

Abstract
Intercellular adhesion molecul-1 (ICAM-1) is a transmembrane glycoprotein belonging to the immunoglobulin superfamily of adhesion molecules and plays perdominant roles in recruitment and trafficking of leucocytes to sites of inflammation. ICAM-1 expression in intestinal epithelial cells (IECs) is enhanced by several stimuli, such as proinflammatory cytokines, bacterial infections or pathogen-associated molecular patterns. One of these stimuli, double-stranded RNA (dsRNA), is a by-product of viral replication and can be recognized by its cognate receptor Toll-like receptor 3 (TLR-3). In spite of expression of both TLR-3 and ICAM-1 in IECs, correlation between TLR-3-signalling and ICAM-1 expression has never been examined in IECs. In the present study, we investigated whether poly I:C, an analogue of dsRNA, can stimulate the expression of ICAM-1 in IEC line, HT-29. Poly I:C-stimulation up-regulated the expression of ICAM-1 mRNA by real-time polymerase chain reaction. Enhanced expression of ICAM-1 was confirmed in protein level by immunofluoresense cell staining and enzyme-linked immunosorbent assay by measuring the released soluble ICAM-1 in culture supernatant. As the stimulation effect was reduced by pre-treatment of the cells with anti-TLR-3 antibody, poly I:C-binding signal was thought to be sensed by TLR-3 on the surface of HT-29. The results of luciferase assay and nuclear factor kappa-b (NF-kB) inhibitor treatment experiments indicated that the downstream signal was mainly transduced by transcription factor, NF-kB. All these results demonstrated the connection between TLR-3 signalling and ICAM-1 expression in HT-29 cells and indicated the importance of coordinated function of both innate and adaptive immunity against viral infections.
Keywords: ICAM-1, IECs, poly I:C, TLR-3
Introduction
Intestinal epithelial cells (IECs) covering the outer surface of the gut play a pivotal role in maintaining intestinal homeostasis [1–3]. They not only form a physical barrier; IECs also drive innate and adaptive immunity against invading pathogens [4,5]. The innate immune system recognizes conserved motifs in pathogens termed ‘pathogen-associated molecular patterns’, and recognition of these motifs is mediated partly by Toll-like receptors (TLRs) [6–8]. To date, 13 TLRs have been identified, all of which are able to trigger innate immunity, followed by adaptive immunity.
The TLR-3 recognizes the double-stranded RNA (dsRNA), a by-product of RNA virus replication [9,10]. IECs express TLR-3 and stimulation with dsRNA can induce anti-viral immunity [11–14].
Intercellular adhesion molecule-1 (ICAM-1) is a cell surface transmembrane glycoprotein belonging to the immunoglobulin superfamily of adhesion molecules with a molecular weight ranging from 80 to 100 kDa [15]. The predominant function of ICAM-1 is the recruitment and trafficking of leucocytes via interaction with the integrins expressed on their cell surface [15]. Thus, the most abundant expression of ICAM-1 is detected in endothelia [15].
The ICAM-1 is also known to be expressed in various epithelial cell lines, including those from intestinal [16,17], pulmonary [18,19] and oral origins [20]. In these cell lines, ICAM-1 expression is induced by several different stimuli, such as proinflammatory cytokines or bacterial infections. ICAM-1 is also utilized by some viruses, such as rhinovirus, as a receptor for cellular entry [15,21,22]. In alveolar epithelial cells, Haemophilus influenzae infection can induce ICAM-1 and results in high susceptibility to influenza virus infection [18]. These co-operative effects are thought to cause frequent peaks of activity in chronic obstructive pulmonary disease. Accordingly, poly I:C, a synthetic analogue of dsRNA, can stimulate the expression of ICAM-1 in respiratory epithelial cells [19]. ICAM-1 is also detected in IECs and its expression up-regulated by several cytokines [23–26], thus enhancing the binding of rhinovirus [23].
These observations suggest that poly I:C can enhance directly the production of ICAM-1 in IECs. Despite the expression of TLR-3 in IECs, the influence of poly I:C-stimulation on the expression of ICAM-1 in IECs has not yet been examined, a fact that prompted us to investigate the relationship between TRL-3 signalling and ICAM-1 induction in IECs. The aim of this study was to examine whether the human colonic adenocarcinoma cell line HT-29 can respond to the TLR-3-ligand poly I:C to produce ICAM-1. Downstream signalling was also examined and poly I:C-induced transcriptional regulation of ICAM-1 is discussed.
Materials and methods
Reagents
Poly I:C, phorbol 12-myristate 13-acetate (PMA) and nuclear factor kappa-β (NF-κB) inhibitor L-1-4′-tosylamino-phenylethyl-chloromethyl ketone (TPCK) was purchased from Sigma (St Louis, MO, USA). Isohelenin, another NF-κB inhibitor, was purchased from Calbiochem (Darmstadt, Germany). Antibodies against human ICAM-1 and human interferon (IFN) regulatory factor-3 (IRF-3) were purchased from SantaCruz Biotechnology (Santa Cruz, CA, USA). Anti-human TLR-3 antibody was purchased from Imgenex (San Diego, CA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunogloulin G (IgG) (H+L) antibody and fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (H+L) antibody were purchased from Zymed Inc. (South San Francisco, CA, USA). Monomeric cyanine nucleic acid stains was purchased from Invitrogen (Tokyo, Japan).
Cell culture
HT-29 cells and Caco-2 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 50 µg/ml streptomycin and 50 U/ml penicillin (10% FCS–DMEM).
DNA construction
For the luciferase assay, 1·4 kb of human ICAM-1 5′-untranslated region was amplified by polymerase chain reaction (PCR) using genomic DNA extracted from HT-29 cells as template. The sequences of primers are listed in Table 1. The amplified PCR product was subcloned into Zero blunt vector (Invitrogen). The 800 base pairs (bp) Nhe I fragment (from −839 bp to −38 bp from the transcription initiation site) was excised and subcloned into pGL4 vector (Stratagene, La Jolla, CA, USA). This plasmid was designated as pGL4-NF-κB. For the NF-κB-binding site deletion mutant (pGL4-non-NF), the Quickchange II site-directed mutagenesis kit (Stratagene) was used to alter specific sequences. The sequences of primers for this construct are also listed in Table 1.
Table 1.
Primers used in this study.
| Name | Sequence |
|---|---|
| 5-UT (1450 bps) | |
| Forward primer | CTCCTCAGCCCCCCAAGAAA |
| Reverse primer | CCTGGGAACAGAGCCCCGAG |
| Non-NF-κB | |
| Forward primer | CCGATTGCTTTAGCTGGAGCTGAAGCGGCC |
| Reverse primer | GGCCGCTTCAGCTCCAGCTAAAGCAATCGG |
| Real-time PCR ICAM-1 (81 bp) | |
| Forward primer | CTCCAATGTGCCAGGCTTG |
| Reverse primer | CAGTGGGAAAGTGCCATCCT |
| Real-time PCR β-actin (225 bp) | |
| Forward primer | TTCCAGCCTTCCTTCCTGG |
| Reverse primer | TTGCGCTCAGGAGGAGCAA |
| RT–PCR TLR-3 (213 bps) | |
| Forward primer | TGCTGCAAATCGAGAATTTCT |
| Reverse primer | TGGATACGTTTCCTTATAAG |
ICAM-1, intercellular adhesion molecule-1; PCR, polymerase chain reaction; TLR, Toll-like receptor; bp, base pairs; NF-κb, nuclear factor kappa-β; RT–PCR, reverse transcription–polymerase chain reaction.
The dsRNA stimulation, real-time PCR and reverse transcription–PCR
Cells were detached from culture dishes using 0·05% trypsin–ethylenediamine tetraacetic acid the day before stimulation and plated at a density of 2 × 105/35 mm dish and cultured for 18 h in a 5% CO2 incubator. The cells were washed twice with 10% FCS–DMEM and stimulated with or without 1–1000 µg/ml of poly I:C for 18 h. At the end of culture, cells were washed once with PBS and total RNA was extracted using an RNeasy mini kit (Qiagen, Tokyo, Japan). reverse transcription–PCR was performed as described previously [27]. Real-time PCR was performed with SYBR premix Ex Taq (Takara, Tokyo, Japan). The primers used in this study are listed in Table 1. For NF-κB inhibition experiments, the cells were incubated with 25 µM of TPCK for 30 min[28] or with 20 µM isohelenin [29] for 2 h in a CO2 incubator. Cells were washed with 10% FCS–DMEM three times and cultured further in the presence or absence of 100 µg/ml of poly I:C. For anti-TLR-3 antibody inhibition, the cells were preincubated with anti-TLR-3 antibody for 2 h and then stimulated with poly I:C. Values presented for the real-time PCR experiments were expressed as fold induction compared with basal levels of non-stimulated cells. The expression levels of ICAM-1 mRNA were normalized with the β-actin mRNA.
Immunoprecipitation
HT-29 cells were prepared and stimulated by poly I:C as described above. After stimulation, the cells were lysed with 1 ml of cell lysis buffer (50 mM Tris-HCl, pH 7·5, 150 mM NaCl, and 0·5% TritonX-100) and subjected to immunoprecipitation followed by Western blotting. Briefly, cell lysates were incubated with or without 5 µl of anti-ICAM-1 or anti-TLR-3 antibodies for 1 h at 4°C respectively. After incubation, 10 µl of protein G-sepharose was added and incubated for a further 1 h. The samples were washed five times with lysis buffer and subjected to Western blot as described previously [30].
Immunofluoresence staining
HT-29 cells were plated on coverslips at a density of 2 × 105. The cells were stimulated with or without 100 µg/ml of poly I:C for 24 h. Immunofluorescent staining was performed as described previously [30]. The primary antibodies against human ICAM-1 or human TLR-3 were diluted 100× or 166× with 1% bovine serum albumin (BSA)–PBS respectively, and incubated with cells for 1 h at room temperature. After washing, the coverslips were incubated with 400× diluted FITC-goat anti-mouse IgG (H+L) for 1 h. The cells were washed three times with PBS and counterstaining was performed by incubating the cells with monomeric cyanine nucleic acid stains (×500 diluted with PBS) for 10 min. After washing, the coverslips were mounted on glass slides using Aqua-Polymount (Polysciences, Warrington, PA, USA). Images were viewed and photographed using a LSM510 confocal laser microscope (Carl Zeiss, Heidelberg, Germany).
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) was performed on 100 µl aliquots of culture supernatant from poly I:C-stimulated or unstimulated HT-29 cells in order to estimate the concentration of soluble ICAM-1 (sICAM-1). The Endogen human sICAM-1 ELISA kit (Pierce, Rockford, IL, USA) was used. The absorbance was measured using a Microplate reader model 3550 (BioRad, Tokyo, Japan).
Assay
HT-29 cells were plated on 24-well culture plates at a density of 2 × 105 the day before transfection. The cells were washed twice with OPTI-MEM (Invitrogen) and a reporter plasmid, pGL4-NF-κB or pGL4-non-NF, was transfected using the lipofectamine plus transfection method. After 6 h of transfection, the cells were washed with 10% FCS–DMEM and cultured for a further 42 h. The cells were washed and stimulated with or without 100 µg/ml of poly I:C for 3 h. At the end of stimulation, the cells were lysed with 1× passive lysis buffer (Promega, Tokyo, Japan) and the cell lysates were collected. Transfection efficiency was normalized to the renilla luciferase activity by co-transfection with pRL/cytomegalovirus vector (Promega). Both firefly and renilla luciferase activities were determined using the Dual-Luciferase Reporter Asay System (Promega). Luminescence was measured on a Lumat LB9507 luminometer (Berthold, Bad Wildbad, Germany).
Small interfering RNA experiment
For small interfering RNA (siRNA) transfection, 2 × 105 of HT-29 cells were plated on 24-well culture plates the day before transfection. The cells were cultured with 10% FCS–DMEM in the absence of antibiotics. Before transfection, the cells were washed three times with OPTI-MEM. siRNA against IRF-3 or control siRNA (Invitrogen) was dissolved in OPTI-MEM at a concentration of 100 nM and incubated with lipofectamine MAX transfection reagent (Invitrogen) for 20 min at room temperature. The transfection mixture was applied to cells and transfection was performed for 6 h in a CO2 incubator. After transfection, the cells were washed with 10% FCS–DMEM once and cultured for a further 42 h. IRF-3 expression levels between siRNA transfected and non-transfected cells were compared by Western blotting as described previously [30]. Anti-IRF-3 antibody (primary) and the HRP-conjugated goat anti-rabbit IgG (H+L) (secondary) were diluted with 1% BSA–PBS to 10× and 5000× respectively. For ELISA assay, the cells were stimulated with 100 µg/ml poly I:C for 24 h after transfection. The culture supernatants were harvested and the amount of sICAM-1 was measured.
Results
Poly I:C-stimulation can induce the expression of ICAM-1 in IEC
To determine whether dsRNA-stimulation can induce the expression of ICAM-1 in the human IEC line, HT-29, was stimulated with 100 µg/ml of poly I:C. Because the expression of ICAM-1 in HT-29 cells may be induced by PMA-stimulation [23], HT-29 cells were also stimulated with 1 µM of PMA and included as a control. After stimulation, total RNA was extracted and the ICAM-1 mRNA expression was compared by real-time PCR. Constitutive low-level expression was observed in non-stimulated HT-29 cells (Fig. 1a). ICAM-1 was up-regulated by PMA-stimulation approximately fivefold. The extent of ICAM-1 up-regulation was much more marked in poly I:C-stimulated HT-29 cells and reached as high as eight- to ninefold. In order to eliminate the possibility that poly I:C-induced ICAM-1 expression was an HT-29 cell-specific phenomenon, another IEC line, Caco-2, was also examined. Although induction efficiency was lower than in HT-29 cells, poly I:C-stimulation resulted in twofold up-regulation in Caco-2 cells (Fig. 1b), indicating that poly I:C-stimulated induction of ICAM-1 in IECs is a common phenomenon. To determine the optimal dose for ICAM-1 induction in HT-29 cells, the cells were stimulated with various concentrations, 0–1000 µg/ml, of poly I:C. A low-level induction was observed at 10 µg/ml and maximum induction was observed at 100 µg/ml of poly I:C (Fig. 2a). The optimal stimulation length was also examined. ICAM-1 expression was up-regulated time-dependently with the most effective induction observed after 3 h of stimulation (Fig. 2b). These results indicate clearly that poly I:C-stimulation can induce expression of ICAM-1 in IECs in a dose- and time-dependent manner.
Fig. 1.

Poly I:C can induce the expression of intercellular adhesion molecule-1 (ICAM-1) mRNA. HT-29 cells, 2 × 105 (a, upper panel) or Caco-2 cells (b, lower panel), were stimulated with 100 µg/ml of poly I:C in 10% fetal calf serum–Dulbecco's modified Eagle's medium (FCS–DMEM) for 18 h in a 5% CO2 incubator. HT29 cells were also stimulated with 1 µM of phorbol 12-myristate 13-acetate for 1 h, washed and cultured further with 10% FCS–DMEM for 16 h. The cells were washed with phosphate-buffered saline and total RNA was extracted. The cDNA was synthesized using random hexamer primers and subjected to real-time polymerase chain reaction. Results are expressed as a mean plus or minus the standard deviation of three different experiments.
Fig. 2.
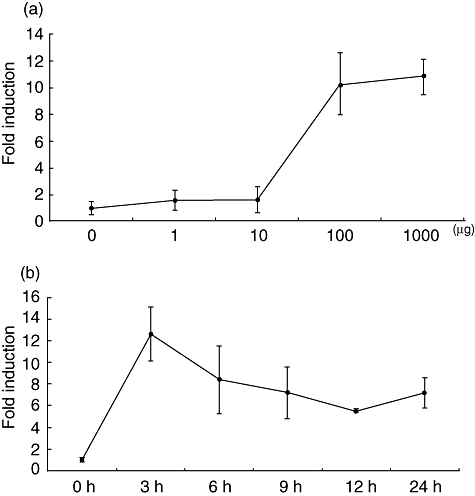
Time- and dose-dependent induction of intercellular adhesion molecule-1 (ICAM-1) mRNA in HT-29 cells. HT-29 cells were stimulated and the real-time polymerase chain reaction was performed by the same protocol described in Fig. 1. Results are expressed as mean plus or minus the standard deviation of three different experiments. (a) The cells were stimulated for 18 h; (b) the cells were stimulated with 100 µg/ml of poly I:C.
Poly I:C-stimulated up-regulation of ICAM-1 protein
Poly I:C-stimulated ICAM-1 production in HT-29 cells was also examined by protein level. First, we attempted immunofluorescence cell staining. After 24 h stimulation, HT-29 cells were incubated with anti-ICAM-1 antibody followed by FITC-conjugated anti-mouse IgG (H+L) antibody. Although only a very faint signal was detected in non-stimulated cells (Fig. 3a, left panel), significant up-regulation was detected clearly in poly I:C-stimulated cells (Fig. 3a, right panel). We also performed an immunoprecipitation experiment followed by Western blotting. Although ICAM-1 was under detectable levels in non-stimulated HT-29 cells, poly I:C-stimulation clearly up-regulated ICAM-1 expression in HT-29 cells (Fig. 3b).
Fig. 3.

Detection of poly I:C-stimulated production of intercellular adhesion molecule-1 (ICAM-1) protein. (a) HT-29 cells, 2 × 105, were plated on coverslips the day before stimulation. After stimulation with or without poly I:C, the cells were stained with anti-ICAM-1 antibody followed by fluorescein isothiocyanate-conjugated goat anti-mouse ICAM-1 monoclonal antibody. Images (×63) were taken using a LSM510 confocal laser microscope (Carl Zeiss, Heidelberg, Germany). (b) HT-29 cells were stimulated with or without 100 µg/ml of poly I:C for 24 h. After stimulation, the cell lysates were prepared and ICAM-1 was immunoprecipitated with anti-ICAM-1 antibody. The immunoprecipitated samples were loaded on 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and subjected to Western blotting. The clear 100 kDa band was detected when HT-29 cells were stimulated with poly I:C. (c) HT-29 cells were stimulated with (+) or without (−) 100 µg/ml of poly I:C for the times indicated. The culture supernatants were collected and the amount of sICAM-1 was measured by enzyme-linked immunosorbent assay. The means of four different experiments are shown.
Next, we performed ELISA to measure sICAM-1 secreted in culture supernatants. sICAM-1 increased time-dependently, and after 48 h of stimulation the amount of sICAM-1 reached 25 ng/ml (Fig. 3c).
All these results indicate that poly I:C-stimulation can induce the production of ICAM-1 protein by HT-29 cells.
Anti-TLR-3 antibody can block the dsRNA-stimulated induction of ICAM-1
The TLR-3 is the cognate receptor for dsRNA [9,10], and ICAM-1 induction by poly I:C may be mediated by TLR-3 in HT-29 cells. To examine the expression of TLR-3 in HT-29 cells, total RNA was extracted and subjected to reverse transcription–PCR. TLR-3 mRNA was clearly detected as a 213 bp band (Fig. 4a). No band was detected when reverse transcription–PCR was performed in the absence of TLR-3-specific primers, indicating the specific amplification of TLR-3 mRNA.
Fig. 4.

Anti-Toll-like receptor (TLR)-3 antibody reduced poly I:C-stimulated intercellular adhesion molecule-1 (ICAM-1) expression and production. (a) Total RNA was extracted from HT-29 cells and subjected to reverse transcription–polymerase chain reaction. The clear 213 base pairs (bp) band was detected (lane 2). This band was not detected in the absence of TLR-3-specific primers (lane 1). The expression of β-actin was shown in the lower panel. (b) The HT-29 cell lysate was prepared and subjected to immunoprecipitation followed by Western blotting. When the cell lysate was incubated with anti-TLR-3 antibody, a clear 115 kDa band was detected (right), but not in non-specific control antibody (left). (c) HT-29 cells were plated on coverslips and incubated with control (left) or anti-TLR-3 antibody (right) followed by fluorescein isothiocyanate–goat anti-mouse immunoglobulin (H+L). Very weak but significant stainings were detected in the anti-TLR-3 antibody staining. (d) HT-29 cells were pretreated with 1µg/ml of anti-TLR-3 antibody for 2 h and stimulated with poly I:C for 3 h. Total RNA was extracted and subjected to real-time polymerase chain reaction. Anti-TLR-3 antibody treatment reduced the induction of ICAM-1 significantly. Addition of control antibody was not affected on ICAM-1 induction. *P < 0·05 for the comparison of anti-TLR-3 plus and minus conditions. (e) After pretreatment with anti-TLR-3 antibody, as above, the cells were stimulated further with poly I:C for 24 h. The culture supernatants were collected and subjected to enzyme-linked immunosorbent assay for estimation of sICAM-1. The amount of sICAM-1 in poly I:C-stimulated HT-29 cells were set as 100% and compared with antibody-treated cells.
Immunoprecipitation experiment followed by Western blotting confirmed further the expression of TLR-3. Namely, TLR-3 was detected as a 115 kDa band by incubating HT-29 cell lysate with anti-TLR-3 antibody (Fig. 4b, right), but not with control antibody (Fig. 4b, left).
To demonstrate the cell surface expression of TLR-3, immunofluorescence stainings were performed. Very weak but significant expression of TLR-3 was detected when HT-29 cells were incubated with anti-TLR-3-specific antibody (Fig. 4c, right), but not with control antibody (Fig. 4c, left). All these data demonstrated the expression of TLR-3 in HT-29 cells.
To confirm the TLR-3-mediated signalling pathway, HT-29 cells were pretreated with specific antibody against TLR-3. After pretreatment, cells were stimulated with or without poly I:C and ICAM-1 expression was compared by real-time PCR. Pretreatment of cells with anti-TLR-3 antibody abrogated poly I:C-stimulated induction of ICAM-1 almost to background levels (Fig. 4d). Non-specific mouse IgG antibody did not affect poly I:C-stimulated ICAM-1 expression [Fig. 4d, control antibody (+)]. The inhibitory effect of anti-TLR-3 antibody on the production of ICAM-1 was also examined by estimating the sICAM-1 in the culture supernatants. sICAM-1 secretion was reduced in anti-TLR-3 antibody-treated cells (Fig. 4e). When the amount of sICAM-1 without antibody treatment was set at 100%, anti-TLR-3 antibody treatment reduced the secretion of sICAM-1 to approximately 40%. Taken together, these results indicate that the poly I:C signal was sensed by TLR-3 and resulted in the production of ICAM-1 in HT-29 cells.
The NF-κB-dependent ICAM-1 induction
To examine the possible involvement of the transcription factor, NF-κB, in the downstream signalling of TLR-3, cells were pretreated with NF-κB-specific inhibitor, TPCK and isohelenin respectively, and stimulated further with poly I:C. Both inhibitors reduced poly I:C-stimulated ICAM-1 expression (Fig. 5a). Inhibitor treatment itself did not affect the expression of ICAM-1 (data not shown).
Fig. 5.
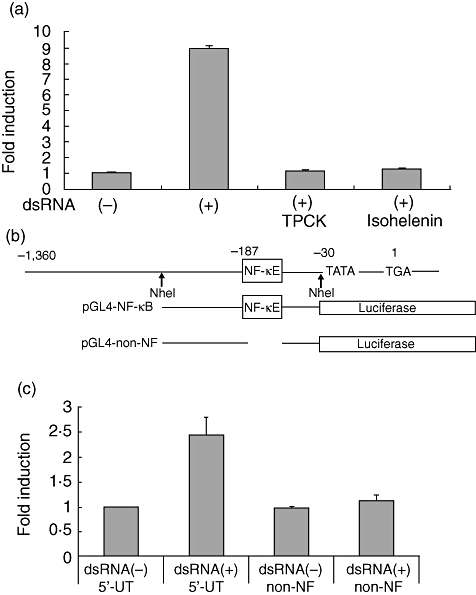
Nuclear factor kappa-B (NF-κB)-dependent induction of intercellular adhesion molecule-1 (ICAM-1). (a) HT-29, 2 × 105, cells were preincubated with L-1-4-tosylamino-phenylethyl-chloromethyl ketone or isohelenin for 30 min and 2 h respectively. After wash, the cells were stimulated with poly I:C for 3 h and the total RNA were extracted and subjected to real-time polymerase chain reaction. The means of three different experiments were shown. (b) Schematic illustration of the reporter construct for the luciferase assay. pGAL4-NF-κB contains 800 base pairs (bp) of ICAM-1 5′-regulatory region. pGAL4-non-NF lacks 10 bp of a NF-κB binding motif. Nucleotide numbering is relative to the transcription initiation site TGA, where G is +1. (c) HT-29 cells, 2 × 105, were plated on a 24-well plate and transfected with reporter plasmids and pRL/cytomegalovirus (CMV) plasmid for 6 h. The cells were washed with 10% fetal calf serum–Dulbecco's modified Eagle's medium and cultured for a further 40 h. The cells were stimulated with poly I:C for 3 h and the cell lysates were prepared. The luciferase activity was measured as described in Materials and methods. The results are expressed as mean plus or minus the standard deviation of three different experiments.
The NF-κB-dependent induction of ICAM-1 in HT-29 cells was examined further by luciferase assay. For this purpose, 1·4 kb fragments of the 5′-untranslated region of the ICAM-1 gene was amplified by PCR. This fragment was sequenced and shown to contain a 10 bp NF-κB-binding motif. The Nhe I fragment containing the NF-κB-binding motif was excised and ligated to the reporter plasmid pGL4 (designated pGL4-NF-κB, Fig. 5b). This plasmid was transfected into HT-29 and the cells stimulated with or without poly I:C for 3 h. Poly I:C-stimulation resulted in a 2·5-fold induction of luciferase activity (Fig. 5c). To confirm further the involvement of NF-κB for poly I:C-stimulated induction of ICAM-1, the 10 bp NF-κB binding site was deleted (Fig. 5b). The resultant plasmid was sequenced and designated pGL4-non-NF. Deletion of the NF-κB site removed luciferase activity completely (Fig. 5c), indicating that poly I:C-stimulated induction of ICAM-1 in HT-29 cells is mediated mainly by NF-κB activation.
Minor contribution of IRF-3 in poly I:C-stimulated induction of ICAM-1
Because the downstream signalling of TLR-3 may also be mediated by IRF-3, we attempted to examine the contribution of IRF-3 in ICAM-1 induction. For this purpose, siRNA experiments were performed. After siRNA transfection, the expression level of IRF-3 was examined by Western blotting. IRF-3 was reduced by approximately 70% in transfected cells (Fig. 6a, lane 2), but not in non-transfected (lane 1) or control siRNA-transfected cells (lane 3). Under these same conditions, cells were stimulated with poly I:C and sICAM-1 levels were measured by ELISA. sICAM-1 secretion was reduced by 20% in IRF-3 siRNA-transfected cells. These results indicate that IRF-3 contributes to poly I:C-induced expression of ICAM-1.
Fig. 6.
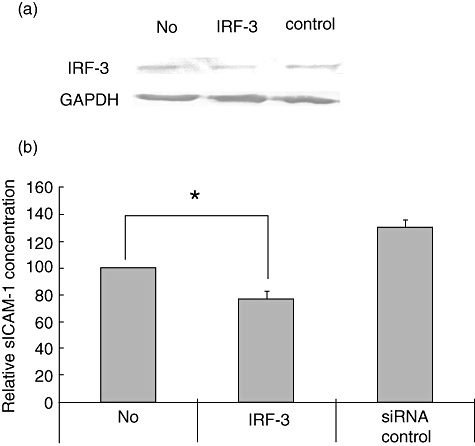
Minor contribution of interferon regulatory factor-3 (IRF-3) on poly I:C-stimulated induction of intercellular adhesion molecule-1 (ICAM-1). HT-29 cells, 2 × 105, were plated on a 24-well plate and transfected with siRNA against IRF-3 or control siRNA for 6 h. The cells were cultured for a further 42 h with 10% fetal calf serum–Dulbecco's modified Eagle's medium. (a) At this time-point, the cell lysates were prepared and 40 µg of protein was subjected to Western blotting. The representative data of three experiments were shown. (b) After transfection, the cells were stimulated with poly I:C for 24 h and the culture supernatants were collected and subjected to enzyme-linked immunosorbent assay. The means of four different experiments were expressed. *P < 0·05 for the comparison of siRNA against IRF-3 transfection plus and minus conditions.
Discussion
The ICAM-1 plays an important role in recruiting leucocytes to sites of inflammation [15]. The existence of ICAM-1 in IECs is well documented and bacterial stimuli and proinflammatory cytokines are known to be potent inducers of ICAM-1 expression [16,17,23].
In addition to these stimulatory factors, ICAM-1 expression has also been reported to be induced by poly I:C in airway epithelial cells [19,31]. The cognate receptor for poly I:C is TLR-3 [9,10] and can be detected in various epithelial cells [9]. Although the existence of TLR-3 can be detected in IECs, the influence of poly I:C-stimulation on the expression of ICAM-1 in IECs has not yet been examined fully. In the present study, we have demonstrated clearly poly I:C-induced expression of ICAM-1 in HT-29 cells. The poly I:C signal was also recognized by Caco-2. HT-29 cells responded to relatively high concentrations of poly I:C, with maximum responsiveness likely to be above 100 µg/ml. This concentration is a magnitude higher than that seen in other cell types [19,32]. An equivalent high concentration was used in poly I:C-induced expression of polymeric immunoglobulin receptor but HT-29 cells could not respond to lower concentrations [13]. This discrepancy might be due to the cell type difference or the localization of TLR-3. It has been reported that TLR-3 localization varies among cell types [33,34]. For instance, in fibroblast, TLR-3 can be detected on the cell surface [34]. Cell surface-expressed TLR-3 is more likely to be able to respond to lower concentrations of poly I:C. There are several reports of intracellular TLR-3 in HT-29 [11,13]; however, our results do not support these findings. In our experiment, HT-29 cells were preincubated with anti-TLR-3 antibody to investigate the specificity of poly I:C signalling; this treatment abolished the poly I:C-stimulated ICAM-1 mRNA expression completely and inhibited the production of sICAM-1 by 40% relative to control antibody-treated cells. As the antibody molecule does not penetrate the cell membrane, we conclude that this inhibitory effect is effected at the cell surface. Moreover, a time-dependent increase of ICAM-1 mRNA was observed with peak induction 3 h post-stimulation. It is not clear whether poly I:C can reach the intracellular space by membrane permeabilization after a few hours' incubation; however, poly I:C-induced expression of IFN-β can be augmented within 1 h of stimulation in murine macrophage cell lines [35]. All these results support cell-surface expression of TLR-3. In fact, our immunofluorescence staining data showed the low-level TLR-3 expression on the cell surface of HT-29 cells. However, TLR-3 is also expected to localize intracellularly. TLR-3s in both locations (cell surface and intracellular) are expected to be responsible for poly I:C, and these possibilities might explain the discrepancy between mRNA inhibition (100% after 3 h stimulation) and sICAM-1 secretion inhibition (40% after 24 h stimulation) in anti-TLR-3 antibody treatment experiments. Further studies will be required in order to elucidate the precise localization of TLR-3 in HT-29 cells.
Another poly I:C-sensing system, the retinoic acid inducible gene I (RIG-I), is important anti-viral machinery [36]. RIG-I can activate type I IFN production in response to poly I:C, introduced into the cells by transfection. The involvement of this mechanism in our experiment is unlikely, because poly I:C was simply added to the culture media and this treatment has been reported not to induce IFN-β induction in HT-29 cells [37]. Although RIG-I signalling and ICAM-1 expression has not yet been reported upon, it would be worth investigating the correlation between intracellular and intercellular systems.
The signalling pathway involving the interaction of poly I:C with TLR-3 was investigated further. TLR-3 has a unique characteristic among TLRs and is known to transduce a ligand-binding signal to the nucleus through a Myd88-independent pathway, via the Toll/interleukin-1 receptor (TIR) domain containing adaptor molecule [9,38,39]. TLR-3 utilizes another adaptor molecule called TIR receptor-related adaptor protein, inducing IFN and culminating in the activation of NF-κB and IRF-3 [38,39]. We examined which transcription factor contributes the most to poly I:C-stimulated induction of ICAM-1 in HT-29 cells. The luciferase reporter construct lacking the NF-κB-binding site and inhibitor treatment abolished poly I:C-induced activity almost completely and suggested that NF-κB is a major contributor to ICAM-1 induction. The IRF-3 knock-down by siRNA resulted in an approximately 20% reduction of poly I:C-stimulated sICAM-1 production, suggesting a less significant role for IRF-3 in this signalling pathway. In the case of TLR-3-mediated type I IFN production, both transcription factors contribute to the effective production of type I IFN [35]. The co-operative function of both transcription factors can also be observed in other genes, such as a chemokine ‘regulated upon activation normal T cells expressed and secreted’[35]. However, in airway epithelial cells, transfecting the siRNA against IRF-3 did not affect poly I:C-stimulated ICAM-1 expression [31]. For the activation of IRF-3, several factors are involved, such as TIR domain-containing adaptor molecules or kinases [9]. Distributional differences between these factors in various epithelial cells are not understood fully. Moreover, there are several fundamental differences between IECs and other epithelial cells. For instances, human beta-defensin 1, a factor of the innate immune system, is expressed constitutively in both IEC and female reproductive tract epithelial cells [14,30]. However, it can be induced in the latter only by specific stimuli [32]. These cell type-specific differences must be considered when interpreting these apparently contradictory results. Moreover, it is possible to speculate that IRF-3 contributes to the induction of ICAM-1 indirectly. ICAM-1 induction is known to be controlled by several cytokines [15] and IRF-3 might influence the induction of these cytokines and exert indirect effects on ICAM-1 induction.
Under normal conditions IECs are devoid of ICAM-1 expression [40], and the biological significance of ICAM-1 in IECs is still controversial. By demonstrating the apical induction of ICAM-1 by bacterial infection, Huang et al. showed the increased adhesion of neutrophils to IEC [16]. Adhered neutrophils prevent further invasion of the mucosa by invading pathogens [16,17]. Moreover, reports revealing ICAM-1 as a receptor for rhinovirus have provided new biological significance for ICAM-1 [21,22]. As demonstrated in this study, a viral replication by-product, poly I:C, can up-regulate the expression of ICAM-1. Viruses might be able to increase infection opportunities by up-regulation of their own receptors. In fact, rhinovirus infection can increase ICAM-1 expression in bronchial epithelial cells [41]. However, we also suggest that virus-infected IECs can mark their own positions by inducing ICAM-1 and facilitate the immunological elimination of infected cells by increasing leucocyte recruitment. The precise functional relevance of the regulated expression of ICAM-1 in IECs awaits further studies.
Acknowledgments
This work was supported by: Grant-in-Aid for Scientific Research (C)(2006–2008), Academic Frontier Project for Private Universities; matching fund subsidy from MEXT 2006–2010 (to Dr Hiroki Nagase), Academic Frontier Project for Private Universities; matching fund subsidy from MEXT 2005–2007 (Ministry of Education, Culture, Sports, Science and Technology); Nihon University Research Grant for Assistants and Young Researchers (2007, 2008); Research Grants from the Sato Fund and the Dental Research Center, Nihon University School of Dentistry for 2008; Nihon University Joint Research Grant for 2006–2007; and the Uemura Fund, Nihon University School of Dentistry (2008).
Disclosure
None.
References
- 1.Kraehenbuhl JP, Neutra MR. Molecular and cellular basis of immune protection of mucosal surfaces. Physiol Rev. 1992;72:853–79. doi: 10.1152/physrev.1992.72.4.853. [DOI] [PubMed] [Google Scholar]
- 2.Lamm ME, Nedrud JG, Kaetzel CS, Mazanec MB. IgA and mucosal defense. APMIS. 1995;103:241–6. doi: 10.1111/j.1699-0463.1995.tb01101.x. [DOI] [PubMed] [Google Scholar]
- 3.Brandtzaeg P. Molecular and cellular aspects of the secretory immunoglobulin system. APMIS. 1995;103:1–19. doi: 10.1111/j.1699-0463.1995.tb01073.x. [DOI] [PubMed] [Google Scholar]
- 4.Mestecky J, Russell MW. Mucosal immunoglobulins and their contribution to defence mechanisms: an overview. Biochem Soc Trans. 1997;25:457–62. doi: 10.1042/bst0250457. [DOI] [PubMed] [Google Scholar]
- 5.Eckmann L, Kagnoff MF. Intestinal mucosal responses to microbial infection. Springer Semin Immunopathol. 2005;27:181–96. doi: 10.1007/s00281-005-0207-5. [DOI] [PubMed] [Google Scholar]
- 6.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 7.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 8.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growthfactor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 11.Fuurie E, Macfarlane S, Thomson G, Macfarlane GT. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology. 2005;115:565–74. doi: 10.1111/j.1365-2567.2005.02200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of Toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–17. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneeman TA, Bruno ME, Schjerven H, Johansen FE, Chady L, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor by signaling through TLRs 3 and 4: linking innate and adaptive immune responses. J Immunol. 2005;175:376–84. doi: 10.4049/jimmunol.175.1.376. [DOI] [PubMed] [Google Scholar]
- 14.O'Neil DA, Porter EM, Elewaut D, et al. Expression and regulation of the human β-defensins hBD-1 and hBD-2 in intestinal epithelium. J Immunol. 1999;163:6718–24. [PubMed] [Google Scholar]
- 15.Hopkins AM, Baird AW, Nusrat A. ICAM-1: targeted docking for exogenous as well as endogenous ligands. Adv Drug Delivery Rev. 2004;56:763–78. doi: 10.1016/j.addr.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 16.Huang GTJ, Eckmann L, Savidge TC, Kagnoff MF. Infection of human intestinal epithelial cells with invasive bacterial upregulates apical intercellular adhesion molecule-1 (ICAM-1) expression and neutrophil adhesion. J Clin Invest. 1996;98:572–83. doi: 10.1172/JCI118825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly CP, O'Keane JC, Orellana J, et al. Human colon cancer cells express ICAM-1 in vivo and support LFA-1-dependent lymphocyte adhesion in vitro. Am J Physiol. 1992;263:G864–70. doi: 10.1152/ajpgi.1992.263.6.G864. [DOI] [PubMed] [Google Scholar]
- 18.Umadevi SS, Yue J, Dawn CN, et al. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 2006;12:2121–3. doi: 10.1096/fj.06-5806fje. [DOI] [PubMed] [Google Scholar]
- 19.Guillot L, Le Goffic R, Bloch S, et al. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;7:5571–80. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 20.Huang GT, Zhang X, Park NH. Increased ICAM-1 expression in transformed human oral epithelial cells: molecular mechanism and functional role in peripheral blood mononuclear cell adhesion and lymphokine-activated-killer cell cytotoxicity. Int J Oncol. 2000;17:479–86. [PMC free article] [PubMed] [Google Scholar]
- 21.Greve JM, Davis G, Meyer AM, et al. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–47. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 22.Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–53. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 23.Kaiserlian D, Rigal D, Abello J, Revillard JP. Expression, function and regulation of the intercellular adhesion molecule-1 (ICAM-1) on human intestinal epithelial cell lines. Eur J Immunol. 1991;21:2415–21. doi: 10.1002/eji.1830211018. [DOI] [PubMed] [Google Scholar]
- 24.Dippold W, Witting B, Schwaeble W, Mayet W, Meyer zum Buschenfelde KH. Expression of intercellular cell adhesion molecule 1 (ICAM-1, CD54) in colonic epithelial cells. Gut. 1993;34:1593–97. doi: 10.1136/gut.34.11.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jobin C, Hellerbrand C, Licato LL, Brenner DA, Sartor RB. Mediation by NF-kB cytokine induced expression of intercellular adhesion molecule 1 (ICAM-1) in an intestinal epithelial cell line, a process blocked by proteasome inhibitors. Gut. 1998;42:779–87. doi: 10.1136/gut.42.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sebastien V, Sullivan L, Francis F, Marie PM, Claude P, Serge C. Cytokine-induced upregulation of NF-κB, IL-8, and ICAM-1 is dependent on colonic cell polarity: implication for PKC delta. Exp Cell Res. 2004;297:165–85. doi: 10.1016/j.yexcr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Suguro H, Asano M, Kaneko Y, et al. Characterization of human dental pulp-derived cell lines. Int Endod J. 2008;41:609–16. doi: 10.1111/j.1365-2591.2008.01409.x. [DOI] [PubMed] [Google Scholar]
- 28.Takenouchi-Ohkubo N, Takahashi T, Tsuchiya M, Mesteckey J, Moldoveanu Z, Moro I. Role of nuclear factor kappa B in the expression by tumor necrosis factor-alpha of the human polymeric immunoglobulin receptor (pIgR) gene. Immunogenetics. 2000;51:289–95. doi: 10.1007/s002510050622. [DOI] [PubMed] [Google Scholar]
- 29.Huamng F, Cao CY, Wachi S, Thai P, Ryu J, Wu R. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-κB activation by IL-17 A in enhancing cytokine expression in human airway epithelial cells. J Immunol. 2007;179:6504–13. doi: 10.4049/jimmunol.179.10.6504. [DOI] [PubMed] [Google Scholar]
- 30.Asano M, Ogura Y, Takenouchi-Ohkubo N, et al. Endoplasmic reticulum resident, immunoglobulin joining chain, can be secreted by perturbation of the calcium concentration in the endoplasmic reticulum. DNA Cell Biol. 2004;23:403–11. doi: 10.1089/1044549041474779. [DOI] [PubMed] [Google Scholar]
- 31.Matsukura S, Kokubu F, Kurokawa M, et al. Synthetic double-stranded RNA induced multiple genes related to inflammation through Toll-like receptor 3 depending on NF-kB and/or IRF-3 in airway epithelial cells. Cin Exp Allergy. 2006;36:1049–62. doi: 10.1111/j.1365-2222.2006.02530.x. [DOI] [PubMed] [Google Scholar]
- 32.Schaefer TM, Fashey JV, Wright JA, Wira CR. Innate immunity in the human female reproductive tract: antiviral response of uterine epithelial cells to the TLR3 agonist poly I:C. J Immunol. 2005;174:992–1002. doi: 10.4049/jimmunol.174.2.992. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto M, Funami K, Tanabe M, et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171:3154–62. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun. 2002;293:1364–69. doi: 10.1016/S0006-291X(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 35.Doyle SE, Vaidya SA, O'Connell R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–63. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 36.Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–37. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 37.Hirata Y, Broquet AH, Menchen L, Kagnoff MF. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in intestinal epithelial cells. J Immunol. 2007;179:5425–32. doi: 10.4049/jimmunol.179.8.5425. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640–43. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 39.Moynagh PN. TLR signaling and activation of IRFs: revisiting old friends from the NF-kB pathway. TRENDS Immunol. 2005;26:469–76. doi: 10.1016/j.it.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 40.Bloom S, Simmons D, Jewell DP. Adhesion molecules intercellular adhesion molecule-1 (ICAM-1), ICAM-3 and B7 are not expressed by epithelium in normal or inflamed colon. Clin Exp Immunol. 1995;101:157–63. doi: 10.1111/j.1365-2249.1995.tb02292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papi S, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-kappaB-mediated transcription. J Biol Chem. 1999;274:9707–20. doi: 10.1074/jbc.274.14.9707. [DOI] [PubMed] [Google Scholar]