

Abstract
Type 1 diabetes (T1D) is caused by T cell-mediated destruction of the pancreatic insulin-producing β cells. While the role of CD4+ T cells in the pathogenesis of T1D is accepted widely, the epitopes recognized by pathogenic human CD4+ T cells remain poorly defined. None the less, responses to the N-terminal region of the insulin A-chain have been described. Human CD4+ T cells from the pancreatic lymph nodes of subjects with T1D respond to the first 15 amino acids of the insulin A-chain. We identified a human leucocyte antigen-DR4-restricted epitope comprising the first 13 amino acids of the insulin A-chain (A1-13), dependent upon generation of a vicinal disulphide bond between adjacent cysteines (A6–A7). Here we describe the analysis of a CD4+ T cell clone, isolated from a subject with T1D, which recognizes a new HLR-DR4-restricted epitope (KRGIVEQCCTSICS) that overlaps the insulin A1-13 epitope. This is a novel epitope, because the clone responds to proinsulin but not to insulin, T cell recognition requires the last two residues of the C-peptide (Lys, Arg) and recognition does not depend upon a vicinal disulphide bond between the A6 and A7 cysteines. The finding of a further CD4+ T cell epitope in the N-terminal A-chain region of human insulin underscores the importance of this region as a target of CD4+ T cell responses in human T1D.
Keywords: CD4, epitope, proinsulin, T cell, type 1 diabetes
Introduction
Type 1 diabetes (T1D) is caused by T cell-mediated destruction of the insulin-producing β cells in the pancreas. Insulin and its parent molecule proinsulin are clearly important in the development of T1D in humans and mice [1–3]. The T cell epitopes recognized by pathogenic CD4+ T cells in T1D are gradually being defined [4], but their detailed molecular analysis has been hindered by a paucity of human T cell clones specific for (pro)insulin.
The non-obese diabetic (NOD) mouse develops T1D spontaneously and is a useful animal model for human T1D [5]. Remarkably, the development of T1D in the NOD mouse appears to be dependent upon a single epitope in the B-chain (B9-23) of insulin [6]. However, the major epitopes in the NOD mouse may not be the same as those targeted by pathogenic human CD4+ T cells. Human CD4+ T cell responses to insulin B9-23 have been reported [7], but to our knowledge T cell clones specific for this epitope have not been reported, although clones specific for B11-27 have been described [8].
Evidence from several groups suggests that CD4+ T cell responses to the N-terminus of the A-chain of insulin are implicated in the pathogenesis of human T1D. CD4+ T cells isolated from human pancreatic lymph nodes of donors who had T1D showed evidence of clonal expansion to a peptide from the N-terminal 15 amino acids of the insulin A-chain [9]. We isolated proinsulin-specific CD4+ T cell clones from the blood of subjects both with and at risk of T1D using a novel cloning strategy [10]. Some of these clones recognized an epitope in the first 13 amino acids of the A-chain of human insulin (GIVEQCCTSICSL). This epitope is generated when a vicinal disulphide bond forms between the adjacent cysteines at positions A6 and A7 [11]. More recently, T cell reactivity to the first 12 amino acids of the A-chain of human insulin was shown to be associated with progression to T1D in children with high diabetes-risk human leucocyte antigen (HLA) haplotypes [12]. Together these data support a role for human CD4+ T cell responses to the N-terminus of the insulin A-chain in the pathogenesis of T1D. Here we describe a new CD4+ T cell epitope in proinsulin, extending from the C-peptide into the insulin A-chain.
Materials and methods
Blood donors and peripheral blood mononuclear cells isolation
Blood was obtained by venepuncture with informed consent and approval from the Royal Melbourne Hospital and Walter and Eliza Hall Institute for Medical Human Research Ethics Committees. The donor, diagnosed with T1D ∼10 years earlier, was HLA-typed as A11, 24; B18, 39; Cw5, w7, DRB1*0301, 0404; DQB1*0201, 0302. Peripheral blood mononuclear cells (PBMC) were isolated over Ficoll/Hypaque (Amersham Pharmacia Biotech AB, Uppsala Sweden) and washed twice in phosphate-buffered saline (PBS). For all experiments, cells were cultured in Iscove's modified Dulbecco's medium (IMDM) (Gibco, Rockville, MD, USA) supplemented with 5% pooled male human serum, 2 mM glutamine (Glutamax; Gibco), 5 × 10−5 M 2-mercaptoethanol (Sigma, St Louis, MO, USA), penicillin (100 U/ml), streptomycin (100 µg/ml) and 100 µM non-essential amino acids (all from Gibco), referred to as 5% PHS/IMDM.
Antigens
Synthetic peptides were purchased from Mimotopes (Clayton, Victoria, Australia) or in some cases synthesized in-house and purified to > 95% purity. A peptide library composed of 15-mer peptides overlapping by 12 amino acids comprising the entire sequence of human proinsulin was used for initial epitope mapping experiments. All peptides were purified to at least 85%, dissolved in 0·5% acetic acid, 40% acetonitrile- water to 5 mM, and stored in aliquots at −70°C. The peptides used in each experiment are shown in the figures. For some experiments, peptides were purified by reverse-phase high-performance liquid chromatography (RP-HPLC) to > 95% purity. Recombinant human proinsulin was produced in-house using a published protocol [13]. After anion exchange chromatography, refolding and RP-HPLC purification, the protein resolved as a single species of correct molecular weight in matrix-assisted laser desorption/ionization-time of flight mass spectrometry. To prepare insulin, clinical grade insulin was dialysed against PBS to remove preservatives and stored in aliquots at −80°C. RP-HPLC and mass spectrometry revealed that the insulin preparation was homogeneous and of the expected mass (5808 Da).
5,6-carboxylfluorescein diacetate succinimidyl ester staining and T cell cloning
Staining with the dye CFSE (5,6-carboxylfluorescein diacetate succinimidylester) (Invitrogen, Carlsbad, CA, USA) was performed as described previously [14]. Briefly, PBMC (1 × 107/ml in PBS) were incubated at 37°C for 5 min with 0·1 µM CFSE. Staining was terminated by adding culture medium containing 5% pooled human serum, the cells were washed once in PBS/1% pooled human serum and resuspended in culture medium at 1·33 × 106/ml. Stained cells (2 × 105/well, 150 µl) were cultured in 96-well round-bottomed plates (Becton Dickinson Labware, Franklin Lakes, NJ, USA) with medium alone, tetanus toxoid (10 LfU/ml) or proinsulin (10 µg/ml). Unstained cells were included in all experiments to set the compensations on the flow cytometer. After 7 days of culture, replicates for each antigen concentration were pooled, washed in PBS and stained on ice with anti-human CD4-phycoerythrin (IgG2a, clone RPA-T4; BD-Pharmingen, San Diego, CA, USA). Optimal compensation and gain settings were determined for each experiment based on unstained and single-stained samples. Propidium iodide staining was used to exclude dead cells. CD4+ T cell clones were isolated as described previously [10]. Briefly, a single CFSEdim, CD4+, propidium iodide-negative cell was sorted into all except the outer wells of a 96-well round-bottomed plate. Each well contained 1 × 105 irradiated (2000 rad), allogeneic PBMC from two unrelated donors and 5 × 104, irradiated (50 Gy) JY Epstein–Barr virus (EBV)-transformed B cell line, interleukin (IL)-2 (10 U/ml), IL-4 (5 ng/ml) (Peprotech, Rocky Hill, NJ, USA) and anti-CD3 monoclonal antibody (30 ng/ml) in 100 µl of culture medium. Fungizone (Amphotericin B; Bristol-Myers Squibb, Princeton, NJ, USA) was added to cultures at a final concentration of 2 µg/ml. All wells were fed with fresh IL-2 and IL-4 every 5–7 days and growing clones were visible after approximately 2 weeks. At this time they were transferred to a 48-well plate and tested for antigen specificity within 2–5 days. For antigen specificity testing, cells were cultured with autologous irradiated PBMC, with or without proinsulin, and their proliferation measured by [3H]-thymidine incorporation as described below.
[3H]-thymidine proliferation assays
All assays were performed in 96-well round bottomed plates in 5% PHS/IMDM. Antigen-presenting cells (APC) were either (i) irradiated (20 Gy) autologous or HLA-matched PBMC (fresh or thawed after storage over liquid nitrogen) or (ii) EBV-transformed B cells from a donor with bare lymphocyte syndrome (BLS) that had been stably transfected with different HLA genes (kindly supplied by Dr Gerald Nepom, Benaroya Institute, Seattle, USA). EBV-transformed B cell lines were irradiated at 50 Gy. In some experiments, APCs were fixed with 1% paraformaldehyde for 20 min at room temperature, washed twice in PBS and once in culture medium before use in proliferation assays. For HLA blocking experiments, protein G-Sepharose-purified monoclonal antibodies to HLA-DR (clone L243, IgG2a), HLA-DQ (clone SPV-L3, IgG2a) and HLA-DP (B7/21, IgG1) were added at the start of the culture at a final concentration of 5 µg/ml. The concentration indicated in the figures refers to the final concentration. In some experiments, freshly prepared Tris (2-carboxyethyl) phosphine hydrochloride (TCEP; Pierce, Rockford, IL, USA) was added to the antigen solution before dilution by medium and addition of APC and T cells, to the final concentration shown in the figures. In all experiments, proliferation was measured by the [3H]-thymidine incorporation. After 2 days, [3H]-thymidine (0·5 µCi/well) was added for 18 h after which the cells were harvested and incorporated radioactivity measured by β-scintillation counting.
T cell receptor analysis
T cell receptor Vα and Vβ usage was determined by sequencing the T cell receptor (TCR) α and TCRβ cDNA. RNA was extracted from 2·5–5 × 106 cloned T cells using RNAeasy columns (Qiagen, Hilden, Germany). RNA (5 µg) was converted to cDNA using M-MLV reverse transciptase (Promega, Madison, USA) and random hexamers (100 µM). cDNA was purified by binding to silica and eluted in 10–20 µl of water and treated with terminal deoxytransferase in the presence of dGTP. TCR-α and -β genes were amplified by anchor polymerase chain reaction (PCR) using a polyC primer (cactcgagcggccgcgtcgaccccccccccccccc) that binds to the 3′-poly G and Vα (cggtgaata ggc aga cag acttgtca)- and Vβ (agaagcctgtggccaggcacaccagt)-specific reverse primers. PCR was performed for 30 cycles, annealing at 62°C. PCR products were cloned into pGEM-T Easy (Promega, Madison, USA) and sequenced using the pGEM T7 and Sp6 primers. TCR genes were analysed using the IMGT website (http://imgt.cines.fr) [15].
Results
A new epitope extends from the C-peptide of proinsulin into the insulin A-chain
Proinsulin-specific CD4+ T cell clones were isolated from the blood of a donor with established T1D. Epitope mapping was performed with a panel of 15-mer proinsulin peptides, each shifted by three amino acids. Pools of three to four peptides were tested, then each individual peptide from the reactive pool was tested (data not shown). In this way several clones were shown to respond to a peptide comprising the final two amino acids in the C-peptide and the first 13 amino acids of the insulin A-chain (KRGIVEQCCTSICSL), referred to as KR-A1-13. All but one of these clones responded to insulin and were shown to recognize an epitope dependent upon a vicinal disulphide bond between adjacent cysteine residues at A6 and A7 [11]. Here we examine the specificity of the remaining clone. This clone responded to proinsulin, but not insulin (Fig. 1a). Testing peptides without the N-terminal lysine revealed that this residue was required for T cell recognition (Fig. 1b). The strongest responses was seen when the C-terminal leucine was present (KR-A1-13). Weaker responses were detected to a truncated peptide without the C-terminal serine (KR-A1-12) (Fig. 1c). No responses were detected to peptide without the C-terminal cysteine, serine and leucine residues (KR-A1-10). Thus, we have defined a previously unknown CD4+ T cell epitope (KRGIVEQCCTSICS) derived from human proinsulin.
Fig. 1.
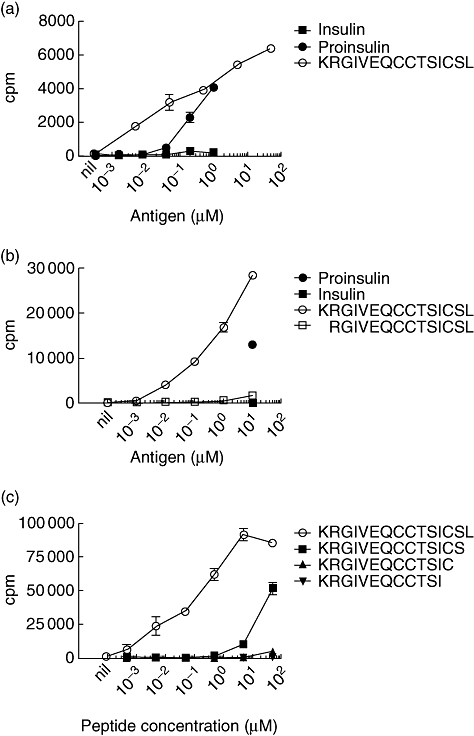
A new epitope extends from the C-peptide of proinsulin into the insulin A-chain. (a) Proinsulin-specific CD4+ T cell clones were tested against varying concentrations of proinsulin, insulin and KR-A1-13 peptide. T cell clones (5 × 104 cells/well) were cultured in the presence of irradiated autologous peripheral blood mononuclear cells (PBMC) (5 × 104/well). (b) Proinsulin-specific CD4+ T-cell clones were tested against varying concentrations of proinsulin, KR-A1-13 (KRGIVEQCCTSICSL) and R-A1-13 (RGIVEQCCTSICSL) peptides. Proinsulin and insulin were present at 10 µM final concentration. T cell clones (3 × 104 cells/well) were cultured in the presence of irradiated autologous PBMC (5 × 104/well). (c) Proinsulin-specific CD4+ T cell clones were tested against varying concentrations of KR-A1-13 (KRGIVEQCCTSICSL) and C-terminal deleted variants, KR-A1-12 (KRGIVEQCCTSICS), KR-A1-11 (RGIVEQCCTSIC) and KR-A1-10 (KRGIVEQCCTSI). T cell clones (4 × 104 cells/well) were cultured in the presence of irradiated autologous PBMC (6 × 104/well). In all experiments, proliferation was measured by the incorporation of 0·5 µCi/well [3H]-thymidine added for the final 18 h of a 72-h culture. The means of triplicates ± standard error of the mean are shown. Data are representative of three (a, b) or two (c) independent experiments.
The KR-A1-13 epitope is presented by HLA-DR4
Proliferation of the clone to proinsulin was prevented by the presence of monoclonal antibody to HLA-DR, but not to HLA-DQ or HLA-DP (Fig. 2a). Presentation by HLA-DR4 was confirmed with a panel of cell lines that express single HLA class II molecules. Cells that express DRB1*0404 (DR4) or DRB1*0405 (DR4) stimulated proliferation of the clone to KR-A1-13, whereas cells that express DQB1*0401, DRB4*0101 (DR52), DQB1*0201 (DQ2), DQB1*0302 (DQ8) did not. Hence, the epitope is presented by HLA-DR4, specifically the subtypes HLA-DRB1*0404 and *0405.
Fig. 2.
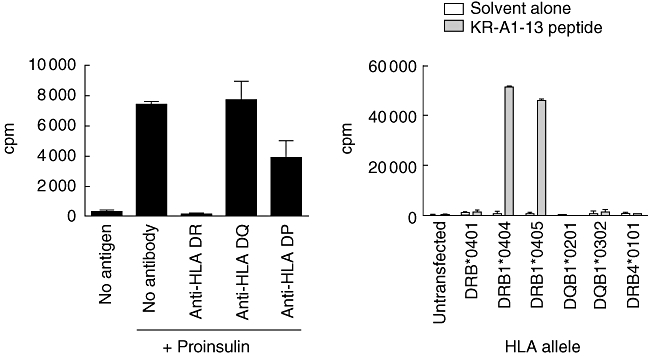
The KR-A1-13-specific clone is restricted by human leucocyte antigen (HLA)-DR4. (a) KR-A1-13-specific T cells (5 × 104 /well) were incubated with irradiated autologous peripheral blood mononuclear cells (PBMC) (1 × 105/well) without antigen (no antigen) or with 10 µg/ml proinsulin. Monoclonal antibodies specific for HLA-DR (L243), HLA-DQ (SPV-L3) or HLA-DP (B7/21) were added to a final concentration of 5 µg/ml. (b) KR-A1-13-specific T cell clone cells (2·5 × 104/well) were incubated with irradiated (50 Gy), HLA-transfected bare lymphocyte syndrome (BLS) cell lines (1 × 104/well), pulsed with 100 µM KR-A1-13 peptide (KRGIVEQCCTSICSL) or an equal volume of peptide solvent. Proliferation was measured by [3H]-thymidine incorporation and proliferation of the BLS cells without T cells (1000–5000 counts per minute) was subtracted. Mean ± standard error of the mean of triplicate wells are shown. Data are representative of two experiments.
Sensitivity to cysteine substitution
To determine if recognition of the new epitope requires a vicinal disulphide bond between the A6 and A7 cysteines, synthetic peptides with each cysteine replaced by a serine were tested for their capacity to stimulate the clone. Unlike the clones specific for the A1-13 epitope [11], the KR-A1-13-specific clone responded to a peptide with serine at position A6. This peptide was not as potent as the native peptide but still stimulated a robust T cell response (Fig. 3a). Substitution of cysteine at position A7 decreased the response markedly. However, in contrast to insulin A1-13-specific clones, when the cysteine at A11 was replaced with serine no response was detected. These data indicate that the new epitope does not depend upon a vicinal disulphide bond between cysteines at A6 and A7, but requires cysteine at positions A7 and A11. A peptide corresponding to the sequence of murine insulin, which has aspartic acid (D) instead of glutamic acid (E) at position A4, was much less potent than the human homologue (Fig. 3a). To investigate the role of disulphide bonds formed by cysteine residues in contributing to the KR-A1-13 epitope, the KR-A1-13 peptide was exposed to the reducing agent TCEP during the proliferation assay. The presence of TCEP, up to 800 µM, had no effect on the response to KR-A1-13 (Fig. 3b), suggesting that this epitope was not redox sensitive.
Fig. 3.
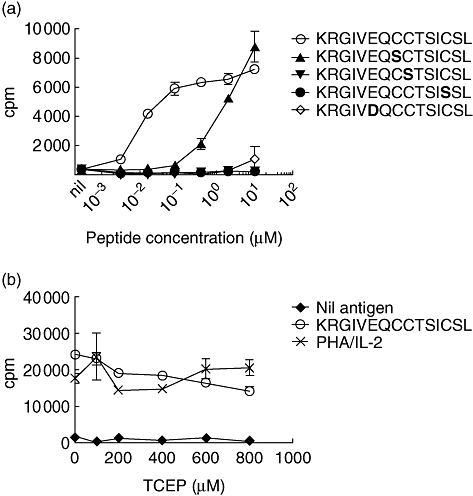
Sensitivity to cysteine substitution. (a) Effect of substituting each cysteine with serine was investigated. KR-A1-13 (KRGIVEQCCTSICSL)-specific T cell clones (2·5 × 104 cells/well) were cultured in the presence of 10–0·0032 µM unmodified KR-A1-13 peptide or variants that have each cysteine (C) substituted with serine (S), or the murine homologue (D for E at position A4). The substituted amino acid is shown in large fount in bold type. (b) Effect of treatment with the disulphide-reducing agent Tris (2-carboxyethyl) phosphine hydrochloride (TCEP). KR-A1-13 (KRGIVEQCCTSICSL) peptide (10 µM final concentration) was treated with freshly prepared TCEP and diluted to the final concentrations shown. Each well included irradiated autologous peripheral blood mononuclear cells (PBMC) (5 × 104) and KR-A1-13 specific T cell clone (2·5 × 104/well). Phytohaemagglutinin (PHA) (1·25 µg/ml) and interleukin (IL)-2 (2·5 U/ml) and solvent alone were added as positive and negative controls respectively. Means ± standard error of the mean of triplicate wells are shown and in each case data are representative of two experiments.
The KR-A1-13-specific clones use different TCR Vα and Vβ genes compared with the insulin A1-13-specific clones
The TCR usage by KR-A1-13 and insulin A1-13-epitope-specific T cell clones was determined by reverse transcription–polymerase chain reaction (RT–PCR). The KR-A1-13-specific clone used TCR Vα4*01, Jα1601, Vβ6-5*01 and Jβ2-5*01. In contrast, all four insulin A1-13-specific clones analysed used Vα13-2*01, Jα4*01, Vβ7-8*01and Jβ1-1*01. Hence, the clone specific for the KR-A-13 epitope uses distinct TCR genes to those of the insulin-specific clones that recognize the overlapping insulin A1-13 epitope.
Discussion
We describe a new human CD4+ T cell epitope that spans the C-peptide-insulin A-chain junction of proinsulin. The N-terminal insulin A-chain region is emerging as a hot spot for CD4+ T cell epitopes in human T1D. Both we [11] and Kent et al.[9] described CD4+ T cell clones specific for an epitope in this region. T cell responses to a peptide comprising the first 12 amino acids of the insulin A-chain have been shown recently to be more frequent in children with recent-onset T1D [12], supporting a role for T cell responses to this region of (pro)insulin in the pathogenesis of T1D. Interestingly, a human CD4+ T cell epitope has been identified in the neighbouring region of the C-peptide [16,17].
The A-chain of human insulin also appears to be a target of autoantibody responses [18]. High-affinity autoantibodies specific for the A-chain of insulin are present in subjects at increased risk of developing T1D [18]. To our knowledge, the role of autoantibodies in modulating the uptake of insulin and presentation of epitopes derived from the A-chain has not been explored. There may be cooperation between insulin-specific antibodies, or the B cells that express them, and T cells specific for epitopes in the A-chain.
The reason that the insulin A-chain is a target of human CD4+ T cell responses in T1D is currently unclear. This region is rich in cysteine residues (3 of 15 amino acids) that could oxidize to create neo-epitopes. For example, the A1-13 epitope we described [11] has a vicinal disulphide bond between the A6 and A7 cysteines that is essential for recognition by insulin A1-13-specific clones. In contrast, this modification is not required in the KR-A-13 epitope because T cell recognition was detected in the absence of a cysteine at A6. However, cysteines at positions A7 and A11 were essential for recognition of the KR-A-13 epitope, whereas the A11 cysteine was dispensable for the A1-13 epitope [11]. Surprisingly, treatment with the disulphide reducing agent TCEP did not prevent recognition of the KR-A1-13 epitope. Furthermore, we tested several peptides with modifications of the A11 cysteine, but none stimulated the clone (data not shown). Hence, we find no evidence of a modification of the A11 cysteine and suggest that this residue is recognized as a free thiol.
Mature insulin is produced in the pancreatic β cells by post-translational excision of the C-peptide from proinsulin to form mature insulin [19], mediated by two proteases PC1/3 and PC2 [20]. PC2 cleaves between the C-terminal arginine residue of the C-peptide and the first glycine of the A-chain, which would destroy the KR-A1-13 epitope. Most proinsulin, estimated to be ∼95%, is converted to insulin [19], making the source for the KR-A1-13 epitope more scarce than that from which the A1-13 epitope is derived. This might explain, at least partially, why T cells specific for this epitope appear to be more rare. Interestingly, an increase in the plasma proinsulin : insulin ratio has been reported in subjects with T1D [21], type 2 diabetes [22] and gestational diabetes [23]. The relative overproduction of proinsulin, under conditions of β cell stress, could promote the activation of KR-A1-13-specific CD4+ T cells and contribute to β cell destruction by increasing the amount of proinsulin available to stimulate potentially pathogenic T cells.
The clinical relevance of this new epitope remains to be determined. The clone was isolated from a subject with established T1D, but T cell responses to the epitope cannot be the result of insulin injection because only proinsulin, not insulin, is recognized. None the less, our finding reveals further diversity of human CD4+ T cell responses to proinsulin and adds to evidence that T cell recognition of the proximal insulin A-chain contributes to the pathogenesis of T1D.
Acknowledgments
S. M. is supported by an Advanced Postdoctoral Fellowship from the Juvenile Diabetes Research Foundation (JDRF). A. W. P. is supported by an NHMRC Senior Research Fellowship. N. M. O. B. S. is supported by the CRC for Oral Health Science. L. C. H. is supported by an NHMRC Senior Principal Research Fellowship. This work was supported by grants from NHMRC and JDRF. We thank Gerry Nepom for providing the HLA-transfected BLS lines and Andrew Lew and Tom Brodnicki for critical feedback on the manuscript.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. 2008;20:111–18. doi: 10.1016/j.coi.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mannering SI, Brodnicki TC. CD4+ T-cell specificity and function in type 1 diabetes. Exp Rev Clin Immunol. 2007;3:557–64. doi: 10.1586/1744666X.3.4.557. [DOI] [PubMed] [Google Scholar]
- 3.Narendran P, Mannering SI, Harrison LC. Proinsulin – a pathogenic autoantigen in type 1 diabetes. Autoimmun Rev. 2003;2:204–10. doi: 10.1016/s1568-9972(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 4.Di Lorenzo TP, Peakman M, Roep BO. Translational Mini-review series on type 1 diabetes: systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. 2007;148:1–16. doi: 10.1111/j.1365-2249.2006.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serreze DV, Leiter EH. Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Curr Dir Autoimmun. 2001;4:31–67. doi: 10.1159/000060527. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama M, Abiru N, Moriyama H, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–3. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alleva DG, Crowe PD, Jin L, et al. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. J Clin Invest. 2001;107:173–80. doi: 10.1172/JCI8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schloot NC, Willemen S, Duinkerken G, de Vries RR, Roep BO. Cloned T cells from a recent onset IDDM patient reactive with insulin B-chain. J Autoimmun. 1998;11:169–75. doi: 10.1006/jaut.1997.0183. [DOI] [PubMed] [Google Scholar]
- 9.Kent SC, Chen Y, Bregoli L, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435:224–8. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 10.Mannering SI, Dromey JA, Morris JS, Thearle DJ, Jensen KP, Harrison LC. An efficient method for cloning human autoantigen-specific T cells. J Immunol Methods. 2005;298:83–92. doi: 10.1016/j.jim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Mannering SI, Harrison LC, Williamson NA, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med. 2005;202:1191–7. doi: 10.1084/jem.20051251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marttila J, Huttunen S, Vaarala O, et al. T-cell reactivity to insulin peptide A1-12 in children with recently diagnosed type 1 diabetes or multiple β-cell antibodies. J Autoimmun. 2008;31:142–8. doi: 10.1016/j.jaut.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 13.Cowley DJ, Mackin RB. Expression, purification and characterization of recombinant human proinsulin. FEBS Lett. 1997;402:124–30. doi: 10.1016/s0014-5793(96)01511-6. [DOI] [PubMed] [Google Scholar]
- 14.Mannering SI, Morris JS, Jensen KP, et al. A sensitive method for detecting proliferation of rare autoantigen-specific human T cells. J Immunol Methods. 2003;283:173–83. doi: 10.1016/j.jim.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Giudicelli V, Chaume D, Lefranc MP. IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic Acids Res. 2004;32:W435–40. doi: 10.1093/nar/gkh412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durinovic-Bello I, Rosinger S, Olson JA, et al. DRB1*0401-restricted human T cell clone specific for the major proinsulin73-90 epitope expresses a down-regulatory T helper 2 phenotype. Proc Natl Acad Sci USA. 2006;103:11683–8. doi: 10.1073/pnas.0603682103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Congia M, Patel S, Cope AP, De Virgiliis S, Sonderstrup G. T cell epitopes of insulin defined in HLA-DR4 transgenic mice are derived from preproinsulin and proinsulin. Proc Natl Acad Sci USA. 1998;95:3833–8. doi: 10.1073/pnas.95.7.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest. 2004;114:589–97. doi: 10.1172/JCI21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dodson G, Steiner D. The role of assembly in insulin's biosynthesis. Curr Opin Struct Biol. 1998;8:189–94. doi: 10.1016/s0959-440x(98)80037-7. [DOI] [PubMed] [Google Scholar]
- 20.Bennett DL, Bailyes EM, Nielsen E, et al. Identification of the type 2 proinsulin processing endopeptidase as PC2, a member of the eukaryote subtilisin family. J Biol Chem. 1992;267:15229–36. [PubMed] [Google Scholar]
- 21.Roder ME, Knip M, Hartling SG, Karjalainen J, Akerblom HK, Binder C. Disproportionately elevated proinsulin levels precede the onset of insulin-dependent diabetes mellitus in siblings with low first phase insulin responses. The Childhood Diabetes in Finland Study Group. J Clin Endocrinol Metab. 1994;79:1570–5. doi: 10.1210/jcem.79.6.7989457. [DOI] [PubMed] [Google Scholar]
- 22.Roder ME, Porte D, Jr, Schwartz RS, Kahn SE. Disproportionately elevated proinsulin levels reflect the degree of impaired B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1998;83:604–8. doi: 10.1210/jcem.83.2.4544. [DOI] [PubMed] [Google Scholar]
- 23.Swinn RA, Wareham NJ, Gregory R, et al. Excessive secretion of insulin precursors characterizes and predicts gestational diabetes. Diabetes. 1995;44:911–15. doi: 10.2337/diab.44.8.911. [DOI] [PubMed] [Google Scholar]