

Abstract
We comprehensively studied the cellular immune response during acute human hepatitis C virus (HCV) infection by monthly prospective sampling of persons with high risk of infection. In 19 of 23 subjects, interferon-gamma secreting T cells specific for one or more peptides spanning the entire HCV polyprotein were detected 1–3 months after infection. The median time to development of interferon gamma responses to HCV peptides was 33 days (range 29 to 50 days), and these responses peaked between 180 and 360 days. Nineteen subjects had sufficient follow-up to determine outcome, with 15 (79%) developing persistent viremia and 4 (21%) clearing viremia spontaneously. Of those with progression to chronic infection and detectable T cell responses, all lost recognition of one or more antigens recognized during acute infection and the median reduction in the magnitude of responses was 85%. Most significantly, despite ongoing viremia those who had persistent infection did not develop new epitope specificities after the first six months of infection. In conclusion, these results suggest that in the majority of individuals, the CD8+ T cell responses generated early in HCV infection decline in peripheral blood and are not replaced with new responses.
Keywords: Viral, Immunology, HCV, CD8, ELISpot
INTRODUCTION
The World Health Organization estimates there are 170 million persons with hepatitis C virus (HCV) infection worldwide, and an estimated 4 million persons are infected with HCV in the United States (1;2). In most countries, HCV infection is found in 1–2% of the general population and may cause cirrhosis or hepatocellular cancer (3–7). Despite the high prevalence of HCV infection, acute HCV infection is difficult to study because it is usually asymptomatic and most infections are due to injection drug use (4). Most studies of acute hepatitis C have investigated infection that occurred from transfusion or, less commonly, from needle stick exposure among health care workers (8–12). Although these studies have provided valuable information about acute HCV infection, they may not be representative of the major mode of acquisition.
Approximately 20% of patients resolve acute HCV infection, but the immune correlates that determine whether a patient resolves infection or proceeds to chronic infection are not well defined. Previous studies have shown that spontaneous clearance of HCV infection occurs in association with a broadly specific and vigorous cellular immune response (13–16). In contrast, chronic infection is characterized by low frequencies of CD8+ T cells in peripheral blood (17–22). Although the cellular immune response may be less vigorous and more narrowly directed in those who fail to clear the infection, a cellular immune response is nonetheless often present in early infection and may even persist into chronic infection. It is unclear why those immune responses fail to control infection, but responses generated in acute infection have been shown to decline in a few subjects who remained persistently infected (23;24). Because of the small numbers of epitopes and subjects tested, previous studies have not fully established the breadth of initial and subsequent cellular responses or the loss of antigen recognition with progression to chronic infection.
To obtain longitudinal data on a large number of subjects representing the most common mode of HCV acquisition, we prospectively studied injection drug users (IDUs) at risk for HCV infection. In this cohort, we sought to define the interval during which new responses are formed, evaluate their breadth and vigor, then assess their durability. We analyzed the cellular immune responses of IDUs to the entire HCV polyprotein from prior to infection through progression to chronicity to maximize the number of epitopes detected in our assessment of the timing of the development of a cellular response and potential loss of antigen recognition over time. Our results demonstrate that cellular immune responses are generated in most individuals during acute HCV infection, but the CD8+ T cell responses generated early in HCV infection decline in peripheral blood and are not replaced with new responses.
METHODS
Participants
The Risk Evaluation Assessment of Community Health (REACH) prospective study of young IDUs in Baltimore, MD examined the incidence and risk factors for HCV infection, as described previously (25). Participants eligible for the study were anti-HCV antibody negative, between 15 to 30 years of age, and acknowledged use of injection drugs. Participants were invited to co-enroll in a substudy of acute hepatitis C and those who consented had blood drawn for isolation of serum, plasma, and peripheral blood mononuclear cells in a protocol designed for monthly follow up. At each visit, participants were provided counseling to reduce the risks of drug use. None of the subjects in our cohort sought medical attention for symptoms during acute infection. All participants with acute HCV infection were offered treatment. None of the subjects in our cohort elected to be treated for HCV infection during the period of follow-up reported in this study. The REACH protocol and the HCV substudy protocols were approved by the institutional review boards of the Johns Hopkins Schools of Medicine and Hygiene and Public Health.
From 1997 to 2002, 179 participants were enrolled and 62 (34.6%) developed anti-HCV antibodies (seroconverted). Beginning in November 2001, acutely infected persons were assessed for donation of ~108 – 1010 PBMC by blood donation or apheresis. HCV-specific lymphocyte responses were studied in detail in 23 acutely infected persons who were assessed frequently during the six-month period following infection and from whom large volumes of PBMC were obtained. The 23 subjects were evaluated at a combined total of 486 (range 8–45) visits.
HCV Testing protocol
Serum or plasma, stored at −80°C, from monthly follow-up visits was tested for the presence of HCV specific antibodies using the commercially available Ortho version 3.0 enzyme-linked immunosorbent assay (ELISA) (Ortho Clinical Diagnostics, Raritan, NJ) according to manufacturer’s instructions. Specimens in which antibodies were detected were retested in duplicate along with the participant’s previous seronegative sample to identify seroconverters.
HCV RNA testing was performed on sera or plasma separated from blood within two hours of collection and stored at −80 °C. For HCV seroconverters, HCV RNA testing was done on samples collected before seroconversion until a negative result was obtained and after seroconversion to evaluate the outcome of infection (viral persistence versus clearance) by using quantitative, and if undetectable, qualitative HCV RNA tests that are described below.
HCV RNA Assays
Qualitative
For detecting HCV RNA, we used the COBAS AMPLICOR™ Hepatitis C Virus Test version 2.0(Roche Molecular Systems, Branchburg, NJ). A limit of detection of 50 (1.7 log10) International Units (IU)/mL at >95% detection is reported for this assay.
Quantitative
To determine concentration of HCV RNA in serum, we used a quantitative RT-PCR assay (COBAS AMPLICOR™ HCV Monitor version 2.0, Roche Molecular Systems). This assay has a lower limit of quantitation of 600 (2.8 log10) IU/mL. When HCV RNA was not detected by using this assay, the sample was retested using the Roche qualitative test.
HCV Genotyping
Genotype was determined by performing phylogenetic analysis on Core-E1 region sequences of HCV obtained from the first viremic specimen. For most specimens, sequences were obtained from cDNA clones generated with a long amplicon RT-PCR method that has been described previously (26). For other specimens, genotype was determined by direct sequencing of RT-PCR products from the same Core-E1 region as previously described (27). Sequences were aligned using ClustalX (28), trimmed to equal length using BioEdit (29). The GTR+I+G analytical model (with parameters pinvar=0.17 and alpha=0.8) was selected using the AIC criterion as implemented in ModelTest version 3.06 (30) and PAUP* version 4b10 (Sinauer Associates, Sunderland, MA). Phylogenetic trees were estimated using the neighbor-joining algorithm implemented in PAUP*, and robustness of clustering was tested using by bootstrap analysis (31).
Biochemistry tests
ALT and total bilirubin measurements were performed using standard assays in The Johns Hopkins Hospital laboratory.
Viral recovery
HCV clearance was defined as the presence of anti-HCV with HCV RNA undetectable by the COBAS AMPLICOR™ qualitative assay in serum or plasma specimens from ≥2 consecutive visits obtained at least 300 days after initial detection of viremia. Persistence was defined as the persistent presence of anti-HCV with HCV RNA detectable by the qualitative or quantitative COBAS AMPLICOR assay in serum or plasma specimens obtained at least 300 days after initial viremia (32).
IFN-γ Elispot assay
Figure 1 illustrates a recursive method for comprehensive screening of large-volume specimens followed by detailed examination of frequently-obtained specimens. HCV-specific CD8+ T-cell responses were quantified by Elispot assay using peptides in a matrix format as previously described (33), except using 523 overlapping peptides (16–22mer peptides overlapping by 10 amino acids and designed to avoid prohibited C-terminal amino acids) spanning the entire HCV-H77 polyprotein (genotype 1a) as well as 73 peptides corresponding to optimal described CTL epitopes (34). The peptides were arrayed in 92 wells with 7 to 12 peptides per well and each peptide contained in two wells of the plate. Ninety-six well polyvinylidene plates (Millipore, Billerica, MA) were coated with 2.5 μg/ml recombinant human anti-IFN-γ antibody (Endogen, Pierce Biotechnology, Rockford, IL) in PBS at 4°C overnight. Previously frozen PBMC were thawed and added at 200,000 cells/well in 140 μl R10 media (RPMI 1640, Sigma-Aldrich Corp., St. Louis, MO; 10% FCS, Sigma-Aldrich; 10mM Hepes buffer, Sigma-Aldrich; 2 mM glutamine; and 50 U/ml penicillin-streptomycin). Peptides were added directly to the wells at a final concentration of 10 μg/ml. The plates were incubated for 20 hours at 37°C, 5% CO2. Plates were then washed, labeled with 0.25 μg/ml biotin-labeled anti-IFN-γ (Endogen), and developed by incubation with streptavidin-alkaline phophatase (Bio-Rad Lab., Hercules, CA) followed by incubation with BCIP/NBT (Bio-Rad) in Tris-buffer (pH 9.5). The reaction was stopped by washing with tap water and the plates were dried then counted on an Immunospot plate reader (Cellular Technology Ltd, Cleveland, Ohio) For quantitation of ex-vivo responses, the assay was performed at least in duplicate and background was not more than 15 spot-forming cells (SFC)/106 PBMC. Responses were considered positive if the number of spots per well minus the background was at least 25 SFC per million PBMC (33). The peptides recognized in the matrix were subsequently tested individually and in at least duplicate to confirm recognition and to measure the number of SFC produced. Intra-assay variability was less than 10%. A control of pooled cytomegalovirus, Epstein-Barr virus, and influenza antigens (CEF control peptide pool) and phytohemmaglutinin (PHA) were used as positive controls (35). Responses to the CEF control peptide pool were quantifiable and varied by no more than 30% among visits, with no temporal trend. Responses to PHA were uniformly positive.
Figure 1.
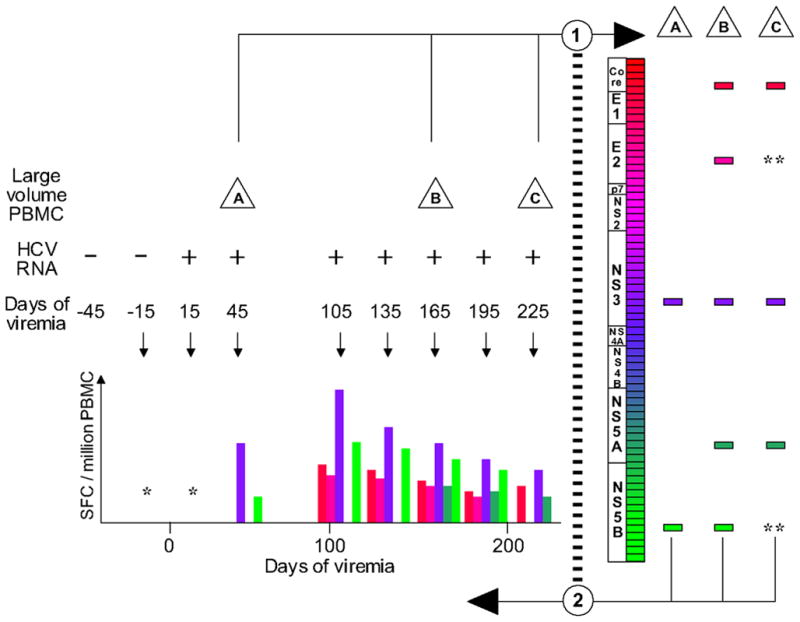
Schema for comprehensive and longitudinal analysis of cellular immune responses to HCV. Prospectively collected specimens were tested for HCV RNA, and the date of onset of viremia was estimated as the midpoint between repeatedly negative and positive specimens. In step 1, large-volume specimens (depicted as triangles A, B, and C) were screened using overlapping peptides, depicted as a series of rectangles along the HCV genome map, and detected responses are depicted as distinct rectangles to the right. In step 2, all peptides eliciting a detectable response were used to determine the temporal course of recognition in monthly smaller-volume PBMC specimens (hypothetical results are depicted, meant to be analogous to figure 2). Occasional missed visits are illustrated by a gap between the visits at 45 and 105 days of viremia; “*” indicates no responses were detected to any peptide when tested in a monthly specimen; “**” indicates a previously-detected peptide response not detected at that screening visit.
Viral sequence analysis
From 140–280 uL of serum or plasma, the 5.2 kb region from the 5′UTR to the NS3/NS4A junction was cloned as previously described.(26) For each specimen, thirty-three clones were assigned to clonotypes by using a previously-described gel shift assay,(36) and 2 clones representing the modal clonotype were sequenced, with a third clone used as needed to resolve discrepancies. Sequences were assembled into contigs using Aligner (CodonCode, Dedham, MA). Sequence data were obtained at the point of initial viremia and approximately six months later.
Statistical analysis
Descriptive statistical methods were used to analyze and present the data. To guide interpretation, statistical inferences were made by using the Chi square or Fisher’s exact tests for categorical variables and by using non-parametric rank-sum tests for continuous variables. A p-value of < 0.05 was considered statistically significant.
RESULTS
Quantitation of Elispot Responses
HCV-specific lymphocyte responses were studied in 23 acutely HCV infected persons who were assessed frequently during the 18 months following infection. The mean duration of follow up, the mean age at seroconversion, the genotypes, and the gender and racial composition of the subjects are shown in Table 1. Cellular immune responses to HCV were examined in the acutely infected subjects using an Elispot assay for the detection of gamma interferon production and a combination of previously identified optimal CD8+ T-cell epitopes and overlapping peptides covering the whole HCV polyprotein as potential antigens. Nineteen subjects (83%) had detectable responses to HCV, and the median number of antigens recognized was 4 (range 0–10), using the time point for each subject at which the largest number of antigens was recognized. Failure to recognize any epitopes was not due to mismatch of HCV subtypes, because three of the four subjects who did not recognize any epitopes were infected with genotype 1a virus.
Table 1.
Characteristics of 23 Subjects with Acute HCV Infection
| Mean age seroconversion in years (range) | 24 (18–31) | |
| Mean duration of follow-up months (range) | 27 (12–47) | |
| Gender n (%) | Male | 16/23 (69) |
| Female | 7/23 (31) | |
| Race n (%) | White | 21/23 (92) |
| African American | 1/23 (4) | |
| Asian | 1/23 (4) | |
| Genotype n (%) | 1a | 18/23 (79) |
| 1b | 2/23 (9) | |
| 3a | 1/23 (4) | |
| Mixed 1a and 1b | 1/23 (4) | |
| No viremia detected* | 1/23 (4) | |
| Outcome** | Spontaneous clearance of viremia | 4/19 (21) |
| Persistent viremia | 15/19 (79) | |
One subject seroconverted with no detectable viremia at any visit and presumably became viremic and cleared in the interval between sampling.
Duration of follow-up was sufficient at the time of analysis to determine outcome for 19 of the 23 subjects.
Nineteen of the 23 subjects tested had sufficient follow-up at the time of analysis to determine the outcome of infection (including 15 of the 19 with and all 4 of the subjects without detectable responses), of whom 15 (79%) remained persistently viremic and 4 (21%) subjects cleared viremia. There were no significant differences detected between those who cleared and those who were persistently infected in duration of follow up, age at seroconversion, gender, race, or genotype. Four subjects with no detectable responses, and 11 subjects with detectable responses, developed persistent infection. The median number of epitopes recognized by those who developed persistent viremia was four (range 0–10), not significantly different from the number recognized by those who cleared viremia spontaneously, which was five (range 4–8). Although some CD4+ T cell responses were detected, over 90% of the responses detected were CD8+ T-lymphocyte derived (data not shown).
Patterns of Cellular Immune Responses, Viremia, and Liver Biochemistry
Patterns of cellular immune responses, viremia, and ALT levels over time are shown for four subjects in Figure 2, and similar results for 5 additional subjects are available in figure S1. Subject 18 cleared viremia more than 500 days after the onset of viremia, associated with a broad and sustained cellular immune response to 7 or 8 peptides (3 adjacent overlapping peptide responses indicate recognition of at least 2, and possibly 3 distinct epitopes). Viral genomic sequences obtained 0, 6, and 12 months after the onset of viremia were much more similar than sequences from distinct study subjects (data not shown), arguing against clearance and repeated infection at 200 days. Subject 17 developed chronic infection with a low level of viremia (300–2500 IU/mL), with transient control of viremia (below the limit of detection) at 138 days associated with an broad and vigorous cellular immune response to 7 peptides, which became progressively narrower and less intense, with the remaining 4 responses only at the limit of detection at 713 days. Subject 24 developed chronic higher-level viremia (400,000–600,000 IU/mL) associated with a delayed and initially narrow cellular immune response, with loss of one of 5 responses at day 572. Subject 29 developed chronic high-level viremia (500,000–2,000,000 IU/mL) associated with a delayed, narrow, and non-sustained cellular immune response.
Figure 2.
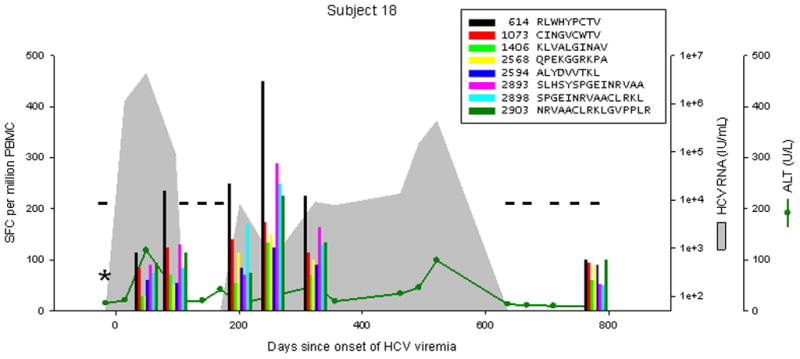
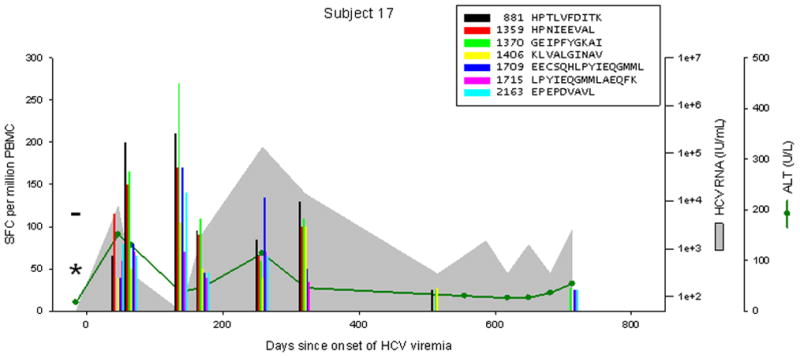
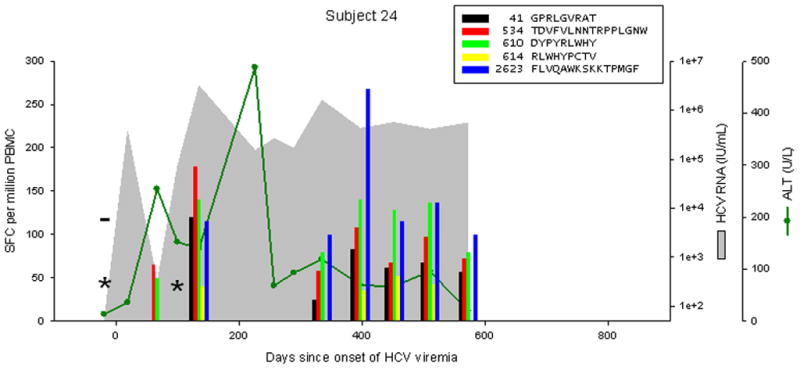
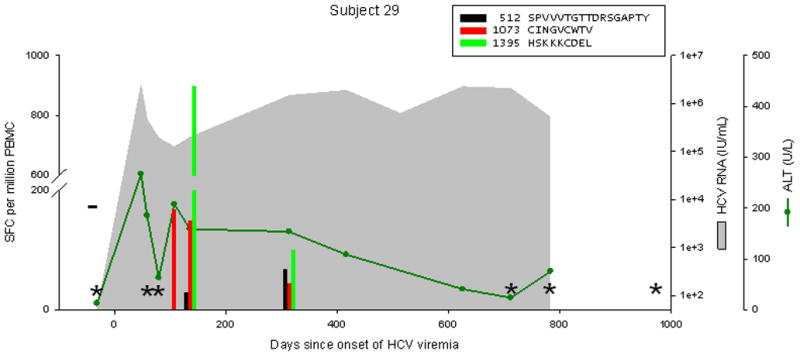
Levels of CTL response, HCV RNA, and ALT during acute HCV infection in 4 individuals. Subject 18 cleared infection after 600 days of viremia. Subjects 17, 24, and 29 progressed to chronic infection. The legend indicates peptide position (relative to H77 polyprotein, Genbank accession #AF009606) and sequence. Responses below 25 SFC per million PBMC are not shown. CTL responses were measured by IFNγ Elispot; “*” indicates no responses were detected to any peptide recognized in other specimens. HCV RNA levels below the limit of quantitation are plotted at 50 IU/mL; “−“ indicates specimens were also negative for HCV RNA by qualitative assay. The point of initial viremia was estimated as the midpoint between the last sample with undetectable HCV RNA and the first sample with detectable HCV RNA.
Anti-HCV cellular immune responses appeared approximately one month after infection. For seven subjects with PBMC available prior to initial viremia, cellular responses were not detected prior to viremia. Thus, all the responses measured were virus infection dependent. Assuming that cellular responses initially occurred midway between the visit with no detectable response and the visit where responses were first detectable, we estimated the median time from onset of viremia to development of detectable responses for these subjects was between 29 and 50 days (mean 38, median 33). Cellular responses were not detected in the first viremic specimen from any subject during monthly follow-up, though it is likely that some of them were viremic for 2–4 weeks prior to this assessment. The lag between HCV infection and detection of functionally active HCV-specific T cells was longer than has been reported for other viruses that infect humans (37–40) and is consistent with the data in a previous report of acute HCV infection (12).
Reduced Peripheral CD8+ T cell Responses Following the Acute Phase
In most subjects who progressed to chronic infection, viremia persisted despite detectable CD8+ lymphocyte responses to multiple epitopes for years. Although CD8+ lymphocyte responses to some epitopes were detectable for years, the level of antigen recognition declined over time. As shown in figure 2, responses peaked between 100 and 400 days following infection and declined from that peak with progression to chronic infection. For seven subjects with detectable cellular immune responses who progressed to chronic infection and were followed for more than 500 days, the Elispot responses at peak recognition and at the last time point tested are shown in Figure 3a. In those who progressed to persistent infection, the median number of SFC per million PBMC at peak was 170 (range 25–900) and was 25 (range 0–380) at the latest time point screened, which occurred at least one year after infection. This represents a decrease in magnitude of recognition of 85% with progression to chronic infection. In subjects who cleared viremia, median peak recognition was similar at 175 SFC per million PBMC (range 90–1135), and decreased to a median of 95 (range 50–1135), a decrease in magnitude of 46%. In addition to a decline in the magnitude of response with progression to chronicity, we also observed declining breadth of response. After an initial increase in the number of antigens recognized in the first 180 days of infection, there was progressive loss in the number of antigens recognized, as shown in Figure 3b. Between 290 and 514 days following infection, complete loss of recognition of one or more antigens recognized during acute infection was observed in all nine subjects who progressed to chronic infection (Table 2). In contrast, neither of the two subjects who cleared infection and were followed for over 500 days lost recognition of any epitopes. It is possible that T cells specific for the antigen were not lost completely, but fell below the limit of detection of our assay. However, we were also unable in two attempts to expand a population of cells specific for the antigen even with prolonged incubation. Interestingly, loss of antigen recognition was not observed until more than 290 days following initial viremia, long after most subjects who clear their viremia have eliminated HCV from the bloodstream (Table 2).
Figure 3.
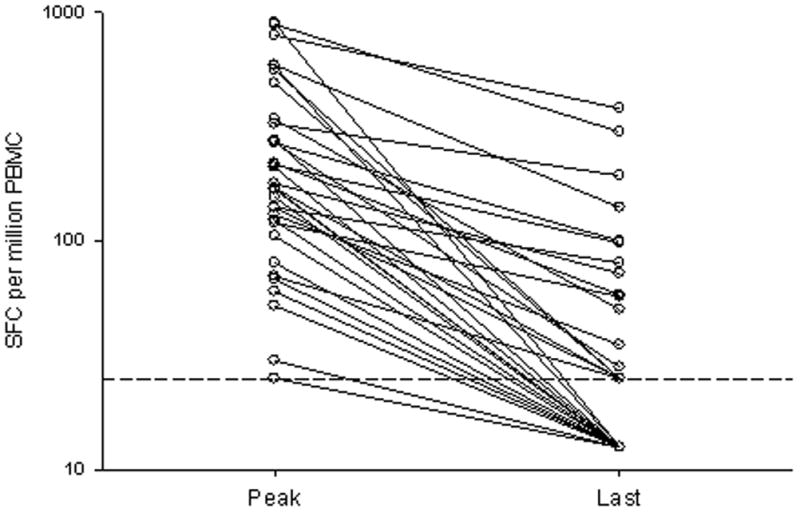

Loss of breadth and magnitude of cellular immune responses during the transition from acute to chronic HCV infection. Results from eight subjects (17, 24, 28, 29, 45, 49, 52, 55) with detectable cellular responses and with sufficient PBMC for frequent study. (A) Magnitude of response, expressed at the number of IFNγ SFC per million PBMC at peak (the specimen in which the maximal response to that peptide was detected, median 213 days and range 106 to 452 after the onset of viremia) and last specimen examined (median 642 and range, 363 to 970 days after the onset of viremia). The horizontal dashed line indicates the limit of detection. (B) Breadth of response, expressed as number of peptides recognized over time.
Table 2.
Loss of antigen recognition with progression to chronic infection in subjects with cellular immune responses during acute infection
| Subject | Number of Responses | Peak of response* | First Ag lost (days) |
|---|---|---|---|
| 17 | 8 | 138 | 290 |
| 24 | 5 | 135 | 541 |
| 28 | 10 | 236 | 425** |
| 29 | 3 | 135 | 513 |
| 30 | 4 | 134 | 400 |
| 45 | 4 | 340 | 363 |
| 49 | 5 | 398 | 514 |
| 52 | 2 | 213 | 321 |
| 54 | 4 | 246 | 376 |
| Median | 4 | 213 | 400 |
Days of viremia at which the sum of the ELISpot responses (expressed as SFC per million PBMC) was maximal.
Ab negative and HCV RNA positive on entry to the study. The estimated initial detection of antigen was determined by subtracting 5 weeks (average time to seroconversion1) from the initial detection of antibody.
No new epitope specificities developed after the first six months of infection, as shown in Figure 4. Of the 38 T cell responses detected in the subjects who developed chronic infection, only one was first identified more than 6 months after initial viremia in any subject despite ongoing viremia. That one outlier was detected in subject 28 at 217 days, after a gap in specimen collection of 120 days, and therefore may have elicited a response at any time between 97 and 217 days after the onset of viremia.
Figure 4.
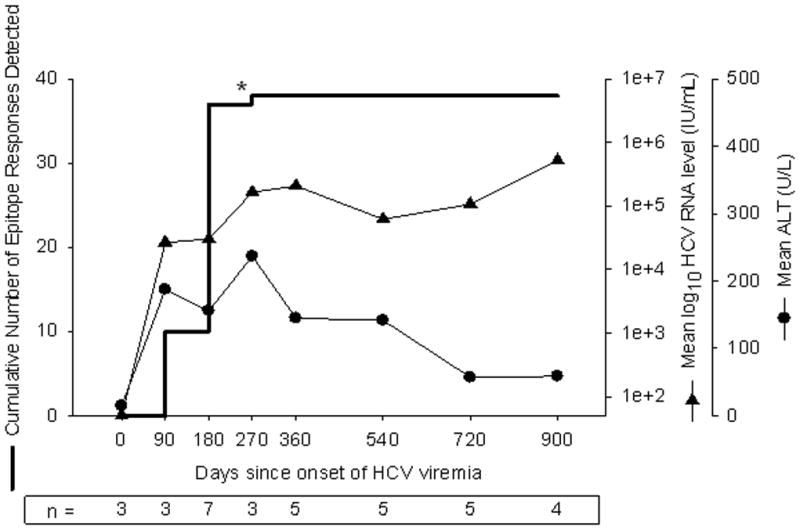
Arrest in development and maintenance of cellular immune responses during the transition from acute to chronic infection. Results from seven subjects with detectable responses and sufficient PBMC for frequent study are summarized with respect to cumulative number of responses detected, geometric mean HCV RNA level, and arithmetic mean ALT. For each time point, the data represent observations during the preceding interval. An outlier event in subject 28 indicated by “*” was detected at 217 days after onset of viremia, and no specimen was available between days 97 and 217; this response might have been detected prior to day 180 if intervening specimens had been available. Because time zero is defined as the midpoint between the last HCV RNA negative specimen and the first HCV RNA positive specimen, the data for time zero represent specimens obtained prior to viremia. The open symbol for HCV RNA level indicates HCV RNA less than 50 IU/mL.
Change in viral amino acid sequence
To address the possibility that the observed failure to develop new T cell specificities might be due to lack of viral sequence evolution, sequencing of most of the HCV genome was performed for eight subjects, using specimens from initial viremia and six months later. Viral amino acid sequence changes were observed in all eight subjects. The median number of amino acid substitutions was 11 (range 2–12) per subject in the 1651 residue region sequenced.
DISCUSSION
This study represents the first assessment for any natural human viral infection of T cell responses against oligopeptides spanning every viral protein from the acute to chronic phase. In this investigation of cellular immune responses in acute HCV infection, the breadth of the response was set early in infection. In addition, even in persons with ongoing infection, no new responses were formed and early responses became undetectable. Those subjects without a detectable cellular immune response uniformly progressed to chronic viremia.
In those with viral persistence despite immune responses directed against HCV epitopes, the mechanisms for immune failure remain unclear. This failure may be due to impaired cellular effector functions (proliferation, cytokine secretion, or cytolytic activity) or T cell exhaustion, as has been suggested by previous data (12;41). We observed a delay in the development of T cell responses to HCV infection relative to other viral infections; however, we also observed this delay in those who cleared infection. There was no qualitative difference between those who cleared infection and those with persistence in terms of the breadth or magnitude of response during the acute phase. We did see decline in and loss of antigen recognition over time in all of the subjects who remained persistently infected, but this decline occurred substantially later than the disappearance of viremia in those with clearance. The earliest point at which we noted complete loss of recognition of any antigen was almost 300 days after initial detection of HCV and most of our subjects who have cleared viremia did so within 90 days of onset. The number of subjects in the study who cleared infection was too small and the sampling around the time of clearance too infrequent to conclude that clearance is associated with preservation of responses while progression to chronicity is associated with declining recognition. In addition, some of the antigens targeted during acute infection were still recognized by chronically infected subjects years after HCV infection. Thus, although exhaustion and impairment of T-cell function may play an important role, they are unlikely to represent the only mechanisms for persistence of HCV infection and may be a consequence of continued antigenic stimulation rather than the cause of persistent viremia. Given that HCV quasispecies complexity increases over time and the finding that viremia persists for years in the presence of HCV-specific T-lymphocytes, it is possible that the T-lymphocytes are no longer effective because the virus has mutated such that the target antigen is no longer present (42).
While viral escape could explain the failure of some T cell specificities, variation in antigen could be expected to generate potential new target antigens; however, such responses were not observed. Despite recognition of a combined 51 antigens and ongoing viremia with sequence evolution, no antigens were first recognized more than 6 months after infection in any subject. In contrast, new responses to HIV-1 and HTLV-1 have been detected after the first six months of infection (43;44), suggesting that HCV may interfere with this process in some way.
There are several possible explanations for the immune system’s failure to generate new responses over the course of infection as viral quasispecies diversification occurs. First, chronic HCV infection may be associated with induction of suppressor T-cells, as has been suggested in a prior study (45). An alternative hypothesis is that chronic HCV infection is associated with dendritic cell dysfunction, as has been shown in other studies (46;47). However, patients with chronic HCV are not demonstrably immunosuppressed, and mount cellular immune responses to other infections. Therefore, the mechanism for the failure to generate new responses to HCV antigens is likely to be HCV-specific. A hypothesized mechanism of HCV-specific immunosuppression is that viral evolution results in sequences that are similar enough to the initial sequence that T lymphocytes recognizing the original sequence are activated rather than naive cells that would potentially generate more effective responses to the new sequence. This concept of “original antigenic sin” has been shown to occur following sequential infection of mice with two similar strains of lymphocytic choriomeningitis virus, but has not been demonstrated in a natural infection (48). It is also possible that anti-HCV immune responses are compartmentalized to the liver, as some studies suggest (49–51), though others have found that specificities in the two compartments are similar, and studies of intrahepatic lymphocyte responses during acute human HCV infection are lacking. Recent evidence suggests that CD4+ T cell help is required for efficient development of effector and memory CD8 T cell populations in HIV infection (52), and when lacking may facilitate viral escape mutation in HCV infection (53).
Although other studies have demonstrated HCV-specific gamma interferon production in IDUs in the absence of HCV antibodies or viremia, we did not find any HCV-specific gamma interferon production before the onset of viremia in our subjects (54;55). However, the number of subjects we assessed for responses prior to viremia was relatively small because the primary purpose of this study was not to assess subjects for cellular responses in the absence of viremia. In addition, all of our subjects were viremic initially and ultimately developed antibodies and most became persistently infected. In contrast, the subjects in whom cellular responses were detected in the other studies failed to seroconvert or acquire chronic HCV infection. It has been hypothesized that the responses seen in subjects who never have detectable antibody or HCV may confer protective immunity (54;55). Failure to find cellular responses prior to infection in our subjects is consistent with that hypothesis since those who progress to chronic HCV infection would not be expected to have markers of protective immunity.
These data are representative of most HCV infections not only because the mode of acquisition for these subjects represents the major mode of HCV infection in the Western world, but also because the subjects were studied prospectively without regard to symptoms before, during, and after infection. The results significantly expand the available data on induction and maintenance of an HCV specific cellular immune response. In addition, this comprehensive longitudinal analysis of CD8+ T cell responses against all viral proteins represents the first such assessment in any human viral infection during the transition from acute to chronic infection and sheds light on evolution of immune responses with prolonged antigenic exposure. Determining the mechanisms that permit viral persistence despite measurable cellular immune responses to multiple epitopes and that interfere with development of new responses could improve efforts to prevent and treat chronic viral infections.
Supplementary Material
Acknowledgments
The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CEF Control Peptide Pool from Dr. Josephine Cox and Dr. Jeffrey Currier. This work was supported by grants from the US Public Health Service K08 DA11880 (to A.L.C.), R01 DK57998 (to S.C.R.), and U19 AI40035 (to D.L.T. and S.C.R.).
References
- 1.World Health Organization. Hepatitis C: global prevalence. Weekly Epidemiological Record. 1997;(72):341–348. [Google Scholar]
- 2.Alter MJ. Epidemiology of hepatitis C. Hepatology. 1997;26(3 Suppl 1):62S–65S. doi: 10.1002/hep.510260711. [DOI] [PubMed] [Google Scholar]
- 3.Alter MJ. Epidemiology of hepatitis C in the West. Semin Liver Dis. 1995;15:5–14. doi: 10.1055/s-2007-1007259. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR. 1998;47(RR19):1–39. [PubMed] [Google Scholar]
- 5.Villano SA, Vlahov D, Nelson KE, Cohn S, Thomas DL. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology. 1999;29(3):908–914. doi: 10.1002/hep.510290311. [DOI] [PubMed] [Google Scholar]
- 6.Cao J, Sullivan N, Desjardin E, Parolin C, Robinson J, Wyatt R, et al. Replication and neutralization of human immunodeficiency virus type 1 lacking the V1 and V2 variable loops of the gp120 envelope glycoprotein. J Virol. 1997;71(12):9808–9812. doi: 10.1128/jvi.71.12.9808-9812.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong MJ, El-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusion-associated hepatitis C. N Engl J Med. 1995;332:1463–1466. doi: 10.1056/NEJM199506013322202. [DOI] [PubMed] [Google Scholar]
- 8.Farci P, Alter HJ, Wong D, Miller RH, Shih JW, Jett B, et al. A long-term study of hepatitis C virus replication in non-A, non- B hepatitis. N Engl J Med. 1991;325:98–104. doi: 10.1056/NEJM199107113250205. [DOI] [PubMed] [Google Scholar]
- 9.Barrera JM, Bruguera M, Ercilla MG, Gil C, Celis R, Gil MP, et al. Persistent hepatitis C viremia after acute self-limiting posttransfusion hepatitis C. Hepatology. 1995;21:639–644. [PubMed] [Google Scholar]
- 10.Garson JA, Tuke PW, Makris M, Briggs M, Machin SJ, Preston FE, et al. Demonstration of viraemia patterns in haemophiliacs treated with hepatitis C virus contaminated factor VIII concentrates. Lancet. 1990;336:1022–1025. doi: 10.1016/0140-6736(90)92487-3. [DOI] [PubMed] [Google Scholar]
- 11.Prince AM, Brotman B, Inchauspe G, Pascual D, Nasoff M, Hosein B, et al. Patterns and prevalence of hepatitis C virus infection in posttransfusion non-A, non-B hepatitis. J Infect Dis. 1993;167:1296–1301. doi: 10.1093/infdis/167.6.1296. [DOI] [PubMed] [Google Scholar]
- 12.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194(10):1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper S, Erickson AL, Adams EJ, Kansopon J, Weiner AJ, Chien DY, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10(4):439–449. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- 14.Gruner NH, Gerlach TJ, Jung MC, Diepolder HM, Schirren CA, Schraut WW, et al. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J Infect Dis. 2000;181(5):1528–1536. doi: 10.1086/315450. [DOI] [PubMed] [Google Scholar]
- 15.Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191(9):1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med. 2000;6(5):578–582. doi: 10.1038/75063. [DOI] [PubMed] [Google Scholar]
- 17.Hiroishi K, Kita H, Kojima M, Okamoto H, Moriyama T, Kaneko T, et al. Cytotoxic T lymphocyte response and viral load in hepatitis C virus infection. Hepatology. 1997;25(3):705–712. doi: 10.1002/hep.510250336. [DOI] [PubMed] [Google Scholar]
- 18.Rehermann B, Chang KM, McHutchison JG, Kokka R, Houghton M, Chisari FV. Quantitative analysis of the peripheral blood cytotoxic T lymphocyte response in patients with chronic hepatitis C virus infection. J Clin Invest. 1996;98(6):1432–1440. doi: 10.1172/JCI118931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerny A, McHutchison JG, Pasquinelli C, Brown ME, Brothers MA, Grabscheid B, et al. Cytotoxic T lymphocyte response to hepatitis C virus-derived peptides containing the HLA A2.1 binding motif. J Clin Invest. 1995;95(2):521–530. doi: 10.1172/JCI117694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He XS, Rehermann B, Lopez-Labrador FX, Boisvert J, Cheung R, Mumm J, et al. Quantitative analysis of hepatitis C virus-specific CD8(+) T cells in peripheral blood and liver using peptide-MHC tetramers. Proc Natl Acad Sci USA. 1999;96(10):5692–5697. doi: 10.1073/pnas.96.10.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shirai M, Okada H, Nishioka M, Akatsuka T, Wychowski C, Houghten R, et al. An epitope in hepatitis C virus core region recognized by cytotoxic T cells in mice and humans. J Virol. 1994;68(5):3334–3342. doi: 10.1128/jvi.68.5.3334-3342.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battegay M, Fikes J, Di Bisceglie AM, Wentworth PA, Sette A, Celis E, et al. Patients with chronic hepatitis C have circulating cytotoxic T cells which recognize hepatitis C virus-encoded peptides binding to HLA-A2.1 molecules. J Virol. 1995;69:2462–2470. doi: 10.1128/jvi.69.4.2462-2470.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75(12):5550–5558. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lechner F, Gruener NH, Urbani S, Uggeri J, Santantonio T, Kammer AR, et al. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur J Immunol. 2000;30(9):2479–2487. doi: 10.1002/1521-4141(200009)30:9<2479::AID-IMMU2479>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 25.Garfein RS, Doherty MC, Monterroso ER, Thomas DL, Nelson KE, Vlahov D. Prevalence and incidence of hepatitis C virus infection among young adult injection drug users. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18 (Suppl 1):S11–S19. doi: 10.1097/00042560-199802001-00004. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Netski DM, Mao Q, Laeyendecker O, Ticehurst JR, Wang XH, et al. Accurate representation of the hepatitis C virus quasispecies in 5.2-kilobase amplicons. J Clin Microbiol. 2004;42(9):4223–4229. doi: 10.1128/JCM.42.9.4223-4229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray SC, Arthur RR, Carella A, Bukh J, Thomas DL. Genetic Epidemiology of Hepatitis C Virus throughout Egypt. J Infect Dis. 2000;182(3):698–707. doi: 10.1086/315786. [DOI] [PubMed] [Google Scholar]
- 28.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998;23(10):403–405. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 29.Hall TA. BioEdit: Biological sequence alignment editor for Windows 95/98/NT version 5.0.7. software. 2001 Distributed by author: http://www.mbio.ncsu.edu/RNaseP/info/programs/BIOEDIT/bioedit.html.
- 30.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 31.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 32.Cox AL, Netski DM, Mosbruger T, Sherman SG, Strathdee S, Ompad DC, et al. Prospective evaluation of community-acquired acute Hepatitis C. Clin Infect Dis. 2004 doi: 10.1086/428578. (in press) [DOI] [PubMed] [Google Scholar]
- 33.Lauer GM, Ouchi K, Chung RT, Nguyen TN, Day CL, Purkis DR, et al. Comprehensive analysis of CD8(+)-T-cell responses against hepatitis C virus reveals multiple unpredicted specificities. J Virol. 2002;76(12):6104–6113. doi: 10.1128/JVI.76.12.6104-6113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward S, Lauer G, Isba R, Walker B, Klenerman P. Cellular immune responses against hepatitis C virus: the evidence base 2002. Clin Exp Immunol. 2002;128(2):195–203. doi: 10.1046/j.1365-2249.2002.01840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S, et al. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J Immunol Methods. 2002;260(1–2):157–172. doi: 10.1016/s0022-1759(01)00535-x. [DOI] [PubMed] [Google Scholar]
- 36.Wang YM, Ray SC, Laeyendecker O, Ticehurst JR, Thomas DL. Assessment of hepatitis C virus sequence complexity by electrophoretic mobilities of both single-and double-stranded DNAs. J Clin Microbiol. 1998;36(10):2982–2989. doi: 10.1128/jcm.36.10.2982-2989.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibility complex-restricted CD8+ T cells. J Exp Med. 1991;174(4):875–880. doi: 10.1084/jem.174.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou S, Doherty PC, Zijlstra M, Jaenisch R, Katz JM. Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J Immunol. 1992;149(4):1319–1325. [PubMed] [Google Scholar]
- 39.Koup RA, Safrit JA, Cao Y, Andrew CA, McLeod G, Borkowsky W, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen TM, O’Connor DH, Jing P, Dzuris JL, Mothe BR, Vogel TU, et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407(6802):386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- 41.Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol. 2002;169(6):3447–3458. doi: 10.4049/jimmunol.169.6.3447. [DOI] [PubMed] [Google Scholar]
- 42.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 2001;15(6):883–895. doi: 10.1016/s1074-7613(01)00245-x. [DOI] [PubMed] [Google Scholar]
- 43.Goulder PJ, Altfeld MA, Rosenberg ES, Nguyen T, Tang Y, Eldridge RL, et al. Substantial differences in specificity of HIV-specific cytotoxic T cells in acute and chronic HIV infection. J Exp Med. 2001;193(2):181–194. doi: 10.1084/jem.193.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGinn TM, Wei Q, Stallworth J, Fultz PN. Immune responses to HTLV-I(ACH) during acute infection of pig-tailed macaques. AIDS Res Hum Retroviruses. 2004;20(4):443–456. doi: 10.1089/088922204323048195. [DOI] [PubMed] [Google Scholar]
- 45.Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. 2003;38(6):1437–1448. doi: 10.1016/j.hep.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 46.Bain C, Fatmi A, Zoulim F, Zarski JP, Trepo C, Inchauspe G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120(2):512–524. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- 47.Kanto T, Hayashi N, Takehara T, Tatsumi T, Kuzushita N, Ito A, et al. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162(9):5584–5591. [PubMed] [Google Scholar]
- 48.Klenerman P, Zinkernagel RM. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature. 1998;394(6692):482–485. doi: 10.1038/28860. [DOI] [PubMed] [Google Scholar]
- 49.Minutello MA, Pileri P, Unutmaz D, Censini S, Kuo G, Houghton M, et al. Compartmentalization of T lymphocytes to the site of disease: intrahepatic CD4+ T cells specific for the protein NS4 of hepatitis C virus in patients with chronic hepatitis C. J Exp Med. 1993;178(1):17–25. doi: 10.1084/jem.178.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomson M, Nascimbeni M, Havert MB, Major M, Gonzales S, Alter H, et al. The clearance of hepatitis C virus infection in chimpanzees may not necessarily correlate with the appearance of acquired immunity. J Virol. 2003;77(2):862–870. doi: 10.1128/JVI.77.2.862-870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grabowska AM, Lechner F, Klenerman P, Tighe PJ, Ryder S, Ball JK, et al. Direct ex vivo comparison of the breadth and specificity of the T cells in the liver and peripheral blood of patients with chronic HCV infection. Eur J Immunol. 2001;31(8):2388–2394. doi: 10.1002/1521-4141(200108)31:8<2388::aid-immu2388>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 52.Kalams SA, Walker BD. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med. 1998;188(12):2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302(5645):659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 54.Freeman AJ, Ffrench RA, Post JJ, Harvey CE, Gilmour SJ, White PA, et al. Prevalence of production of virus-specific interferon-gamma among seronegative hepatitis C-resistant subjects reporting injection drug use. J Infect Dis. 2004;190(6):1093–1097. doi: 10.1086/422605. [DOI] [PubMed] [Google Scholar]
- 55.Mizukoshi E, Eisenbach C, Edlin B, Weiler C, Carrington M, O’Brien T, et al. HCV-specific cellular immune responses in subjects who are anti-HCV-negative, HCV RNA-negative despite long term (>15 years) injection drug use (AASLD Annual Meeting 2003 abstract #111) Hepatology. 2003;38(4 supp):210A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.