC Duplex Stealth RNAi Negative Control (12935‐200; Invitrogen) or with CDK1 siRNA (12935‐006; Invitrogen). Twenty‐four hours after transfection, some cells were subsequently treated with 10 µM Nutlin‐3, and p21, Bcl‐2, or survivin expression levels were analyzed.
Cell cycle and apoptosis analysis. For cell cycle analysis, cells were fixed in 70% ice‐cold ethanol and stained with propidium iodide (PI) solution (25 µg/mL PI, 180 U/mL RNase, 0.1% Triton X‐100, and 30 mg/mL polyethylene glycol in 4 mM citrate buffer, pH 7.8; Sigma). The DNA content was determined using a FACSCalibur flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA). Data were gated on the FL2‐area versus FL2‐width cytogram to exclude doublets and aggregates, and a minimum of 3 × 104 gated cells was analyzed per sample. Cell cycle distribution was analyzed using ModFit LT software (Verity Software House, Topsham, ME, USA). Cells with a hypodiploid DNA content were counted as apoptotic based on DNA fragmentation. For Annexin V binding studies, cells were washed twice with binding buffer (10 mM HEPES, 140 mM NaCl, and 5 mM CaCl2 at pH 7.4; Sigma) and incubated with a 1 : 500 solution of FITC‐conjugated Annexin V (Roche Diagnostic, Indianapolis, IN, USA) for 15 min at room temperature. Stained cells were analyzed by flow cytometry and membrane integrity was simultaneously assessed by PI exclusion. To measure mitochondrial membrane potential (Δψm), cells were loaded with MitoTracker Red CMXRos (300 nM) and MitoTracker Green (100 µM, both from Molecular Probes, Eugene, OR, USA) for 1 h at 37°C. The Δψm was then assessed by measuring CMXRos retention (red fluorescence) while simultaneously adjusting for mitochondrial mass (green fluorescence). All experiments were conducted in triplicate.
Western blot analysis. Equal amounts of protein lysate were separated by SDS‐PAGE (12% gel) for 2 h at 80 V. Proteins were transferred to Hybond‐P membranes (Amersham Biosciences, Piscataway, NJ, USA), immunoblotted with appropriate antibodies, and reacted with enhanced chemiluminescence reagent (Amersham Biosciences). Signals were detected by Light‐Capture AE‐6981FC (Atto, Tokyo, Japan). An anti‐β‐actin blot was used in parallel as a loading control. Visualized blots were analyzed using ImageJ 1.32 software (National Institutes of Health, Bethesda, MD, USA), and Bcl‐2/Bax ratios were calculated using the software.
Quantitation of intracellular proteins by flow cytometry. Involvement of Bax conformational change was analyzed by means of an antibody directed against the NH2‐terminal region of Bax (YTH‐6A7; Trevigen, Gaithersburg, MD, USA). Cellular fixation, permeabilization, and staining with primary antibody or an isotypic control were carried out using the Dako IntraStain kit (Dako Cytomation), according to manufacturer's instructions. After washing, cells were incubated with Alexa Fluor 488 chicken antimouse secondary antibodies (Molecular Probes) for 30 min at 4°C. Total Bax levels were determined using polyclonal anti‐Bax antibodies (Cell Signaling Technology) and Alexa Fluor 488 chicken antirabbit secondary antibodies (Molecular Probes).
Real‐time quantitative PCR. RNA was prepared from cells using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and first‐strand cDNA was generated using random hexamers (SuperScript III First‐Strand Synthesis SuperMix; Invitrogen) from 1 µg total RNA. The mRNA expression levels of p21, Bcl‐2, survivin, and 18S were quantified using TaqMan gene expression assays (p21, Hs99999142_m1; Bcl‐2, Hs00608023_m1; survivin, Hs00153353_m1; 18S, Hs99999901_s1; Applied Biosystems, Foster City, CA, USA) on an ABI Prism 7000 Sequence Detection System (Applied Biosystems).
RT‐PCR and DNA sequencing of p53. Total RNA was extracted from bone marrow mononuclear cells with the RNeasy Mini Kit. First‐strand cDNA synthesis was carried out with oligo(dT) as primer (Superscript II System; Invitrogen). PCR for p53 gene expression followed by direct sequencing was carried out as described previously.( 16 )
Statistical analysis. The statistical analysis was carried out using the two‐tailed Student t‐test. Statistical significance was considered when P < 0.05. Average values were expressed as mean ± SD. Synergism, additive effects, and antagonism were assessed using the Chou–Talalay method and Calcusyn software (Biosoft, Ferguson, MO, USA).( 16 ) The extent of apoptosis was quantified as the percentage of Annexin V‐positive cells, and the extent of drug‐specific apoptosis was assessed by the formula: % specific apoptosis = (test – control) × 100/(100 – control). In the formula, the numerator is the actual amount of killing that occurred and the denominator is the potential amount of killing that could occur. The combination index (CI), a numerical description of combination effects, was calculated using the more stringent statistical assumption of mutually nonexclusive modes of action. The averaged CI values were calculated from the values for effective dose (ED)50, ED75, ED90, and ED95. When CI = 1, this equation represents the conservation isobologram and indicates additive effects. CI values less than 1.0 indicate a more than expected additive effect (synergism), whereas CI values greater than 1.0 indicate antagonism between the two drugs.
Results
CDK1 inhibition by RO‐3306 induces cell cycle arrest and apoptosis in AML cell lines. We first examined the effect of the CDK1 inhibitor RO‐3306 on the growth and viability of cultured AML cell lines. As shown in Figure 1(a), RO‐3306 treatment of cells resulted in a dose‐dependent accumulation of cells with 4 N and <2 N sub‐G1 DNA content, suggesting induction of G2/M‐phase cell cycle arrest and apoptosis. This effect was seen in both p53 wild‐type OCI‐AML‐3 and MOLM‐13 cells and p53‐deleted HL‐60 cells, implying that RO‐3306 would not primarily require functional p53 to induce cell cycle arrest and apoptosis. To further investigate if apoptosis induced by CDK1 inhibition is independent of p53 activation, OCI‐AML‐3 cells stably infected with retroviruses encoding either scrambled or p53‐specific siRNA were exposed to 5 µM RO‐3306 for 24 h. Knockdown of p53 in OCI‐AML3 cells did not significantly affect their susceptibility to apoptosis induced by RO‐3306 (27.1 ± 2.9 vs 25.8 ± 3.4% specific Annexin V‐positive cells), suggesting that p53 activity is dispensable for apoptosis induced by CDK1 inhibition. Similarly, p53 status did not significantly affect cell cycle arrest induced by RO‐3306 (72.1 ± 4.1 vs 76.6 ± 3.2% G2/M‐phase cells). These findings are consistent with previously published data in solid tumor cells.( 9 )
Figure 1.

Cyclin‐dependent kinase (CDK) 1 inhibition induces cell cycle arrest and apoptosis in a dose‐ and time‐dependent manner in AML cells. (a) AML cell lines with wild‐type p53 (OCI‐AML‐3 and MOLM‐13) or deleted p53 (HL‐60) were incubated with the indicated concentrations of RO‐3306 for 24 h and stained for DNA content. Treatment of cells with RO‐3306 induced G2/M‐phase cell cycle arrest and apoptosis in a dose‐dependent manner, as evidenced by increased 4 N and <2 N DNA content. (b) Time course of effects induced in MOLM‐13 cells by 5 µM RO‐3306 on cell cycle and apoptosis. The Annexin V‐positive fractions in correlation with DNA content were measured by flow cytometry. Data were gated on the FL2‐area versus FL2‐width cytogram to exclude doublets. Cells were gated on dot plots (middle row), based on Annexin staining. The Annexin V‐positive fraction (AnV+) corresponds to apoptotic cells, whereas the Annexin V‐negative fraction (AnV−) corresponds to viable cells. Histograms indicate the distribution of DNA content in the gated populations (AnV+, upper row; AnV−, lower row) of the preceding dot plots. The percentages of sub‐G1 cells in the total cells accounted for <1.5% in the first 8 h, indicating minimal DNA fragmentation. At 16 h after exposure, the sub‐G1 DNA content increased modestly to 4%. RO‐3306 caused G2/M cell cycle arrest in the AnV− fraction in a time‐dependent manner. In contrast, the cell cycle effect was not apparent in the AnV+ fraction in the first 8 h, suggesting that apoptosis induction by RO‐3306 does not necessarily require cell cycle arrest. Cell cycle distribution was analyzed using ModFit LT software. Results are representative of three independent experiments. (c) Apoptosis induction by RO‐3306 does not require G2/M cell cycle arrest. Four hours after release of HL‐60 cells from G2/M synchronization by removal of nocodazole, cells were incubated in the absence or presence of 5 µM RO‐3306 for 8 h. The Annexin V‐positive fractions in correlation with DNA content were measured by flow cytometry. Cell cycle distribution was analyzed using ModFit LT software. Results are expressed as mean ± SD. *Significance at P < 0.01.
We next asked whether prolonged cell cycle arrest initiates apoptosis in cells treated with RO‐3306. RO‐3306 at a consistent concentration (5 µM) induced G2/M‐phase cell cycle arrest (Fig. 1b, lower row) and apoptosis (Fig. 1b, upper row) of AML cells in a time‐dependent manner. Phosphatidylserine externalization was detectable as early as 4 h after exposure to RO‐3306. Although it was anticipated that apoptosis induction secondary to G2/M‐phase cell cycle arrest would accumulate apoptotic cells exclusively into a 4 N DNA content, the cell cycle distribution changes of apoptotic cells were similar to those of viable cells. Because apoptosis induction was not restricted to G2/M‐phase cells, apoptosis induction by RO‐3306 may not necessarily require cell cycle arrest. To examine this issue further, HL‐60 cells were synchronized in G2/M phase by treatment with nocodazole (200 ng/mL) for 16 h, resulting in cell accumulation (74.1 ± 4.8%) in a G2/M state with a 4 N DNA content. OCI‐AML‐3 and MOLM‐13 cells were susceptible to nocodazole treatment, resulting in cell death in the majority of cells. Four hours after release of the cells from G2/M synchronization by removal of nocodazole, cells were exposed to 5 µM RO‐3306 for 8 h. RO‐3306 treatment significantly increased the percentage of Annexin V‐positive cells in G1‐phase cells without affecting the cell cycle distribution (P < 0.01, Fig. 1c), indicating that cell cycle arrest would be dispensable for apoptosis induced by CDK1 inhibition.
RO‐3306 enhances p53‐mediated apoptosis. To clarify if RO‐3306 enhances p53‐mediated apoptosis, we next combined RO‐3306 and the MDM2 antagonist Nutlin‐3. OCI‐AML‐3 and MOLM‐13 cells have wild‐type p53 and HL‐60 cells have deleted p53. We have shown that Nutlin‐3 treatment induces apoptosis in OCI‐AML‐3 and MOLM‐13 cells, but not in HL‐60 cells.( 16 ) As shown in Figure 2(a), RO‐3306 augmented Nutlin‐induced phosphatidylserine externalization in wild‐type p53 cells. The calculated CI values were 0.85 for ED50, 0.76 for ED75, 0.69 for ED90, and 0.66 for ED95 in OCI‐AML‐3 cells, and 0.63, 0.49, 0.38, and 0.32, respectively, in MOLM‐13 cells (Fig. 2b). The averaged CI values were 0.74 in OCI‐AML‐3 cells and 0.45 in MOLM‐13 cells, suggesting that the combination is additive or slightly synergistic to induce apoptosis. A similar potentiating effect was not seen in p53‐deleted, Nutlin‐resistant HL‐60 cells (Fig. 2a). These findings suggest that RO‐3306 enhances p53‐mediated apoptosis.
Figure 2.
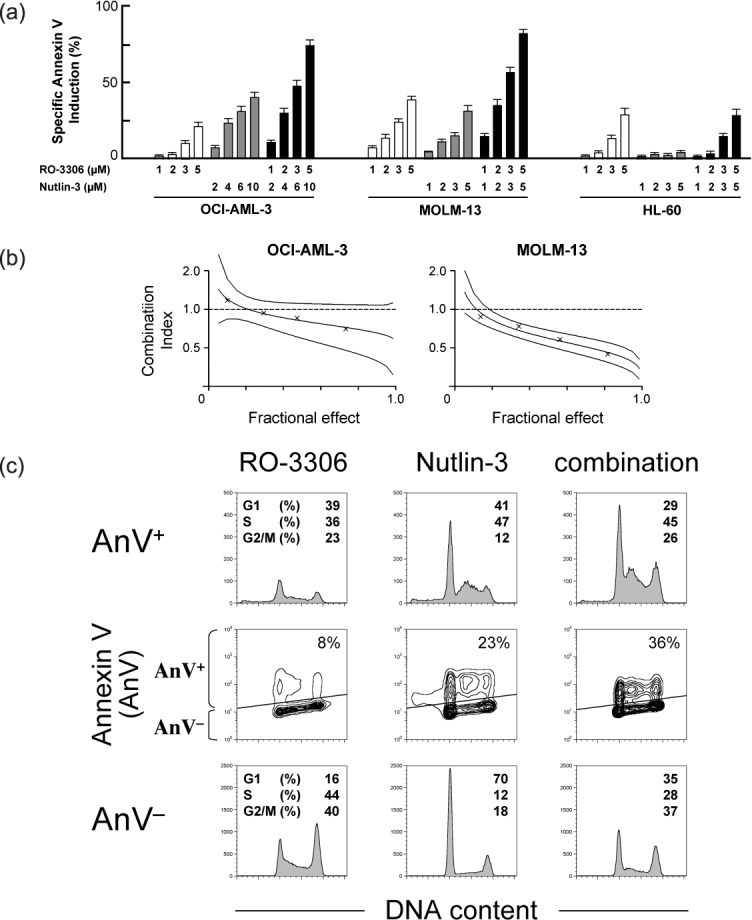
Cyclin‐dependent kinase (CDK) 1 enhances p53‐mediated apoptosis in AML cells. (a) AML cells were incubated with the indicated concentrations of RO‐3306 or Nutlin‐3 for 18 h, and the Annexin V‐positive fractions were measured by flow cytometry. Results are expressed as mean ± SD. RO‐3306 enhanced Nutlin‐induced apoptosis in p53 wild‐type OCI‐AML‐3 and MOLM‐13 cells but not in mutant p53 HL‐60 cells. (b) Combination index plots as mean values ± 2SD generated using CalcuSyn software (crosses represent actual data points for the RO‐3306 and Nutlin‐3 combination). Combination index values of less than 1.0 indicate synergism. (c) Cell cycle‐independent synergistic apoptosis induction by the RO‐3306 and Nutlin‐3 combination. MOLM‐13 cells were treated for 8 h with 5 µM RO‐3306 or 2.5 µM Nutlin‐3. The Annexin V‐positive fractions in correlation with DNA content were measured by flow cytometry. Data were gated on the FL2‐area versus FL2‐width cytogram to exclude doublets. Cells were gated on dot plots (middle row), based on Annexin staining. The Annexin‐V‐positive fraction (AnV+) corresponds to apoptotic cells, whereas the Annexin‐V‐negative fraction (AnV−) corresponds to viable cells. Histograms indicate the distribution of DNA content in the gated populations (AnV+, upper row; AnV−, lower row) of the preceding dot plots.
As shown in Figure 2(c), induction of apoptosis by the RO‐3306 and Nutlin‐3 combination was not associated with a cell cycle‐specific mechanism. p53‐mediated apoptotic pathways converge upon interaction between antiapoptotic and proapoptotic Bcl‐2 family proteins, resulting in activation and conformational change of the proapoptotic Bax. The degree of Bax conformational change was analyzed by means of an antibody directed against the NH2‐terminal region of Bax (clone YTH‐6A7). The epitope‐specific antibody can react only with Bax in the active conformation because the NH2‐terminal region is occluded in unstressed intact cells. AML cells were preincubated for 1 h in the presence of 50 µM Z‐VAD‐FMK before the addition of RO‐3306, Nutlin‐3, or both to inhibit caspase activation‐mediated Bax cleavage. As shown in Figure 3(a), few control cells were stained with this antibody, and 8‐h treatment with RO‐3306 induced a small degree of conformational change of Bax. Nutlin‐3 treatment also induced Bax activation in p53 wild‐type OCI‐AML‐3 and MOLM‐13 cells but not in p53‐deleted, Nutlin‐resistant HL‐60 cells. Interestingly, RO‐3306 enhanced Bax conformational changes by Nutlin‐3 in p53 wild‐type cells. As reported previously,( 18 ) no differences in the fluorescence pattern between control and drug‐treated cells were observed when Bax antibodies that detect total Bax were used (not shown). Nutlin‐3 rapidly accumulates p53 (~1 h) that can directly activate Bax, and our results suggest that CDK1 inhibition actively enhances the p53‐mediated mitochondrial apoptotic pathway. As a consequence of Bax conformational change, RO‐3306 enhanced p53‐mediated mitochondrial membrane permeabilization in p53 wild‐type AML cells (Fig. 3b).
Figure 3.
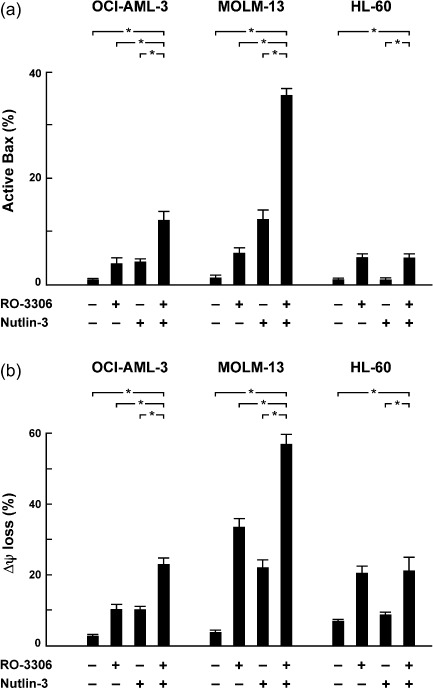
RO‐3306 cooperated with Nutlin‐3 in activating Bax and inducing mitochondrial apoptosis. AML cells were treated for 8 h with 5 µM RO‐3306 or 2.5 µM Nutlin‐3 (5 µM in OCI‐AML‐3). (a) The percentages of Bax in its active conformation were determined by staining with the active conformation‐specific anti‐Bax antibody YTH‐6A7 (shaded histogram) or a corresponding isotype control (open histogram). To block caspase activation‐mediated conformational change of Bax, cells were preincubated for 1 h with 50 µM Z‐VAD‐FMK. (b) Δψm was assessed by flow cytometry. The results are expressed as the mean ± SD of triplicate measurements. *Significance at P < 0.01.
RO‐3306 downregulates antiapoptotic p21, Bcl‐2, and survivin protein expression in AML. To investigate the molecular events that contribute to apoptosis induced by RO‐3306 and Nutlin‐3 treatment, MOLM‐13 cells were treated with 5 µM RO‐3306 or 2.5 µM Nutlin‐3, either as individual agents or in combination, and p53‐related protein levels were investigated (Fig. 4a). RO‐3306 treatment did not significantly alter basal and Nutlin‐induced p53 levels, but it blocked p53‐mediated induction of MDM2 and p21. Although RO‐3306 or Nutlin‐3 as individual agents modestly reduced Bcl‐2 levels, the reduction was prominent when cells were simultaneously treated with RO‐3306 and Nutlin‐3, resulting in a >75% decrease in the Bcl‐2/Bax ratio at 6 h. The RO‐3306 and Nutlin‐3 combination also reduced levels of survivin. We found that survivin protein diminished within 2 h of RO‐3306 treatment, preceding the obvious onset of cell cycle arrest. The levels of other p53‐related proteins did not change significantly. p53‐related protein expression after RO‐3306 and Nutlin‐3 treatment was also investigated in OCI‐AML‐3 and HL‐60 cells. Downregulation of p21, MDM2, Bcl‐2, and survivin protein expression by RO‐3306 was found in OCI‐AML‐3 (Fig. 4b). RO‐3306 treatment reduced Bcl‐2 and survivin levels in p53‐deleted HL‐60 cells, implying that regulation of Bcl‐2 and survivin expression by RO‐3306 is p53‐independent. Expression of p21 and MDM2 was poorly detected in HL‐60 cells.
Figure 4.
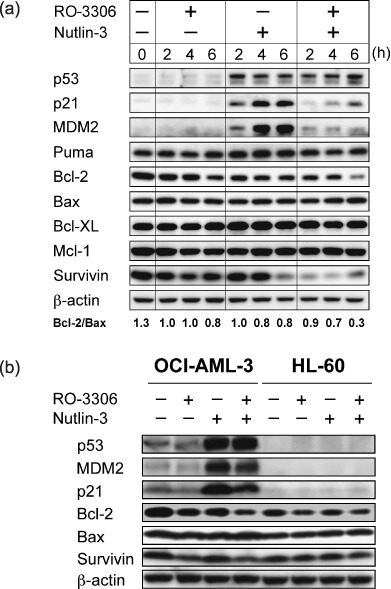
RO‐3306 downregulates the expression of antiapoptotic Bcl‐2 and survivin and blocks p53‐mediated induction of murine double minute 2 homolog (MDM2) and p21. (a) Protein expression in MOLM‐13 cells, which were treated for up to 6 h with 5 µM RO‐3306 or 2.5 µM Nutlin‐3 either as individual agents or in combination. β‐Actin was used to confirm equal loading of proteins. (b) Protein expression in OCI‐AML‐3 and HL‐60 cells, which were treated for 6 h with RO‐3306 (5 µM) or Nutlin‐3 (5 µM in OCI‐AML‐3 and 2.5 µM in HL‐60) either as individual agents or in combination. β‐Actin was used to confirm equal loading of proteins. Results are representative of three independent experiments.
RO‐3306 inhibits p53‐induced p21 synthesis. It has been suggested that p21 not only mediates cell cycle arrest but also prevents apoptosis.( 19 ) To monitor the fate of previously stabilized pools of p21, MOLM‐13 cells were preincubated for 1 h with 70 µM cycloheximide prior to addition of 5 µM RO‐3306. Cycloheximide did not affect the rate of decrease in p21 levels after RO‐3306 treatment (Fig. 5a), suggesting that RO‐3306 reduces p21 levels via blockage of new protein synthesis. We next investigated if the proteasome inhibitor MG132 prevents the reduction of p21 levels. After 1 h preincubation with 0.2 µM MG132, MOLM‐13 cells were treated with 5 µM RO‐3306. MG132 did not block p21 reduction after RO‐3306 treatment (Fig. 5b), indicating that the mechanism of p21 decrease is independent of ubiquitination. We treated MOLM‐13 cells with RO‐3306 and/or Nutlin‐3 for 4 h and monitored the expression of p21 mRNA by real‐time PCR. RO‐3306 transiently reduced basal p21 mRNA levels at 1 h (Fig. 5c), suggesting that CDK1 inhibition may block transcription and that there may be compensatory mechanisms for the blockade. RO‐3306 treatment significantly inhibited Nutlin‐induced transcriptional activation of p21, suggesting that CDK1 inhibition blocks the transcriptional activity of p53. We also monitored Bcl‐2 and survivin mRNA levels. As shown in Figure 5(d), RO‐3306 significantly repressed Bcl‐2 transcription but it did not affect survivin mRNA levels. Finally, CDK1 levels were reduced in HCT116 colon cancer cells using siRNA. The gene silencing was unsuccessful in MOLM‐13 cells because of the low transfection efficiency. HCT116 cells have been shown to have intact p53–p21 signaling.( 15 ) CDK1 siRNA led to a significant inhibition of CDK1 expression relative to cells transfected with control siRNA, and did not interfere with β‐actin synthesis (Fig. 5e). CDK1 siRNA led to reduced basal and p53‐induced p21 protein expression, whereas it did not affect Bcl‐2 and survivin levels (Fig. 5e). At the mRNA level, CDK1 siRNA blocked Nutlin‐induced transcriptional activation of p21 (Fig. 5f) but it did not reduce Bcl‐2 or survivin mRNA levels (not shown). As Bcl‐2 and survivin levels in HCT116 cells were not significantly affected by a 6‐h treatment with 5 µM RO‐3306 (not shown), one explanation could be that associations of CDK1 with Bcl‐2 or survivin differ in each cancer cell type. Taken together, our findings suggest that RO‐3306 can inhibit p21 and Bcl‐2 synthesis at the transcriptional level in AML cells.
Figure 5.
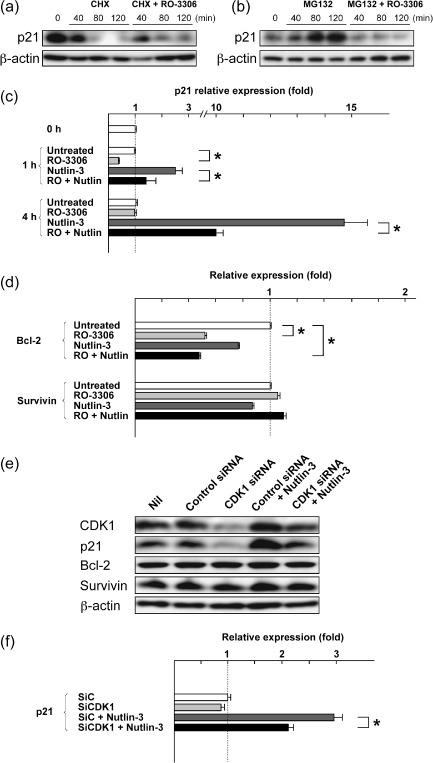
Cyclin‐dependent kinase (CDK) 1 inhibition prevents p53‐induced p21 synthesis. (a,b) MOLM‐13 cells were preincubated for 1 h with (a) 70 µM cycloheximide or (b) 0.2 µM MG132, and p21 levels after the addition of 5 µM RO‐3306 were monitored. Results are representative of three independent experiments. β‐Actin was used to confirm equal loading of proteins. (c) MOLM‐13 cells were treated with 5 µM RO‐3306 or 5 µM Nutlin‐3 for 4 h, either as individual agents or in combination, and p21 transcripts were quantitated by real‐time RT‐PCR. Each real‐time PCR was carried out in triplicate, and the average fold induction relative to time 0 is shown with SD. *Significance at P < 0.05. (d) MOLM‐13 cells were treated with 5 µM RO‐3306 or 5 µM Nutlin‐3 for 4 h, either as individual agents or in combination, and Bcl‐2 and survivin transcripts were quantitated by real‐time RT‐PCR. Each real‐time PCR was carried out in triplicate, and the average fold induction relative to untreated cells is shown with SD. *Significance at P < 0.05. (e) Western blot analysis of HCT116 cells transfected with either control or CDK1 siRNA. CDK1 siRNA led to reduced basal and p53‐induced p21 expression. (f) HCT116 cells were transfected with either control (SiC) or CDK1 siRNA (SiCDK1), and were incubated for 4 h in the absence or presence of 10 µM Nutlin‐3. p21 transcripts were quantitated by real‐time RT‐PCR. Each real‐time PCR was carried out in triplicate, and the average fold induction relative to untreated cells is shown with SD. *Significance at P < 0.05.
RO‐3306 does not inhibit RNA polymerase II C‐terminal domain phosphorylation. The transcriptional proteins CDK7 and CDK9 phosphorylate the C‐terminal domain (CTD) of RNA polymerase II, facilitating the initiation of transcription and efficient elongation.( 6 ) Because flavopiridol and roscovitine (CYC202) have been reported to block CDK7 and CDK9 in addition to CDK1/2,( 20 , 21 ) we investigated if RO‐3306 could affect RNA polymerase II CTD phosphorylation. As shown in Figure 6(a), RO‐3306 treatment did not cause dephosphorylation of RNA polymerase II CTD at Ser2 or Ser5. It is therefore unlikely that RO‐3306 affects the transcriptional activities of CDK7 or CDK9.
Figure 6.
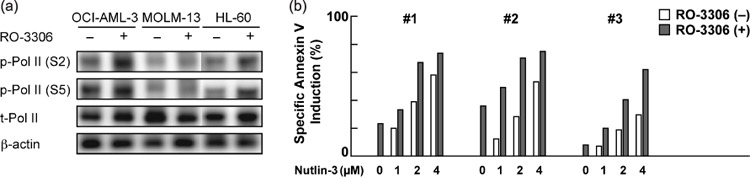
(a) RO‐3306 does not affect the transcriptional activities of cyclin‐dependent kinase (CDK) 7 or CDK9. The phosphorylation status of the RNA polymerase II C‐terminal domain at Ser2 or Ser5 along with total RNA polymerase II expression were determined in AML cells, which were treated for 6 h with RO‐3306 (5 µM). β‐Actin was used to confirm equal loading of proteins. The results are representative of three independent experiments. (b) Nutlin‐induced apoptosis is enhanced by combination with RO‐3306 in primary AML cells. Primary AML cells from three AML patients were incubated for 24 h with 5 µM RO‐3306 or the indicated concentrations of Nutlin‐3, either as individual agents or in combination, and the Annexin V‐positive fractions were measured by flow cytometry.
RO‐3306 augments p53‐mediated apoptosis in primary AML cells. We cultured primary cells from three AML patients with more than 70% blasts with 5 µM RO‐3306 and/or a range of concentrations of Nutlin‐3, and evaluated apoptosis after 24 h. TP53 mutation was not detected in these cases. The cooperative nature of the RO‐3306–Nutlin‐3 interaction was seen in all samples (Fig. 6b).
Combination of RO‐3306 and the DNA damaging agent doxorubicin has additive apoptotic effects in p53 wild‐type AML cells. Doxorubicin is one of the most active chemotherapeutic agents for the therapy of AML and it remains the backbone of induction and consolidation regimens. We investigated if CDK1 inhibition potentiates the apoptotic effect of doxorubicin, a DNA damaging agent that activates p53. The results indicated an additive interaction of RO‐3306 and doxorubicin in induction of apoptosis in OCI‐AML‐3 and MOLM‐13 cells (Table 1). RO‐3306 and doxorubicin showed an antagonistic effect on apoptosis induction in HL‐60 cells. Because doxorubicin has p53‐independent mechanisms to induce apoptosis, it is possible that p53‐independent apoptotic mechanisms of doxorubicin could antagonize RO‐3306 to induce apoptosis.
Table 1.
Combination index (CI) values for the apoptotic effects of RO‐3306 and doxorubicin
| Cell line | ED50 | ED75 | ED90 | Averaged CI † |
|---|---|---|---|---|
| OCI‐AML‐3 | 1.1 | 1.1 | 1.2 | 1.1 |
| MOLM‐13 | 1.0 | 1.0 | 1.0 | 1.0 |
| HL‐60 | 1.6 | 1.5 | 1.5 | 1.5 |
The averaged CI values were calculated from the ED50, ED75, and ED90.
Discussion
The potential therapeutic efficacy of non‐genotoxic p53 activation by a small‐molecule antagonist of MDM2 has been reported in hematological malignancies.( 22 , 23 , 24 , 25 ) The advantage of MDM2 inhibition as an anticancer modality is supported by the finding that transient induction of the p53 response by disruption of the MDM2–p53 interaction in normal somatic cells results in reversible growth arrest but not apoptosis.( 26 ) Furthermore, treatment of nude mice with MDM2 inhibitors at therapeutic doses is well tolerated without significant adverse side effects.( 15 , 26 ) It has been reported that the sensitivity of cells to MDM2 inhibitors is significantly dependent on functional p53 and high MDM2 basal expression.( 16 , 26 ) Considering the rare incidence of TP53 mutations, frequent MDM2 overexpression, and intact downstream p53 signaling toward apoptosis prevalent in leukemias, targeting the MDM2–p53 interaction in these tumors has been recognized as a promising therapeutic strategy. As many signaling pathway components are frequently affected in AML, however, other effective targeted therapies need to be developed that synergize with p53‐mediated apoptosis.( 11 , 12 , 13 , 14 ) We found that CDK1 inhibition by RO‐3306 actively enhances downstream p53 signaling and that a combination strategy aimed at inhibiting CDK1 as well as activating p53 signaling deserves further investigation in AML.
Until recently, CDK1 has not received attention as a cancer therapy target because it has been assumed that tampering with non‐redundant CDK1 would result in intolerable toxic effects. However, multi‐CDK inhibitors that also target CDK1 have been found to be well tolerated in cancer patients.( 27 , 28 , 29 ) The high homology of CDK1 with CDK2 and the intense search for CDK2 inhibitors in recent years has resulted in a number of small molecules that inhibit both CDK2 and CDK1.( 6 , 7 ) Furthermore, CDK1 inhibition has been reported to be more proapoptotic in cancer cells than normal cells.( 9 , 30 ) Vassilev et al. have reported that CDK1 inhibition for 72 h is well tolerated in immortalized non‐tumorigenic epithelial cell lines, but inhibition for more than 24 h in colon cancer cell lines HCT116 and SW480 induces significant apoptosis.( 9 ) We found that AML cells are susceptible to RO‐3306‐induced apoptosis. Phosphatidylserine externalization was detectable as early as 4 h after exposure and, more importantly, apoptosis induction appeared to occur in a cell cycle‐independent manner. Induction of apoptosis unrelated to cell cycle arrest has also been reported in tumor cell lines treated with flavopiridol, a small molecule inhibitor of CDK1, CDK2, CDK4 and CDK7.( 31 ) In accordance with our findings, other studies have reported that CDK1 inhibition induces apoptosis that is independent of cell cycle regulation.( 32 , 33 )
The molecular mechanisms of apoptosis induced by CDK1 inhibition remain largely unclear, though recent studies have shown that CDK1 can protect cells from apoptosis by stabilization of mitochondria,( 3 ) inhibition of the forkhead transcription factor FOXO1,( 34 , 35 ) or by blocking initiator protease caspase‐9.( 36 ) It has been reported that Bax activation participates in apoptosis induction by roscovitine (CYC202).( 37 ) We found that the cooperative apoptotic induction between RO‐3306 and Nutlin‐3 was associated with both Bax activation and mitochondrial outer membrane permeabilization, probably resulting from disruption of the balance of antiapoptotic and proapoptotic Bcl‐2 family proteins (decreased Bcl‐2/Bax ratios), as well as with downregulation of p21 and survivin. Bcl‐2 and survivin are targets of p53 repression,( 38 , 39 ) and survivin is also a substrate of CDK1.( 40 ) Our data suggest that RO‐3306 reduces p21 and Bcl‐2 levels at least partially by transcriptional repression. On the other hand, survivin mRNA levels were not affected by RO‐3306 treatment, suggesting a transcription‐independent mechanism in survivin downregulation. Survivin expression levels have been reported to fluctuate through the cell cycle,( 41 ) raising the possibility that the changes in survivin level could be indicative of its narrow expression around M phase, reflecting cell cycle modulation by RO‐3306. However, the immediate (≤2 h) decrease in survivin after the RO‐3306 and Nutlin‐3 combination makes the possibility unlikely. Further studies are required to clarify the mechanism by which RO‐3306 downregulates survivin. It has been reported that the CDK inhibitor p21 protects cells from p53‐mediated apoptosis.( 19 , 42 , 43 , 44 , 45 ) Direct mechanisms by which p21 inhibits cell death include inhibition of initiator caspase‐8 cleavage, interaction with procaspase‐3 masking the serine proteinase‐cleaving site, and stabilization of cellular inhibitor of apotosis protein 1 (c‐IAP1). We suggest that CDK1 inhibition augments p53‐mediated apoptosis partially by repressing p21 transcription. Although some CDK inhibitors have been reported to inhibit the RNA transcription regulators CDK7 and CDK9,( 20 , 21 ) transcriptional repression after RO‐3306 treatment appears to be associated with CDK1 inhibition but not with CDK7 or CDK9 inhibition. Therefore it remains unknown how RO‐3306 could repress p21 and Bcl‐2 transcription.
In conclusion, the CDK1 inhibitor RO‐3306 downregulates expression of Bcl‐2 and survivin as well as blocks p53‐mediated induction of p21, thereby enhancing p53‐mediated apoptosis. Our data support a mechanistic rationale by which inhibition of CDK1 can augment p53‐mediated apoptosis in AML.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health (AML‐PO1) CA‐55164, CA‐49639, and CA‐89346 (to M. Andreeff) and by the Yasuda Medical Foundation and Japan Leukaemia Research Fund (K. Kojima). The authors acknowledge Dr Maria Soengas of the Department of Dermatology, University of Michigan, Ann Arbor, MI, USA, for kindly providing retrovirus encoding p53‐specific shRNA.
References
- 1. Malumbres M, Barbacid M. Mammalian cyclin‐dependent kinases. Trends Biochem Sci 2005; 30: 630–41. [DOI] [PubMed] [Google Scholar]
- 2. Santamaría D, Barrière C, Cerqueira A et al . Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007; 448: 811–15. [DOI] [PubMed] [Google Scholar]
- 3. Shapiro GI. Cyclin‐dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24: 1770–83. [DOI] [PubMed] [Google Scholar]
- 4. De Cárcer G, De Castro IP, Malumbres M. Targeting cell cycle kinases for cancer therapy. Curr Med Chem 2007; 14: 969–85. [DOI] [PubMed] [Google Scholar]
- 5. Aleem E, Kiyokawa H, Kaldis P. Cdc2‐cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol 2005; 7: 831–6. [DOI] [PubMed] [Google Scholar]
- 6. Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over‐expressing MYC. Nat Med 2007; 13: 820–7. [DOI] [PubMed] [Google Scholar]
- 7. Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res 2003; 63: 230–5. [PubMed] [Google Scholar]
- 8. Pennati M, Campbell AJ, Curto M et al . Potentiation of paclitaxel‐induced apoptosis by the novel cyclin‐dependent kinase inhibitor NU6140: a possible role for survivin down‐regulation. Mol Cancer Ther 2005; 4: 1328–37. [DOI] [PubMed] [Google Scholar]
- 9. Vassilev LT, Tovar C, Chen S et al . Selective small‐molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA 2006; 103: 10 660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faderl S, Kantarjian HM, Estey E et al . The prognostic significance of p16 (INK4a)/p14 (ARF) locus deletion and MDM‐2 protein expression in adult acute myelogenous leukemia. Cancer 2000; 89: 1976–82. [DOI] [PubMed] [Google Scholar]
- 11. Kornblau SM, Womble M, Qiu YH et al . Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006; 108: 2358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong S, McLaughlin J, Cheng D, Zhang C, Shokat KM, Witte ON. Sole BCR‐ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations. Proc Natl Acad Sci USA 2004; 101: 17 456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kojima K, Konopleva M, Samudio IJ, Ruvolo V, Andreeff M. MEK inhibition enhances nuclear proapoptotic function of p53 in AML cells. Cancer Res 2007; 67: 3210–19. [DOI] [PubMed] [Google Scholar]
- 14. Secchiero P, Zerbinati C, Di Iasio MG et al . Synergistic cytotoxic activity of recombinant TRAIL plus the non‐genotoxic activator of the p53 pathway nutlin‐3 in acute myeloid leukemia cells. Curr Drug Metab 2007; 8: 395–403. [DOI] [PubMed] [Google Scholar]
- 15. Vassilev LT, Vu BT, Graves B et al . In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science 2004; 303: 844–8. [DOI] [PubMed] [Google Scholar]
- 16. Kojima K, Konopleva M, Samudio IJ et al . MDM2 antagonists induce p53‐dependent apoptosis in AML: implications for leukemia therapy. Blood 2005; 106: 3150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kojima K, Shimanuki M, Shikami M et al . The dual PI3 kinase/mTOR inhibitor PI‐103 prevents p53 induction by Mdm2 inhibition but enhances p53‐mediated mitochondrial apoptosis in p53 wild‐type AML. Leukemia 2008; 22: 1728–36. [DOI] [PubMed] [Google Scholar]
- 18. Kojima K, Konopleva M, Tsao T, Nakakuma H, Andreeff M. Concomitant inhibition of Mdm2‐p53 interaction and Aurora kinases activates the p53‐dependent postmitotic checkpoints and synergistically induces p53‐mediated mitochondrial apoptosis along with reduced endoreduplication in AML. Blood 2008; 112: 2886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gartel AL, Tyner AL. The role of the cyclin‐dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1: 639–49. [PubMed] [Google Scholar]
- 20. Chen R, Keating MJ, Gandhi V, Plunkett W. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood 2005; 106: 2513–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alvi AJ, Austen B, Weston VJ et al . A novel CDK inhibitor, CYC202 (R‐roscovitine), overcomes the defect in p53‐dependent apoptosis in B‐CLL by down‐regulation of genes involved in transcription regulation and survival. Blood 2005; 105: 4484–91. [DOI] [PubMed] [Google Scholar]
- 22. Saddler C, Ouillette P, Kujawski L et al . Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood 2008; 111: 1584–93. [DOI] [PubMed] [Google Scholar]
- 23. Secchiero P, Barbarotto E, Tiribelli M et al . Functional integrity of the p53‐mediated apoptotic pathway induced by the nongenotoxic agent nutlin‐3 in B‐cell chronic lymphocytic leukemia (B‐CLL). Blood 2006; 107: 4122–9. [DOI] [PubMed] [Google Scholar]
- 24. Coll‐Mulet L, Iglesias‐Serret D, Santidrián AF et al . MDM2 antagonists activate p53 and synergize with genotoxic drugs in B‐cell chronic lymphocytic leukemia cells. Blood 2006; 107: 4109–14. [DOI] [PubMed] [Google Scholar]
- 25. Stühmer T, Chatterjee M, Hildebrandt M et al . Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood 2005; 106: 3609–17. [DOI] [PubMed] [Google Scholar]
- 26. Shangary S, Qin D, McEachern D et al . Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA 2008; 105: 3933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Byrd JC, Lin TS, Dalton JT et al . Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high‐risk chronic lymphocytic leukemia. Blood 2007; 109: 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tibes R, Jimeno A, Von Hoff DD et al . Phase I dose escalation study of the oral multi‐CDK inhibitor PHA‐848125. J Clin Oncol 2008; 26: 160s. [Google Scholar]
- 29. Mahadevan D, Plummer R, Squires MS et al . A dose escalation, pharmacokinetic, and pharmacodynamic study of AT7519, a cyclin‐dependent kinase inhibitor in patients with refractory solid tumors. J Clin Oncol 2008; 26: 161s. [DOI] [PubMed] [Google Scholar]
- 30. Bayart E, Grigorieva O, Leibovitch S, Onclercq‐Delic R, Amor‐Guéret M. A major role for mitotic CDC2 kinase inactivation in the establishment of the mitotic DNA damage checkpoint. Cancer Res 2004; 64: 8954–9. [DOI] [PubMed] [Google Scholar]
- 31. Mayer F, Mueller S, Malenke E, Kuczyk M, Hartmann JT, Bokemeyer C. Induction of apoptosis by flavopiridol unrelated to cell cycle arrest in germ cell tumour derived cell lines. Invest New Drugs 2005; 23: 205–11. [DOI] [PubMed] [Google Scholar]
- 32. Castedo M, Perfettini J‐L, Roumier T et al . Cyclin‐dependent kinase‐1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ 2002; 9: 1287–93. [DOI] [PubMed] [Google Scholar]
- 33. Golsteyn RM. Cdk1 and Cdk2 complexes (cyclin dependent kinases) in apoptosis: a role beyond the cell cycle. Cancer Lett 2005; 217: 129–38. [DOI] [PubMed] [Google Scholar]
- 34. Yuan Z, Becker EB, Merlo P et al . Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science 2008; 319: 1665–8. [DOI] [PubMed] [Google Scholar]
- 35. Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 2008; 27: 4733–44. [DOI] [PubMed] [Google Scholar]
- 36. Allan LA, Clarke PR. Phosphorylation of caspase‐9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol Cell 2007; 26: 301–10. [DOI] [PubMed] [Google Scholar]
- 37. Hahntow IN, Schneller F, Oelsner M et al . Cyclin‐dependent kinase inhibitor Roscovitine induces apoptosis in chronic lymphocytic leukemia cells. Leukemia 2004; 18: 747–55. [DOI] [PubMed] [Google Scholar]
- 38. Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM. Negative regulation of bcl‐2 expression by p53 in hematopoietic cells. Oncogene 2001; 20: 240–51. [DOI] [PubMed] [Google Scholar]
- 39. Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti‐apoptotic survivin gene by wild type p53. J Biol Chem 2002; 277: 3247–57. [DOI] [PubMed] [Google Scholar]
- 40. O’Connor DS, Grossman D, Plescia J et al . Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci USA 2000; 97: 13 103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin‐proteasome pathway regulates survivin degradation in a cell cycle‐dependent manner. J Cell Sci 2000; 113: 4363–71. [DOI] [PubMed] [Google Scholar]
- 42. De la Cueva E, García‐Cao I, Herranz M et al . Tumorigenic activity of p21Waf1/Cip1 in thymic lymphoma. Oncogene 2006; 25: 4128–32. [DOI] [PubMed] [Google Scholar]
- 43. Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and Bcl‐2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle 2006; 5: 2778–86. [DOI] [PubMed] [Google Scholar]
- 44. Xu SQ, El‐Deiry WS. p21WAF1/CIP1 inhibits initiator caspase cleavage by TRAIL death receptor DR4. Biochem Biophys Res Commun 2000; 269: 179–90. [DOI] [PubMed] [Google Scholar]
- 45. Steinman RA, Johnson DE. p21WAF1 prevents down‐modulation of the apoptotic inhibitor protein c‐IAP1 and inhibits leukemic apoptosis. Mol Med 2000; 6: 736–49. [PMC free article] [PubMed] [Google Scholar]