

Abstract
Recruitment of immune cells to infection sites is a critical component of the host response to pathogens. This process is facilitated partly through interactions of chemokines with cognate receptors. Here, we examine the importance of fractalkine (CX3CL1) receptor, CX3CR1, which regulates function and trafficking of macrophages and dendritic cells, in the host's ability to control respiratory infections with Mycobacterium tuberculosis or Francisella tularensis. Following low-dose aerosol challenge with M. tuberculosis, CX3CR1−/− mice were no more susceptible to infection than wild-type C57BL/6 mice as measured by organ burden and survival time. Similarly, following inhalation of F. tularensis, CX3CR1−/− mice displayed similar organ burdens to wild-type mice. CX3CR1−/− mice had increased recruitment of monocytes and neutrophils in the lung; however, this did not result in increased abundance of infected monocytes or neutrophils. We conclude that CX3CR1-deficiency affects immune-cell recruitment; however, loss of CX3CR1 alone does not render the host more susceptible to M. tuberculosis or F. tularensis.
Keywords: CX3CL1, CX3CR1, fractalkine, F. tularensis, M. tuberculosis
Introduction
Respiration moves 15 000 litres of air over the extensive epithelial surface of the human airway each day [1]. As a result, the lung environment is prone to routine exposure to foreign particles and potentially harmful microorganisms. Because respiratory function is essential for life, it is critical that the host responds to foreign material and maintains a sterile environment in the lung. The host accomplishes this through a combination of physical barriers, phagocytic cells, microbicidal molecules and molecular signals that facilitate communication among these various components. Often the initial response to an invading pathogen within the lung is recognition and phagocytosis by macrophages and dendritic cells (DCs). If these cells are unable to dispose efficiently of the pathogenic invader, cytokine signals are produced which lead to the recruitment of monocytes and neutrophils from the bloodstream to infected tissues. Monocytes are the precursors to mature macrophages [2–4] and DCs [5], so during an immune response they replenish apoptotic or necrotic cells as well as provide increased numbers of phagocytic cells to aid in controlling infection. Neutrophils are terminally differentiated, highly phagocytic cells that are among the first circulating cells to migrate into infected tissues and they utilize both oxygen-dependent and oxygen-independent mechanisms to combat invading microorganisms.
The chemokine receptor CX3CR1 and its ligand, fractalkine (CX3CL1), are important for directing cell migration from the bloodstream into tissues. During infection, endothelial cells respond to inflammatory signals such as tumour necrosis factor-α, interleukin-1, lipopolysaccharide and interferon-γ by activating expression and secretion of fractalkine, a transmembrane protein [6]. Through the interaction of fractalkine with CX3CR1 on the surface of circulating cells, particularly monocytes and T cells, capture and migration of these cells into infected tissues is facilitated [6–8].
Interestingly, CX3CR1 and its ligand fractalkine are also implicated in the pathogenesis of atherosclerosis. Atherosclerotic plaques are associated with increased infiltration of monocytes and T cells [9,10], and individuals with coronary artery disease display increased expression of CX3CR1 and CX3CL1 [11]. Mice lacking CX3CR1 have decreased development of atherosclerosis [12–14]. Importantly, a CX3CR1 mutation is associated with protection from coronary artery disease [15]. Thus, CX3CR1 has been proposed as a therapeutic target for the prevention of atherosclerosis. One caveat of such treatment is the potential for increased susceptibility to infectious diseases.
Monocyte, neutrophil and lymphocyte recruitment is often a critical component of the immune response to pulmonary bacterial pathogens. Here we examine the impact of CX3CR1-deficiency during infection with the respiratory pathogens, Mycobacterium tuberculosis or Francisella tularensis. Both organisms are intracellular pathogens that cause pulmonary disease; however, the nature of infection and host response of the two bacteria differs. M. tuberculosis establishes a chronic infection in which host control depends on an adaptive T cell response and granuloma formation [16]. The granulomas are comprised primarily of macrophages and T cells and they serve to control M. tuberculosis replication and spread [17]. In M. tuberculosis infection, chemokines could play important roles in the early innate immune response, development of an adaptive immune response and/or formation of granulomas. F. tularensis causes an acute respiratory infection where innate immune cell recruitment is a significant component of host control [18]. F. tularensis infection leads to delayed recruitment of monocytes and neutrophils which become the predominant host cell during the later stages of disease [18]. In this study, we examine disease outcomes following inhalation of M. tuberculosis and F. tularensis in mice deficient for CX3CR1 and assess the contribution of CX3CR1 to monocyte and neutrophil migration to the lung during the disease course.
Materials and methods
Bacterial strains
The M. tuberculosis H37Rv was used in this study. To prepare the strain for infection, H37Rv was grown in Middlebrook 7H9 broth (Becton, Dickinson and Company, Sparks, MD, USA) with 0·2% glycerol, 1× albumin dextrose saline (ADS) and 0·05% Tween 80. F. tularensis live vaccine strain (LVS) was obtained from the Centers for Disease Control and Prevention (Atlanta, GA, USA). LVS was maintained on chocolate agar supplemented with Isovitalex (BD Biosciences, San Jose, CA, USA). Green fluorescent protein (GFP)-expressing strains contained a modified pKK214gfp plasmid [18].
Mouse strains
Female CX3CR1−/− mice 10–20 weeks old were bred at the University of North Carolina (UNC). These animals were bred 12 generations onto C57BL/6 [13]. Splenocytes from these mice did not contain CX3CR1 transcript and did not bind CX3CL1 [13]. C57BL/6 mice aged 38–45 days were purchased from Charles River Laboratories−/− or bred at UNC. The mice were maintained in sterile microbarrier cages with autoclaved chow and water.
Aerosol infection with M. tuberculosis
The bacteria were cultured to mid-log phase [optical density (OD) ∼ 0·5–1·0] in the standard media. The cultures were washed once and resuspended in phosphate-buffered saline (PBS) with 0·05% Tween 80 to a concentration of 1e7 colony-forming units (CFU)/ml. Fifteen ml of the bacterial suspension was placed into the nebulizer jar of a whole body exposure aerosol chamber (Mechanical Engineering Workshop, Madison, WI, USA). Mice were exposed for 15 min. This resulted in delivery of approximately 250 CFU to each mouse. At each time-point, four mice were killed from each infected group by CO2 asphyxiation and the organs removed and homogenized in PBS 0·05% Tween 80 with 100 ng/ml cycloheximide and 50 µg/ml carbinicillin. The homogenates were plated onto 7H10 ADS Gly plates with 10 mg/ml cycloheximide for CFU enumeration.
Intranasal infection with F. tularensis
C57BL/6 or CX3CR1−/− mice were inoculated with Francisella diluted in sterile PBS and enumerated by Klett reading or OD600. Target inoculation dose of 1000 CFU was verified by plating and counting inoculum on chocolate agar. Mice were anaesthetized with avertin until unresponsive to a toe pinch, and 50 µl of bacterial suspension was dispensed onto anterior nares of the mouse. Previous results suggest that this is an effective procedure for establishing a pulmonary Francisella infection [18]. All experiments involving animals were conducted in accordance with animal care and use guidelines, and animal protocols were approved by the Institutional Animal Care and Use Committee at UNC Chapel Hill.
Lung cell isolation
Mice were anaesthetized with avertin + heparin (1000 U/ml) and perfused with 4–7 ml of PBS + heparin (200 U/ml) until lungs were fully blanched. The trachea was cannulated using a 16-gauge blunt-tipped needle, and lungs were inflated with approximately 1 ml of the neutral protease, dispase (BD Biosciences). The trachea was tied off with surgical sutures and lungs were removed and incubated in 3·0 ml dispase at room temperature for 45 min. Tracheas were removed, and lungs were transferred to a Petri plate along with 7 ml of PBS + DNaseI (250 µg/ml) and tissue was teased apart using forceps. Cells were swirled gently in the Petri plate for 1–2 min, and the suspension was filtered through 40-µm mesh. Filtered suspensions were pelleted by centrifugation at 300 g for 5 min at 4°C and resuspended in 1 ml red blood cell lysis solution for 2 min at room temperature before adding 9 ml PBS to neutralize osmolarity. Cells were pelleted and resuspended in PBS and enumerated.
Staining of lung cells for flow cytometry
Cells were kept on ice and all incubations were performed at 4°C. Lung cells were incubated in 2.4G2 culture supernatant for 20 min to block Fc receptors. Cells (106) were stained with the following fluorescently labelled antibodies to cell surface components, F4/80 phycoerythrin (PE) (clone BM8; eBioscience, San Diego, CA, USA), GR-1 peridinin chlorophyll (PerCP) (clone RB6–8C5; BD Biosciences), CD11b PE-Cy7 (clone M1/70; eBioscience) and CD11c Alexa 647 (clone N418; eBioscience) in flow buffer (1% bovine serum albumin and 0·09% sodium azide in PBS) for 30 min. Cells were washed with PBS and fixed with 4% paraformaldehyde in PBS for 30 min. After fixation, cells were washed and resuspended in PBS and stored at 4°C until analysis by flow cytometry.
Flow cytometry of lung cells and data analysis
Cells were analysed using a CyAn™ ADP LX 9 colour flow cytometer (Dako, Glostrup, Denmark). Data were analysed using Summit version 4.3 (Dako). Compensation was performed using lung cells stained with each labelled antibody individually, and gates were drawn based on n–1 controls. Each data bar represents the mean of six mice for inoculations of wild-type C57BL/6 mice, four mice for LVS-infected CX3CR1−/− mice and two mice for PBS-inoculated CX3CR1−/− mice with error bars representing the standard deviation of the mean. Significance was determined using an unpaired two-tailed t-test. P-values of <0·05 were characterized as significant.
Identifying lung cell types
Cell types were identified based on differential staining with fluorescently labelled antibodies to F4/80, CD11b, CD11c and GR-1, as described previously [18]. Monocytes were defined as F4/80low, CD11bmid, CD11clow and GR-1low/mid with low forward- and side-scatter. Neutrophils were defined as F4/80low, CD11bhigh, CD11clow and GR-1high with low forward-scatter and heterogeneous side-scatter properties. F4/80+ cells that were CD11chighCD11blow were classified as alveolar macrophages while F4/80highCD11bhigh cells were classified as CD11bhigh macrophages. F4/80lowCD11chigh cells were classified as DCs and subdivided into CD11blow/mid DCs and CD11bhigh DCs.
Results
Aerosol infection of CX3CR1−/− mice with M. tuberculosis
Using CX3CR1−/− mice and wild-type C57BL/6 mice, the role of CX3CR1 in controlling M. tuberculosis infection was tested using a low-dose aerosol infection model. Both strains of mice were infected via the aerosol route with the virulent M. tuberculosis H37Rv strain (∼250 CFU). In three independent experiments, groups of mice were killed at various times post-infection and the bacterial burden in the lungs, livers and spleens was determined by plating organ homogenates and enumerating CFU (Fig. 1a).
Fig. 1.
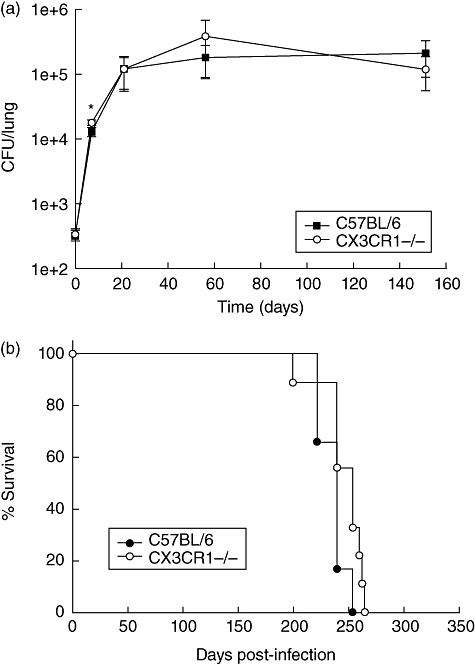
(a) Bacterial burden in lungs of wild-type (C57BL/6) and CX3CR1−/− mice infected with Mycobacterium tuberculosis H37Rv. Error bars represent standard deviation of the mean. A representative experiment of the three performed is presented. *P < 0·05. (b) Long-term survival of M. tuberculosis infected wild-type (C57BL/6) and CX3CR1−/− mice. The data reflect percentage survival over time of a group of six C57BL/6 mice and nine CX3CR1−/− mice.
As expected for H37Rv infection of C57BL/6 mice, we observed significant growth in lungs during the early phase of infection followed by a period of persistence in which the number of bacilli remained constant over time. This latter phase of infection correlates with the onset of an effective T cell-mediated immune response [19]. The same growth pattern was observed in lungs of H37Rv-infected CX3CR1−/− mice infected in parallel. However, in all three experiments the lungs of CX3CR1−/− mice exhibited a slightly higher number of CFU than the lungs of C57BL/6 mice (1·9-fold ± 0·3 standard error) at the day 7 time-point (P < 0·05). This modest increase in bacterial burden was restricted to this early time-point and observed only in lungs. By day 21 post-infection, no significant difference existed between the bacterial burden of lungs of C57BL/6 and CX3CR1−/− mice. Similarly, there was no statistically significant difference in bacterial recovery at days 56 or 154. At all time-points, histological examination of infected lung tissue revealed no difference in severity, extent of inflammation or granuloma formation (data not shown). There were also no differences in liver or spleen bacterial burdens at any time-point tested (data not shown). Long-term survival of infected animals was also monitored and revealed no difference between CX3CR1−/− and C57BL/6 mice. The mean survival time of C57BL/6-infected animals was 235 days versus 245 days for CX3CR1−/− mice (Fig. 1b). Together, these experiments demonstrate that in a low-dose aerosol infection CX3CR1 deficiency does not change tuberculosis disease outcome.
Inhalation tularaemia in CX3CR1-deficient mice
To assess the importance of CX3CR1 during pneumonic tularaemia, CX3CR1−/− mice were inoculated intranasally with 1000 CFU of F. tularensis LVS. At this dose, C57BL/6 mice develop disease, but will recover and clear the infection (data not shown) [20]. In this way, we can assess the importance of CX3CR1 in the host's ability to control the infection. Three and 7 days post-inoculation, lungs, liver and spleen were harvested from infected mice to determine bacterial organ burdens in wild-type C57BL/6 versus CX3CR1−/− mice. On day 3, logarithmic bacterial growth was evident in the lungs of both wild-type and CX3CR1−/− mice (Fig. 2) and similar numbers of bacteria were recovered from each mouse strain. Dissemination from the lung to liver and spleen, a hallmark of tularaemia, had also occurred to a similar extent by day 3 for wild-type and CX3CR1−/− mice.
Fig. 2.
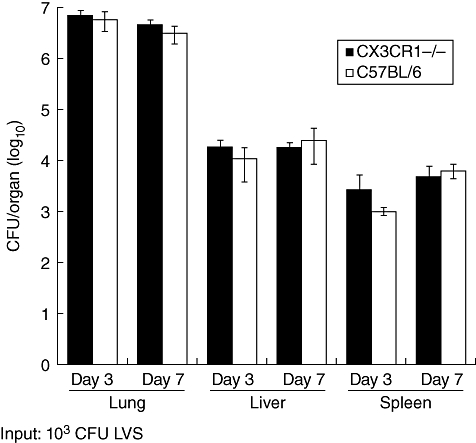
Bacterial burden in lungs, liver and spleen from wild-type and CX3CR1−/− mice infected with Francisella tularensis live vaccine strain (LVS). Error bars represent standard deviation of the mean.
Bacterial burdens in the lung did not continue to increase from days 3 to 7, indicating that the immune system was controlling the infection (Fig. 2). During a lethal infection, logarithmic bacterial growth continues in the lungs between days 3 and 7 [21]. Bacterial burdens in all organs were similar between mouse strains at day 7. These data indicated that the absence of CX3CR1 did not alter disease progression of pulmonary tularaemia, and mice deficient for CX3CR1 controlled Francisella infection similarly to wild-type mice.
Cellular response to Francisella in the lungs of CX3CR1-deficient mice
Given that CX3CR1-fractalkine interactions play a role in monocyte recruitment to infected tissues, we tested whether cell recruitment to the lung in response to pulmonary infection was altered in CX3CR1−/− mice. Using fluorescently labelled antibodies specific for F4/80, CD11b, CD11c and GR-1, we identified and analysed populations of monocytes, neutrophils, macrophages and DCs using flow cytometry. No increase in monocyte number was observed on day 1 following inhalation of Francisella by wild-type or CX3CR1−/− mice (Fig. 3a). On day 3, monocyte recruitment to the lung was evident for both wild-type and CX3CR1−/− mice; however, the degree of recruitment differed between mouse strains. There was a 2·2-fold increase in monocyte number between days 1 and 3 post-inhalation in wild-type mice and a 4·9-fold increase in CX3CR1−/− mice. At both time-points there were more monocytes in mock-inoculated CX3CR1−/− mice than in wild-type mice (P < 0·05), indicating that CX3CR1−/− mice had more resident monocytes within the lung.
Fig. 3.
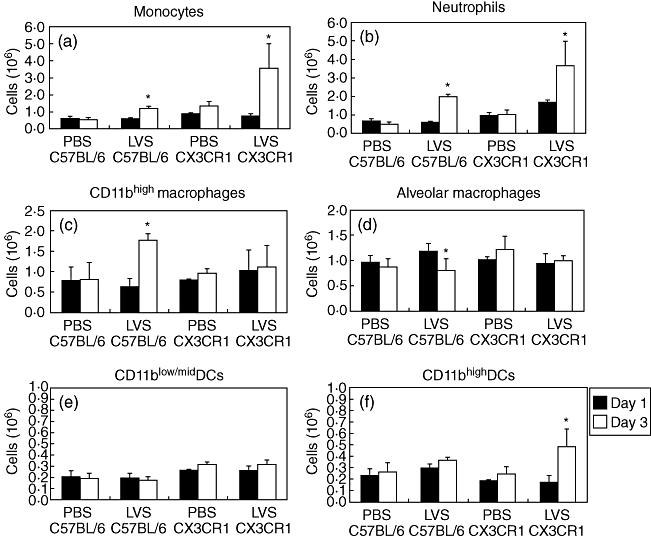
Abundance of cell populations in the lungs of wild-type and CX3CR1−/− mice following inoculation with Francisella tularensis live vaccine strain (LVS). Lungs were digested to single cell suspensions and were stained with fluorescently labelled antibodies to surface markers to enable identification of specific cell types. Statistical difference was determined between days 1 and 3 time-points. *P < 0·05 unpaired two-tailed t-test assuming unequal variance.
There was no increase in neutrophil numbers in the lung at day 1 for infected wild-type mice compared with PBS-inoculated mice (Fig. 3b). However, infected CX3CR1−/− mice displayed significantly more neutrophils at day 1 than mock-infected CX3CR1−/− mice or LVS-infected wild-type mice (P < 0·05). From days 1 to 3, the number of neutrophils increased by 3·5-fold in wild-type mice and an additional 2·2-fold for CX3CR1−/− mice. Also, there were more neutrophils in the lungs of CX3CR1-deficient mice on day 3 than in wild-type mice (P < 0·05). These data demonstrate that in CX3CR1-deficient mice there is earlier and increased neutrophil influx in response to inhaled Francisella in comparison with wild-type mice.
Once monocytes enter into tissue, they have the ability to differentiate into macrophages [4] or DCs [5]. Therefore, we also examined the number of these cell types within infected wild-type or CX3CR1−/− mice. In response to Francisella infection, CX3CR1−/− mice did not have increased numbers of CD11bhigh macrophages from days 1 to 3, whereas wild-type mice did (Fig. 3c). Alveolar macrophage numbers also stayed constant within CX3CR1−/− mice, whereas wild-type mice exhibited a decrease in this population during disease (P < 0·05) (Fig. 3d). With regard to DCs, CX3CR1−/− mice had similar numbers of CD11blow/mid DCs in the lung at days 1 and 3 compared with wild-type mice; however, CD11bhigh DC numbers increased significantly during these time-points only in CX3CR1−/− mice (P < 0·05) (Fig. 3e and f).
Francisella-infected cells following inhalation by CX3CR1−/− mice
Following inhalation, Francisella survives and replicates within multiple cell types in the lung including epithelial cells, macrophages, DCs, monocytes and neutrophils [18]. Because CX3CR1−/− mice had higher numbers of monocytes and neutrophils recruited to the lung in response to Francisella infection, this could result in a greater number of these cells becoming infected in CX3CR1−/− mice compared with wild-type mice. We infected C57BL/6 and CX3CR1−/− mice with LVS expressing GFP, which enabled the identification of infected lung cells. On day 1 following inhalation, we detected < 10 GFP+ neutrophils in the lungs of either mouse strain (Fig. 4b). By day 3 there were approximately 100 000 infected neutrophils in the lungs of each mouse strain, indicating that neutrophils become infected to the same extent in CX3CR1-deficient mice as in wild-type mice. It also indicates that despite there being increased neutrophil numbers in the lungs of CX3CR1−/− mice early during infection, there are no increased numbers of infected neutrophils. Similarly, there was no significant difference in the number of infected monocytes in the lungs of CX3CR1−/− mice when compared with wild-type mice, despite there being increased monocyte recruitment during infection in a CX3CR1-deficient background (Fig. 4a).
Fig. 4.
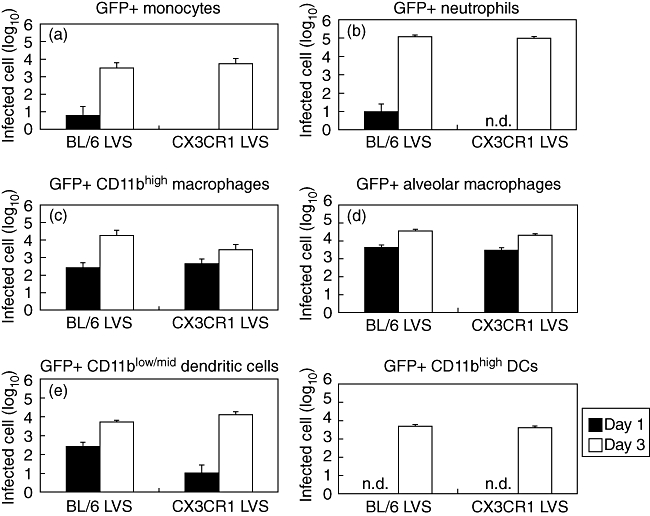
Abundance of infected cell types in the lungs of wild-type and CX3CR1−/− mice following inoculation with Francisella tularensis live vaccine strain (LVS). Mice were inoculated with green fluorescent protein (GFP)-expressing LVS to identify infected cells and lung digests were stained with fluorescently labelled antibodies to enable identification of specific cell types; n.d.: none detected at this time-point.
Absence of CX3CR1 had no impact on the number of Francisella-infected alveolar macrophages (Fig. 4d). However, CX3CR1−/− mice had fewer infected CD11bhigh macrophages on day 3 (Fig. 4c). This may be related to the observation that there was no increase in this population in the lungs of infected CX3CR1−/− mice during disease. There were initially fewer infected CD11blow/mid DCs in CX3CR1-deficient mice than wild-type; however, by day 3 similar numbers of infected DCs were found in the lungs of each mouse strain (Fig. 4e). Infected CD11bhigh DC numbers were similar between mouse strains (Fig. 4f).
These data, taken together, indicate that the absence of CX3CR1 impacts the recruitment of monocytes and neutrophils from the bloodstream into the lung, and this alteration could impact the population dynamics of macrophages and DCs during infection of bacterial pathogens. However, these dissimilarities did not result in an appreciable difference in disease outcome following inhalation of the pulmonary bacterial pathogens, M. tuberculosis and F. tularensis.
Discussion
An important component of the innate immune response is the recruitment and extravasation of circulating inflammatory cells into infected tissues. Here we demonstrate that the absence of the fractalkine receptor, CX3CR1, does not have an appreciable impact on disease outcome during pulmonary infections with M. tuberculosis or F. tularensis. While this indicates that CX3CR1 is not essential for host control of these infections, it does not rule out any contribution for CX3CR1 as there may exist functional redundancy with other chemokine receptors [22]. Similar to our results, when a low-dose M. tuberculosis infection model is employed, mice deficient in chemokine receptors CCR2 and CCR5 exhibit no defect in the control of infection [23,24]. The true contribution of chemokine receptors to disease outcome may be revealed only once mice lacking multiple chemokine receptor genes are tested.
As an alternative approach to investigate a role for CX3CR1 in host response to pulmonary infection, we evaluated cell recruitment to the lungs during infection with Francisella. The absence of CX3CR1 resulted in elevated numbers of monocytes and neutrophils being present in the lungs of infected animals during the later stages of pulmonary tularaemia. This is somewhat surprising, as the known function of CX3CR1 is to facilitate binding of monocytes and other CX3CL1-expressing cells to endothelial cells which precedes migration into infected tissues. Therefore, we might have expected to observe fewer monocytes recruited to the lung during pulmonary tularaemia in CX3CR1-deficient mice. However, there is precedence for increased cell recruitment in the absence of chemokine receptors. In studies with CCR5-deficient mice following infection with M. tuberculosis, increased lymphocyte migration is observed [24].
We were also surprised to observe an effect of CX3CR1-deficiency on neutrophil influx. CX3CR1 is not expressed highly on neutrophils, nor does fractalkine stimulate migration of these cells [6,7]. Therefore, it is unclear why the absence of CX3CR1 would impact upon the kinetics or magnitude of the neutrophil response. One explanation for the increased inflammatory-cell recruitment observed in CX3CR1-deficient mice is that mice lacking chemokine receptors might have higher serum or tissue levels of corresponding chemokines that may act similarly on analogous receptors. CX3CR1−/− mice have 300-fold higher levels of fractalkine circulating in the blood than wild-type mice [25], which could support this hypothesis. Elevated fractalkine levels could lead to increased interactions with other chemokine receptors important for cell recruitment and extravasation such as CCR2 and CCR5. Additionally, it is plausible that the levels of other chemokines, such as CCL2 or CCL5, or cytokine profiles could be altered in CX3CR1-deficient mice which could impact upon cell recruitment in response to infection.
After receiving an intranasal dose of 1000 CFUs of Francisella LVS, C57BL/6 mice will recover and eventually clear the bacterial infection. During a fatal infection, the bacterial burden in lungs, liver and spleen continues to increase exponentially from days 3 to 7, whereas in recovering animals the number of Francisella decreases during this time-point [20,26]. In mice lacking CX3CR1, the Francisella organ burden declined from days 3 to 7, indicating that mice could recover from tularaemia similarly to mice with wild-type levels of the receptor. Despite differences in the abundance of various cell populations between CX3CR1 expressing and deficient strains, the make-up of Francisella-infected cells was not affected. These data indicate that CX3CR1 plays a role in regulating inflammatory cell recruitment to the lung; however, it is not critical in the disease outcome of F. tularensis.
Studies by Liu et al. demonstrated that CX3CR1-deficient mice are less likely to develop intimal hyperplasia following femoral artery injury because of decreased monocyte infiltration at the perturbation site [27]. CX3CR1-deficiency has been implicated similarly in protection from coronary artery disease and internal carotid artery occlusive disease [15]. CX3CR1−/− mice display reduced atherosclerotic plaque burden [28]. Therefore, targeting CX3CR1 has been proposed as a potential drug target for acute and chronic vascular injuries, such as restenosis and atherosclerosis. One potential problem with this treatment would be the potential increased susceptibility to infectious pathogens because of decreased or altered immune cell infiltration. CX3CR1-deficient mice have demonstrated increased susceptibility to Salmonella infections [29]. Results presented here demonstrate that, at least for the respiratory human pathogens M. tuberculosis and F. tularensis, the ability to control infection is not impeded by a deficiency of CX3CR1, indicating that targeting CX3CR1 for the treatment of heart disease may be a viable method.
Acknowledgments
This work was supported by a Southeast Regional Center of Excellence in Biodefense and Emerging Infections grant (NIH/NIAID U54-AI057157) and by the National Institutes of Health (R01-HL077406, R21-AI053399) and UNC.
References
- 1.McCormack FX, Whitsett JA. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J Clin Invest. 2002;109:707–12. doi: 10.1172/JCI15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. 1968;128:415–35. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van oud Alblas AB, van Furth R. Origin, kinetics, and characteristics of pulmonary macrophages in the normal steady state. J Exp Med. 1979;149:1504–18. doi: 10.1084/jem.149.6.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Oud Alblas AB, van der Linden-Schrever B, Van Furth R. Origin and kinetics of pulmonary macrophages during an inflammatory reaction induced by intravenous administration of heat-killed bacillus Calmette–Guérin. J Exp Med. 1981;154:235–52. doi: 10.1084/jem.154.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–61. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 6.Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–4. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 7.Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–30. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 8.Fong AM, Robinson LA, Steeber DA, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–19. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–8. doi: 10.1161/01.atv.6.2.131. [DOI] [PubMed] [Google Scholar]
- 10.Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 11.Damas JK, Boullier A, Waehre T, et al. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, is elevated in coronary artery disease and is reduced during statin therapy. Arterioscler Thromb Vasc Biol. 2005;25:2567–72. doi: 10.1161/01.ATV.0000190672.36490.7b. [DOI] [PubMed] [Google Scholar]
- 12.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1–/– mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–40. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Combadiere C, Potteaux S, Gao JL, et al. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–16. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 14.Liu P, Yu YR, Spencer JA, et al. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol. 2008;28:243–50. doi: 10.1161/ATVBAHA.107.158675. [DOI] [PubMed] [Google Scholar]
- 15.McDermott DH, Fong AM, Yang Q, et al. Chemokine receptor mutant CX3CR1-M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest. 2003;111:1241–50. doi: 10.1172/JCI16790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders BM, Britton WJ. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol. 2007;85:103–11. doi: 10.1038/sj.icb.7100027. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Juarrero M, Turner OC, Turner J, Marietta P, Brooks JV, Orme IM. Temporal and spatial arrangement of lymphocytes within lung granulomas induced by aerosol infection with Mycobacterium tuberculosis. Infect Immun. 2001;69:1722–8. doi: 10.1128/IAI.69.3.1722-1728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall JD, Woolard MD, Gunn BM, et al. Repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–52. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloom BR. Tuberculosis: pathogenesis, protection, and control. Washington, DC: ASM Press; 1994. [Google Scholar]
- 20.Wu TH, Hutt JA, Garrison KA, Berliba LS, Zhou Y, Lyons CR. Intranasal vaccination induces protective immunity against intranasal infection with virulent Francisella tularensis biovar A. Infect Immun. 2005;73:2644–54. doi: 10.1128/IAI.73.5.2644-2654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall JD, Craven RR, Fuller JR, Pickles RJ, Kawula TH. Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect Immun. 2007;75:1034–9. doi: 10.1128/IAI.01254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Algood HM, Chan J, Flynn JL. Chemokines and tuberculosis. Cytokine Growth Factor Rev. 2003;14:467–77. doi: 10.1016/s1359-6101(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 23.Scott HM, Flynn JL. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect Immun. 2002;70:5946–54. doi: 10.1128/IAI.70.11.5946-5954.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Algood HM, Flynn JL. CCR5-deficient mice control Mycobacterium tuberculosis infection despite increased pulmonary lymphocytic infiltration. J Immunol. 2004;173:3287–96. doi: 10.4049/jimmunol.173.5.3287. [DOI] [PubMed] [Google Scholar]
- 25.Cardona AE, Sasse ME, Mizutani M, et al. Scavenging roles of chemokine receptors: chemokine receptor deficiency is associated with increased levels of ligand in circulation and tissues. Blood. [DOI] [PMC free article] [PubMed]
- 26.Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, Kawula TH. RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect Immun. 2008;76:4934–43. doi: 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu P, Patil S, Rojas M, Fong AM, Smyth SS, Patel DD. CX3CR1 deficiency confers protection from intimal hyperplasia after arterial injury. Arterioscler Thromb Vasc Biol. 2006;26:2056–62. doi: 10.1161/01.ATV.0000234947.47788.8c. [DOI] [PubMed] [Google Scholar]
- 28.Teupser D, Pavlides S, Tan M, Gutierrez-Ramos JC, Kolbeck R, Breslow JL. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci USA. 2004;101:17795–800. doi: 10.1073/pnas.0408096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–8. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]