

Abstract
Inherited deficiencies in components of the classical complement pathway are strong disease susceptibility factors for the development of systemic lupus erythematosus (SLE) and there is a hierarchy among deficiency states, the strongest association being with C1q deficiency. We investigated the relative importance of the different complement pathways regarding clearance of apoptotic cells. Phagocytosis of labelled apoptotic Jurkat cells by monocyte-derived macrophages in the presence of sera from individuals with complement deficiencies was studied, as well as C3 deposition on apoptotic cells using flow cytometry. Sera from individuals deficient in C1q, C4, C2 or C3 all showed decreased phagocytosis. Mannose binding lectin (MBL) and the alternative pathway did not influence phagocytosis. Notably, the components of the complement classical pathway, including C1q, were equally important in clearance of apoptotic cells. This indicates that deposition of C3 fragments is of major significance; we therefore studied C3 deposition on apoptotic cells. Experiments with MBL-deficient serum depleted of C1q or factor D confirmed the predominance of the classical pathway. At low dilution, sera deficient of C1q, C4 or C2 supported C3 fragment deposition demonstrating alternative pathway activation. In conclusion, we have found that complement-mediated opsonization and phagocytosis of apoptotic cells, particularly those undergoing secondary necrosis, are dependent mainly upon an intact classical pathway. The alternative pathway is less important, but may play a role in some conditions. C1q was not more important than other classical pathway components, suggesting a role in additional pathogenetic processes in SLE other than clearance of apoptotic cells.
Keywords: apoptosis, complement, opsonization, phagocytosis, SLE
Introduction
Clearance of apoptotic cells is a complex process involving many different ligands and receptors on the phagocyte and dying cells, and apoptotic cells expose ‘eat me’ signals that flag them for their removal from tissues by phagocytes. Components of the complement system appear to play important roles in clearance of dying cells [1], and involvement of the classical, alternative and lectin pathways of complement have all been suggested [1,2]. Inherited deficiencies of the components of the classical pathway are strong risk factors for the development of the autoimmune disease, systemic lupus erythematosus (SLE), and there is a hierarchy of disease susceptibility and severity according to the position of the missing protein in the classical pathway of complement activation, with the strongest association with C1q [3]. A similar hierarchical role for classical pathway proteins in the clearance of apoptotic cells has been suggested [4]. However, investigations of the relative importance of the different complement pathways regarding clearance of apoptotic cells, and also specifically within the classical pathway, are warranted because of the conflicting results reported in the literature. In the present investigation we study apoptotic cells defined as either early apoptotic cells with maintained membrane integrity or late apoptotic cells undergoing secondary necrosis. We demonstrate that activation of the classical pathway is most important for complement deposition on late apoptotic cells and for the clearance of these cells in an in vitro model. Moreover, under certain conditions the alternative pathway may play a role. We also demonstrate that components of the classical pathway appear to be equally important in clearance of apoptotic cells. Our findings do not support the idea that the hierarchical role of deficiency stated within classical pathway of complement proteins, in relation to SLE susceptibility, is dependant solely upon their role in clearance of apoptotic cells.
Materials and methods
Serum samples and complement reagents
Normal human sera (NHS), used in the phagocytosis experiments, were obtained from two healthy individuals. Pooled normal human serum (PNHS) used in the C3 deposition experiments was obtained from 10 healthy laboratory staff members. Sera collected from individuals deficient in the complement components C1q, C2, C4, C3, properdin and mannose binding lectin (MBL) were used; the clinical phenotypes of these individuals are summarized in Table 1. In addition, one of the C2-deficient individuals and the properdin-deficient individual were also deficient in MBL.
Table 1.
Clinical phenotypes and serum concentrations of complement protein in the complement-deficient individuals.
| Deficient individuals | Clinical findings | C1q (%)† (78–131)‡ | C2 (%) (77–159) | C3 (g/l) (0·77–1·38) | C4 (g/l) (0·12–0·33) | C5 (%) (72–171) | P (%) (54–157) | FB (%) (59–154) | MBL (g/l) (0·1–6·0) | C4BP (%) (58–102) |
|---|---|---|---|---|---|---|---|---|---|---|
| C1qD-1 | Recurrent infections | <0·1 | 84 | 1·15 | 0·19 | 75 | 128 | 95 | 4·2 | 152 |
| C1qD-2 | SLE | <6 | n.d.§ | 1·59 | 0·39 | n.d. | n.d. | n.d. | n.d. | n.d. |
| C4D | SLE | 91 | 98 | 1·37 | 0 | 145 | 137 | 189 | 1·0 | 190 |
| C2D-1 | Pneumonia and UCTD | 133 | 0 | 1·48 | 0·37 | 125 | 198 | 86 | 0·2 | 175 |
| C2D-2 | Septicaemia | 97 | 0 | 1·47 | 0·41 | 135 | 129 | 80 | <0·1 | 143 |
| C3D | Recurrent meningitis and septicaemia | 167 | 100 | <0·06 | 0·16 | 95 | 123 | 132 | 2·6 | 97 |
| PD | Healthy | 186 | 185 | 1·62 | 0·29 | 130 | 0 | 147 | <0·1 | 167 |
| MBLD | Healthy | 122 | 105 | 1·11 | 0·2 | 127 | 79 | 82 | <0·1 | 112 |
% of normal serum pool;
reference interval;
not done; SLE, systemic lupus erythematosus; UCTD, undifferentiated connective tissue disease; MBLD, mannose binding lectin-deficient; FB, factor B; C4BP, C4-binding protein; PD, properdin.
Serum reagents lacking C1q and factor D or factor D only were prepared. MBL-deficient (MBLD) serum from a healthy donor was depleted of C1q and factor D as described previously [5]. Serum from one healthy individual was used for the preparation of factor D-depleted serum by affinity chromatography on an anti-D-Sepharose column. Polyclonal rabbit anti-human factor D [6] was coupled to CNBr-activated Sepharose CL-4B (Amersham Pharmacia Biotech AB, Uppsala, Sweden) according to the manufacturer's protocol. The serum was applied to the column in veronal-buffered saline (VBS) pH 7·4 containing 10 mM ethylenediamine-tetraacetic acid (EDTA). The collected fraction was dialysed against VBS containing 0·15 mM CaCl2 and 0·5 mM MgCl2 before being used. All sera and reagents were stored at –80°C.
Complement-deficient and complement-depleted sera were characterized with regard to concentrations of several complement proteins by electroimmunoassay, including C1q, C3, C4, C5, factor B (FB), properdin [7] and C4-binding protein (anti-human C4-binding protein; The Binding Site, Birmingham, UK) (Table 1). Measurements of MBL and MBL genotyping were performed according to published methods [8], and both the MBLD donor and the combined C2- and MBLD donor had the MBL genotype ALX/BLY. Heat-inactivated serum (NHS-HI) was produced by incubating NHS in 56°C for 30 min.
The study was conducted within the framework of a multi-centre project approved by the Lund University Research Ethics Committee (LU 513-01).
Purified complements proteins
Recombinant MBL (rMBL) was kindly provided by Professor J. C. Jensenius (Aarhus, Denmark). MBL activity was analysed by C4 deposition in MBLD serum to which rMBL was added [9]. The following complement proteins were available in the laboratory and published methods were used for purification of C1q [10], C4 [11], factor D [6] and properdin [12]. Purification of C2 was a modification of published protocol [13], where the separation of C2 from FB was achieved by affinity chromatography using NHS-activated Sepharose (Amersham Pharmacia Biotech AB) coupled with rabbit anti-C2 immunoglobulin G (IgG). Electroimmunoassay was used for determinations of C1q, factor D, properdin and C2 concentrations [7] with dilution of purified proteins in appropriate complement-deficient or complement-depleted sera. The pooled serum used as reference was assumed to contain C1q at 70 mg/l, factor D at 1 mg/l, properdin at 25 mg/l and C2 at 26 mg/l [14].
Generation of macrophages
Peripheral blood mononuclear cells were obtained from fresh heparinized blood samples from two different donors (healthy laboratory personnel). The mononuclear cells were isolated by density gradient centrifugation on Lymphoprep™ (Axis Shield Poc AS, Oslo, Norway) at 752 g for 30 min. Cells were suspended in RPMI-1640 medium with L-glutamine (PAA Laboratories GmbH, Linz, Austria) (RPMI) containing 15% NHS from one single donor, added to cell density of 1·5 × 106 cells/ml. In order to separate lymphocytes from monocytes, 0·4 ml of the cell suspension was added to an eight-well plate (Nalgene Nunc International, Naperville, IL, USA) and incubated for 2 h. This enabled monocytes to adhere, and lymphocytes were removed by washing three times with RPMI. The adherent monocytes were incubated with 10% NHS in RPMI containing 50 µg/ml gentamicin and 2·5 µg/ml amphotericin B (medium), and were allowed to differentiate into macrophages (MDM) during 5 days at 37°C, 96% humidity with 5% CO2. The medium was changed once on day 3.
Apoptotic cells
Apoptotic cells were obtained by incubating Jurkat cells (TIB-152; ATCC, Manassas, VA, USA) in RPMI at cell density of 2 × 106 cells/ml with staurosporine (Sigma-Aldrich, St Louis, MO, USA) at a final concentration of 1 µM at 37°C for 4 h, yielding approximately 50% early apoptotic cells and 50% late apoptotic cells, which are cells undergoing secondary necrosis. This enabled measurement of C3 deposition on both early and late apoptotic cells in the complement deposition experiments. After staurosporine treatment, the cells were used immediately in the experiments. Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) (Apoptosis Detection Kit; BD Biosciences PharMingen, San Diego, CA, USA) staining, measured by flow cytometry (Epics XL-MCL; Beckman-Coulter, Fullerton, CA, USA) as described earlier [15], was used to monitor percentage of early apoptosis, defined as annexin V-positive/PI-negative cells versus late apoptosis or secondary necrosis, defined as annexin V-positive/PI-positive cells. In order to detect Jurkat cells in the phagocytosis assay (see below), the cells were incubated for 15 min with 5,6 carboxyfluorescein diacetate succinimidyl ester (CFS-E) (Sigma-Aldrich) at a final concentration of 2·5 µM [16], before exposure to staurosporine.
Phagocytosis assay
The CFS-E labelled apoptotic Jurkat cells, 3 × 105, were added to each well with adherent MDM together with 20% of serum from the complement-deficient individuals or as a control NHS in RPMI. The apoptotic cells were allowed to interact with the MDM for 30 min at 37°C before washing with RPMI. MDM were then detached from the glass slide by adding 0·2 ml of 0·5 mM EDTA in phosphate-buffered saline pH 7·2 (6·2 mM sodium phosphate, 0·15 M NaCl, pH 7·2) to each well for 3 min. After washing, R-phycoerythrin cytosolic extract (RPE-cy5)-labelled anti-CD14 (Serotec, Kidlington, Oxford, UK) was added and the cell suspension was incubated for 30 min at room temperature. The solution, consisting of CD14-positive MDM with and without CFS-E-labelled apoptotic cells, was analysed by flow cytometry. Cells positive for both CD14 and CFS-E were considered as apoptotic cells engulfing MDM, and the percentage of such cells of the total number of CD14-positive cells was calculated. In each experiment, the percentage of phagocytic CD14-positive cells in the presence of complement-deficient test serum or complement reconstituted deficient test serum was compared with the corresponding number of cells in the presence of NHS. Results were given as a phagocytosis index (PhI): double-positive cells in test serum/double-positive cells in NHS. The experiments were performed at least three times.
Complement deposition experiments
Apoptotic cells were applied in round-bottomed microtitre plate wells (Nunclon Microwell Plates, 143761; Nunc, Kamstrup, Denmark), with 2 × 105cells/well. The supernatant was removed after centrifugation at 350 g for 2 min. The cells were resuspended in 0·1 ml of serum diluted in RPMI with the addition of 0·1% human serum albumin (RPMI-HSA) (Sigma-Aldrich). After incubation at 37°C for 30 min, the cells were centrifuged and washed with 0·2 ml of RPMI-HSA and resuspended in 0·1 ml RPMI-HSA. Complement activation was assessed by detection of C3 deposition using monoclonal antibodies against C3d (A207; Quidel Corporation, Santa Clara, CA, USA). The cells were incubated with antibodies for 15 min at room temperature. After centrifugation the cell pellet was resuspended in binding buffer (10 mM Hepes/NaOH pH 7·4, 140 mM NaCl, 2·5 mM CaCl2), supplied with the apoptosis detection kit (BD Biosciences Pharmingen). This was followed by a second incubation step with RPE-conjugated F(ab′)2 fragments of rabbit anti-mouse Igs (Dako A/S, Glostrup, Denmark) for 15 min at room temperature before analysis by flow cytometry. In each series of experiments, one well with apoptotic Jurkat cells and one well with Jurkat cells not treated with staurosporine were incubated with annexin V-FITC and PI.
Statistical analysis
The Wilcoxon matched-pairs rank test was used and only differences with a P value <0·05 were considered significant.
Results
Results from the phagocytosis experiments are summarized in Fig. 1. A PhI was calculated by comparing the percentage of phagocytizing CD14-positive cells in the presence of test serum with the corresponding number of cells in the presence of NHS. The experiments were performed in 20% serum, which allows activation of all three complement pathways [17]. A test serum with the same capacity as NHS for facilitating phagocytosis was consequently given a PhI value of 1, and heat-inactivated serum PhI was given a value of 0·5. Thus, in these experiments approximately 50% of the phagocytosis capacity of apoptotic cells was dependent upon the complement system, and 50% upon other factors.
Fig. 1.
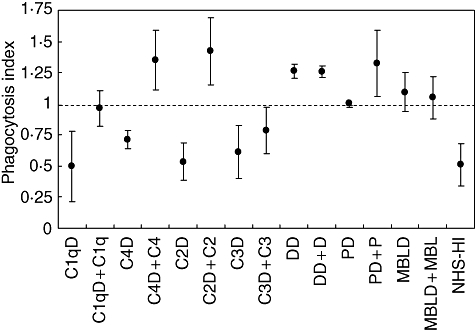
Clearance of apoptotic 5,6 carboxyfluorescein diacetate succinimidyl ester (CFS-E)-labelled Jurkat cells by macrophages in the presence of sera deficient in selected complement components was studied using flow cytometry. A phagocytosis index (PhI) was calculated by comparing the percentage of phagocytizing CD14-positive cells in the presence of test serum with the presence of normal human sera (NHS). A test serum that had the same capacity of facilitating phagocytosis as NHS was consequently given the value 1. Sera from individuals deficient in C1q (C1qD; patient C1q-D1), C4 (C4D), C2 (C2D; patient C2D-1) and C3 (C3D) all showed decreased phagocytosis. Addition of C1q, C4 or C2 all increased the PhI by the same magnitude, while addition of C3 gave less increase. Sera lacking factor D (DD), properdin (PD) or mannose binding lectin-deficient (MBLD) did not show low PhI and only addition of properdin increased PhI further. Mean and standard deviation from three experiments are shown.
The lectin and the alternative pathways did not influence phagocytosis of apoptotic cells in our experimental system because phagocytosis in the presence of sera lacking MBL, properdin or factor D did not differ from NHS. Addition of the lacking complement component was performed as a control experiment. These results showed that the PhI was unchanged when sera lacking factor D or MBL were restored with the missing component. An increase from normal level to a slightly higher level was seen when properdin was added to a properdin-deficient serum.
Phagocytosis index in sera from individuals deficient in the classical pathway components C1q, C4, C2 and C3 all showed decreased PhI levels. Addition of C1q, C4 and C2 to the corresponding complement-deficient serum increased PhI to the level of NHS or above. Reconstitution with C3 to the C3-deficient serum increased PhI, but not quite to the level of NHS.
Thus, the results so far showed that clearance of apoptotic cells was dependent upon the classical pathway, whereas the lack of lectin or alternative pathway activity had no measurable influence. Notably, components of the complement classical pathway, including C1q, were equally important in clearance of apoptotic cells. This indicates that deposition of C3 fragments on apoptotic cells is of major significance; therefore we studied C3 deposition on apoptotic cells by flow cytometry.
Initial activation experiments were performed with Jurkat cells that had been triggered to undergo apoptosis by incubation with staurosporine. Cells were analysed by flow cytometry and divided into two populations based on forward-scatter (size) and side-scatter (granularity). One population (G1) contained cells of the same size and granularity as vital cells. These cells were annexin V-positive and PI-negative (early apoptotic cells). The other population (G2) contained cells of smaller size and higher granularity and stained positive for both annexin V and PI (late apoptotic or secondary necrotic cells). Each population contained approximately 50% of the total number of cells (Fig. 2a).
Fig. 2.
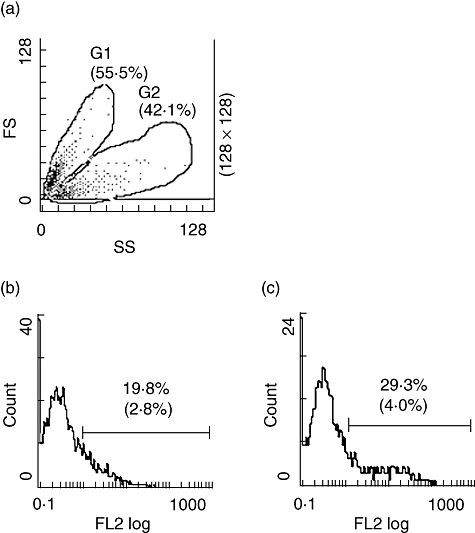
Complement activation on apoptotic Jurkat cells analysed by flow cytometry. Apoptotic cells were incubated with serum or with medium alone and analysed by flow cytometry. (a) Forward-scatter (FS) and side-scatter (SS) of apoptotic cells. Cells were gated into two regions, G1 containing early apoptotic cells and G2 containing late apoptotic cells. Results with apoptotic cells incubated with 10% normal human serum are shown. (b) Deposition of C3 in region G1 and (c) deposition of C3 in region G2 using antibodies against C3d. Percentages of positive cells are shown with percentages from control experiments within parenthesis. Histograms from one experiment typical of at least five are shown.
To assess complement activation, cells were incubated at 37°C with serial dilutions of PNHS and analysed for deposition of C3 using monoclonal antibodies recognizing C3d epitopes. Binding to each of the two cell populations (G1 and G2) was analysed. The number of cells that bound C3 increased when the system was supplemented with higher serum concentrations (Fig. 3a). Both early apoptotic cells and late apoptotic cells stained positive for C3 fragments in a dose-dependent manner, but late apoptotic cells bound C3 fragments to a greater extent than early cells. The mean fluorescence intensity (MFI) on the early apoptotic cells showed only a minor increase (Fig. 3). Histograms from a representative experiment are shown in Fig. 2b and c.
Fig. 3.
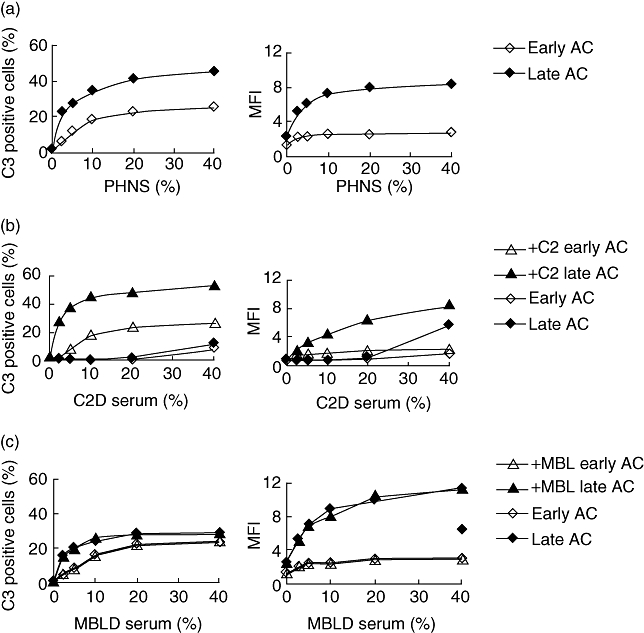
Deposition of C3 on apoptotic Jurkat cells after incubation in serum analysed with antibodies against C3d. Apoptotic cells were incubated in serial dilutions of (a) pooled normal human sera (PNHS); (b) C2-deficient serum (C2D serum; patient C2D-2) or with C2 added (+C2) at physiological concentration (26 mg/l); (c) mannose binding lectin (MBL)-deficient (MBLD serum or after addition of recombinant MBL at a concentration of 2 mg/l (+MBL). Results from one experiment of three experiments performed are shown. Deposition of C3 was given as the percentage of positive cells and as mean fluorescence intensity (MFI). Early apoptotic and late apoptotic cells are given as early AC and late AC respectively.
In order to clarify the respective roles of the classical and the alternative pathways we used MBLD serum depleted of C1q and factor D. By adding back purified C1q or factor D we had a tool to examine the two pathways separately. The experiments were performed in the presence of 25% serum, allowing activation of both these pathways. In the depleted serum, C3 fragments deposition on apoptotic cells was very low before adding back purified C1q. Reconstitution of the classical pathway was achieved by the addition of C1q at concentrations ranging from 0·1% (0·07 mg/l) to 100% (70 mg/l) (Fig. 4a). Very low doses of C1q (1%) had a clear impact on C3 deposition on late apoptotic cells. A plateau was reached in the interval between 10% and 100%. There was a clear difference between early and late apoptotic cells; C3 deposition on early apoptotic cells increased only slightly when C1q was added, not reflected in the MFI values. Reconstitution with factor D at 0·1% (0·001 mg/l) to 100% (1 mg/l) did not influence C3 deposition (Fig. 4b). With the addition of both C1q and factor D no significant (P > 0·05) increase of C3 deposition was found when compared with the addition of C1q alone. This suggested that deposition of C3 fragments on apoptotic cells is dependent upon activation of the classical pathway, and the alternative pathway-mediated amplification was limited under the prevailing conditions.
Fig. 4.
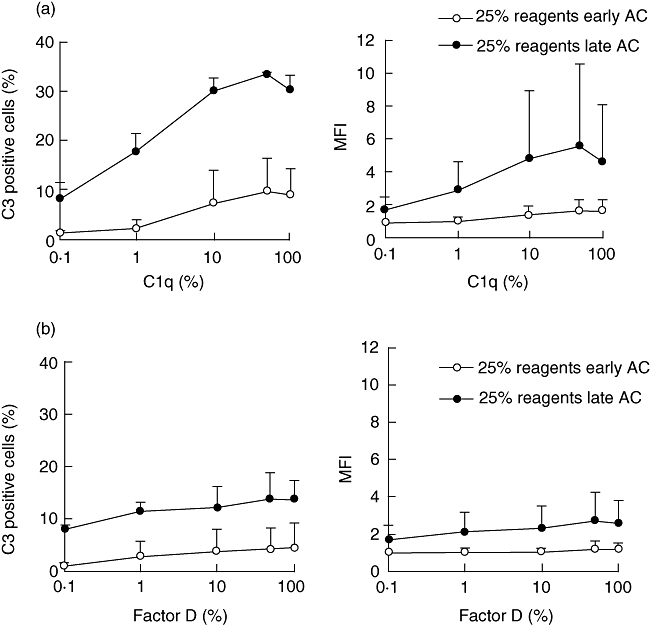
Influence of the classical and the alternative pathway on deposition of C3 on apoptotic Jurkat cells. Purified C1q (a) or factor D (b) was added to a mannose binding lectin (MBL)-deficient serum depleted of C1q and factor D. Cells were incubated with the C1q- and D-depleted serum at serum concentration of 25%. Deposition of C3 is given as the percentage of positive cells and as mean fluorescence intensity (MFI) using antibodies against C3d. (a) Deposition of C3 after addition of C1q at 0·1% (0·7 mg/l) to 100% (70 mg/l); (b) deposition of C3 after addition of factor D at 0·1% (0·001 mg/l) to 100% of normal (1 mg/l). Mean values and the standard deviation of three experiments are shown. Early apoptotic and late apoptotic cells are given as early AC and late AC respectively.
Using another approach, we investigated a C2-deficient serum that was also deficient in MBL (C2D:2). At low serum concentrations the serum did not support C3 deposition on apoptotic cells, but at the highest serum concentration (40%) measurable levels of C3 deposition could be detected (Fig. 3b). The addition of purified C2 at a physiological concentration of 100% (26 mg/l) to the C2-deficient serum promoted C3 deposition, so that the results resembled those obtained with NHS. Thus, C2 but not MBL was needed for an efficient C3 deposition on apoptotic cells.
For additional analyses of the role of the alternative pathway we assessed C3 deposition on apoptotic cells, using sera at different concentrations from individuals deficient in C1q, C4 or C2. Results from experiments with serum concentrations of 10%, 20% and 40% are shown in Fig. 5. In the experiments with C2-deficient serum, deposition of C3 fragments was found only at the highest serum concentration, 40%, in accordance with the results shown in Fig. 3b. C3 deposition measured in C1q-deficient (C1qD-2) and C4-deficient serum could be detected at 20% serum, and somewhat higher at 40%. Thus, these experiments showed involvement of the alternative pathway at high serum concentrations. As expected, no C3 deposition could be detected in C3-deficient serum.
Fig. 5.
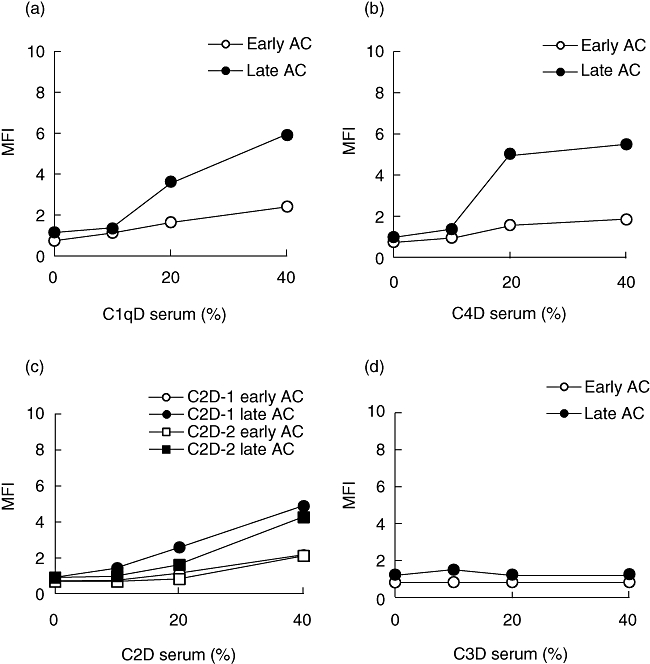
C3 deposition on apoptotic Jurkat cells after incubation with complement-deficient sera at different concentrations. (a) C1q-deficient serum (C1qD serum; patient C1qD-2); (b) C4-deficient serum (C4D serum); (c) C2-deficient serum (C2D serum; patient C2D-1 and C2D-2); (d) C3-deficient serum (C3D serum). C3 deposition was measured by flow cytometry using antibodies to C3d, and deposition of C3 is given as mean fluorescence intensity (MFI).Early apoptotic and late apoptotic cells are given as early AC and late AC respectively.
C3 deposition in the presence of different concentrations of MBLD serum was similar to normal human serum, and was not changed when MBL was added (Fig. 3c). In summary, deposition of C3 fragments on apoptotic cells, preferentially the late apoptotic cells, depends predominantly upon the activation of the classical pathway; however, involvement of the alternative pathway was possible if the classical pathway was not functional. Under the prevailing conditions, the lectin pathway did not influence C3 deposition on apoptotic cells.
Discussion
In the present investigation, clearance of apoptotic cells by uptake in phagocytes was shown to be dependent upon the classical pathway of complement activation, whereas neither a functional lectin nor alternative pathway was necessary. These findings are in accordance with most other reports within this research area [1,4,18–20]. However, in our experimental setup, where phagocytosis of apoptotic cells was measured in the presence of sera from complement-deficient individuals, we could see no difference between serum from individuals deficient in C1q, C4 or C2. Thus, we have found that components of the complement classical pathway appear to be equally important in clearance of apoptotic cells, which contradicts the hypothesis of a hierarchical role for classical pathway complement proteins with regard to their role in clearance of apoptotic cells [4].
The results obtained in this study using complement-deficient sera and sera depleted of C1q and factor D showed that activation of complement by apoptotic cells resulted in deposition of C3 fragments, which was dependent mainly upon the classical pathway. Deposition of C3 fragments through classical pathway activation was seen at low serum concentrations, and was most pronounced on secondary necrotic cells. This is in agreement with results from other investigators [19,21].
The C1q binding pattern seen by confocal microscopy, as described by others, indicated that C1q bound preferentially to the surface blebs on late apoptotic cells in a clustered distribution and that the binding of C1q to normal and early apoptotic cells showed a more even distribution [2]. In our experiments, which allowed for measurement of C3 deposition on both early and late apoptotic cells, a more prominent C3 deposition was seen on late apoptotic cells. This may indicate a more efficient complement activation of the classical pathway when C1q molecules are present at increased density. It is most probable that natural IgM antibodies recognizing lysophosphatidylcholine exposed during apoptosis account for most of the C1q binding by apoptotic cells [22], but it has been shown recently that C1q binds phosphatidylserine (PS) at the early stage of apoptosis [23]. This binding of C1q to PS is in competition with annexin V, but whether this binding initiates complement activation is still unknown. Mevorach et al. have shown a reduction of C3bi deposition on apoptotic cells preincubated with annexin V, suggesting that exposure of PS can influence complement activation [1], and in addition C1q can activate complement through binding to DNA [24,25]. However, our results demonstrate that the classical pathway is involved in clearance of late apoptotic cells, which is of major importance mainly because of the proinflammatory situation that will occur if these secondary necrotic cells are not handled properly [26].
When using a higher serum concentration, which allows activation of the alternative pathway, the deficient sera were shown to support some C3 deposition. However, only low levels were detected compared with the levels of C3 deposition seen using serum with an intact classical pathway. The main role of the alternative pathway in deposition of C3 fragments on apoptotic cells may be to amplify activation of the classical pathway. It has been described previously that properdin is able to bind to apoptotic cells independently of C3b and activate the alternative pathway [27,28]. This may be an explanation for our finding regarding the increase from normal level to a slightly higher level of the PhI index when properdin was added to a properdin-deficient serum. The alternative pathway may be important for opsonization of early apoptic cells as well as of secondary necrotic cells, as shown with classical pathway-deficient sera. When testing C2-deficient sera, C3 deposition was seen only at the highest serum concentration tested. This might be explained by low levels of FB in these sera [29]. With C1q- or C4-deficient sera we could see involvement of activation via the alternative pathway, when less diluted serum was used, and preferentially on late apoptotic cells. This was most pronounced with the C4-deficient serum already showing C3 fragment deposition at a 10% serum concentration. This C4-deficient serum had a very high concentration of FB (189% of the normal), and in previous work by Selander et al.[30] this serum showed C3 deposition on serogroup O antigen-specific Salmonella oligosaccharides via the lectin pathway, suggesting a C4-bypass mechanism. However, we found no evidence of C3 deposition through the lectin pathway, as suggested previously by Nauta et al.[18].
The partly discrepant findings compared with previous studies with regard to complement pathways in opsonization and phagocytosis of apoptotic cells [1,18,19,27,28,31] could be due to the serum concentration used, but also to different cell types and cells at different stages of apoptosis. In our experiments human sera from complement-deficient individuals were used, which could be another important difference. Regardless, our experiments indicate strongly that activation of the classical pathway is beneficial for the phagocytosis of cells undergoing apoptosis, especially of those also being secondary necrotic.
The SLE is an autoimmune disease characterized by involvement of many different organ systems and by immunological abnormalities, notably hyperactive B cells producing various autoantibodies. It is also a disease where the role of apoptosis in pathogenesis has been acknowledged increasingly. It is believed that many lupus autoantigens are derived from apoptotic cells [32], and defect clearance of apoptotic cells seems to be of major importance in SLE [33,34].
Homozygous deficiency of the classical pathway components C1q, C4 or C2 is associated with an increased susceptibility to SLE. Nearly all individuals with C1q deficiency develop SLE, often at a young age, whereas C4-deficient individuals develop SLE less often, with individuals deficient in C2 even less so. Deficiency of C3 is associated mainly with severe infections, not SLE [3,35]. Accordingly, there is a clear hierarchy of disease susceptibility regarding deficiency states involving the classical pathway, and it has been suggested that a hierarchy could also be applied on phagocytic clearance of apoptotic cells [4,36]. Taylor et al. used gene-targeted C1q- or C4-deficient mice in an in vivo model of apoptotic cell clearance during sterile inflammatory peritoneal macrophages. These authors could demonstrate less efficient apoptotic cell clearance in both C1q-deficient and C4-deficient mice, but it was more evident in C1q-deficient than in C4-deficient mice [4]. We could not confirm this hierarchy in our in vitro model using human sera with specific complement factor deficiencies. Notably, we have included sera from C2-deficient individuals which were not included by Taylor et al.
The first observations with the gene-targeted C1q-deficient mice were the development of a lupus-like disease and glomerulonephritis with anti-nuclear antibodies and the presence of apoptotic bodies in glomeruli [37]. C1q has also been shown to bind specifically to the surface blebs of apoptotic keratinocytes, where common SLE autoantigens are localized in high concentrations. These findings have pointed towards C1q deficiency predisposing to SLE as a consequence of impaired phagocytosis of apoptotic cells. More recently, it has become evident that the genetic background in the C1q-deficient mice is more important than the C1q gene deletion itself [38]. In humans, however, the fact remains that homozygous C1q deficiency is almost always associated with SLE and C1q is often markedly decreased during severe lupus flares. The C1q molecule is, thus, of major importance for the development of SLE, but not necessarily only because of its role in apoptotic cell clearance. Other properties of this molecule, such as cytokine regulation, might also be of importance [39].
In conclusion, complement-dependent opsonization of dying cells by deposition of C3 fragments was needed for efficient phagocytosis. The classical pathway of activation is the most important, and C1q, C4 and C2 all appear to be equally important. Our findings suggest that the strong association between C1q deficiency and SLE is explained only partly by the role of C1q in the clearance of apoptotic cells. Ongoing studies by us and others will clarify alternative explanations such as cytokine regulation by C1q. However, these results support the hypothesis that a functional complement system is of vital importance in the protection against development of SLE.
Acknowledgments
We would like to express our gratitude to our dear friend and mentor Anders G. Sjöholm, who initiated the work regarding complement deposition on apoptotic cells before he sadly passed away in June 2006. We also would like to acknowledge Ingrid Johansson, Annica Andreasson and Kristina Törnqvist for their contribution and for laboratory work. The study was supported by grants from the Swedish Research Council (grants 13489 and 15092), the Medical Faculty at Lund University, the Swedish National Association against Rheumatism, Alfred Österlund's Foundation, The Crafoord Foundation, Greta and Johan Kock's Foundation, King Gustaf V's 80th Birthday Fund and Lund University Hospital.
References
- 1.Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. 1998;188:2313–20. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogden CA, deCathelineau A, Hoffmann PR, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–95. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40:560–6. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- 4.Taylor PR, Carugati A, Fadok VA, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–66. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Praz F, Barreira MC, Lesavre P. A one-step procedure for preparation of classical pathway (C1q) and alternative pathway (factor D) depleted human serum. J Immunol Methods. 1982;50:227–31. doi: 10.1016/0022-1759(82)90229-0. [DOI] [PubMed] [Google Scholar]
- 6.Truedsson L, Sturfelt G. Human factor D of the alternative pathway: purification and quantitation by enzyme amplified electroimmunoassay. J Immunol Methods. 1983;63:207–14. doi: 10.1016/0022-1759(83)90424-6. [DOI] [PubMed] [Google Scholar]
- 7.Johnson U, Truedsson L, Gustavii B. Complement components in 100 newborns and their mothers determined by electroimmunoassay. Acta Pathol Microbiol Immunol Scand [C. 1983;91:147–50. [PubMed] [Google Scholar]
- 8.Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC. Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods. 2000;241:33–42. doi: 10.1016/s0022-1759(00)00198-8. [DOI] [PubMed] [Google Scholar]
- 9.Petersen SV, Thiel S, Jensen L, Steffensen R, Jensenius JC. An assay for the mannan-binding lectin pathway of complement activation. J Immunol Methods. 2001;257:107–16. doi: 10.1016/s0022-1759(01)00453-7. [DOI] [PubMed] [Google Scholar]
- 10.Tenner AJ, Lesavre PH, Cooper NR. Purification and radiolabeling of human C1q. J Immunol. 1981;127:648–53. [PubMed] [Google Scholar]
- 11.Klint C, Truedsson L, Sturfelt G. Binding to erythrocyte complement receptor type 1 of BSA/anti-BSA complexes opsonized by C4A3 or C4B1 in the presence of serum. Scand J Immunol. 1995;42:425–32. doi: 10.1111/j.1365-3083.1995.tb03676.x. [DOI] [PubMed] [Google Scholar]
- 12.Fredlund H, Sjoholm AG, Selander B, Holmstrom E, Olcen P, Danielsson D. Serum bactericidal activity and induction of chemiluminescence of polymorphonuclear leukocytes: complement activation pathway requirements in defense against Neisseria meningitidis. Int Arch Allergy Immunol. 1993;100:135–43. doi: 10.1159/000236400. [DOI] [PubMed] [Google Scholar]
- 13.Williams SC, Sim RB. Dye–ligand affinity purification of human complement factor B and beta 2 glycoprotein I. J Immunol Methods. 1993;157:25–30. doi: 10.1016/0022-1759(93)90066-g. [DOI] [PubMed] [Google Scholar]
- 14.Cooper N. Laboratory investigation of complement proteins and complement receptors. pp. 263–93. Bailliéres Clin Immunol Allergy.
- 15.Bengtsson AA, Sturfelt G, Gullstrand B, Truedsson L. Induction of apoptosis in monocytes and lymphocytes by serum from patients with systemic lupus erythematosus – an additional mechanism to increased autoantigen load? Clin Exp Immunol. 2004;135:535–43. doi: 10.1111/j.1365-2249.2003.02386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods. 2000;243:147–54. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 17.Söderström C, Braconier JH, Kayhty H, Sjöholm AG, Thuresson B. Immune response to tetravalent meningococcal vaccine: opsonic and bactericidal functions of normal and properdin deficient sera. Eur J Clin Microbiol Infect Dis. 1989;8:220–4. doi: 10.1007/BF01965264. [DOI] [PubMed] [Google Scholar]
- 18.Nauta AJ, Raaschou-Jensen N, Roos A, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33:2853–63. doi: 10.1002/eji.200323888. [DOI] [PubMed] [Google Scholar]
- 19.Nauta AJ, Trouw LA, Daha MR, et al. Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol. 2002;32:1726–36. doi: 10.1002/1521-4141(200206)32:6<1726::AID-IMMU1726>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 20.Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol. 2008;45:1199–207. doi: 10.1016/j.molimm.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Gaipl US, Kuenkele S, Voll RE, et al. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ. 2001;8:327–34. doi: 10.1038/sj.cdd.4400826. [DOI] [PubMed] [Google Scholar]
- 22.Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. 2002;196:655–65. doi: 10.1084/jem.20020542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paidassi H, Tacnet-Delorme P, Garlatti V, et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. 2008;180:2329–38. doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang H, Cooper B, Robey FA, Gewurz H. DNA binds and activates complement via residues 14–26 of the human C1q A chain. J Biol Chem. 1992;267:25597–601. [PubMed] [Google Scholar]
- 25.Paidassi H, Tacnet-Delorme P, Lunardi T, Arlaud GJ, Thielens NM, Frachet P. The lectin-like activity of human C1q and its implication in DNA and apoptotic cell recognition. FEBS Lett. 2008;582:3111–16. doi: 10.1016/j.febslet.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA. 2008;105:9023–8. doi: 10.1073/pnas.0801015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu W, Berger SP, Trouw LA, et al. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. 2008;180:7613–21. doi: 10.4049/jimmunol.180.11.7613. [DOI] [PubMed] [Google Scholar]
- 29.Truedsson L, Gullstrand B, Jönsson T, Klint C. Serum concentrations of C4 isotypes and factor B in type I C2 deficiency suggest haplotype-dependent quantitative expression of MHC class III complement genes. Exp Clin Immunogenet. 1995;12:66–73. [PubMed] [Google Scholar]
- 30.Selander B, Martensson U, Weintraub A, et al. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J Clin Invest. 2006;116:1425–34. doi: 10.1172/JCI25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsui H, Tsuji S, Nishimura H, Nagasawa S. Activation of the alternative pathway of complement by apoptotic Jurkat cells. FEBS Lett. 1994;351:419–22. doi: 10.1016/0014-5793(94)00897-3. [DOI] [PubMed] [Google Scholar]
- 32.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–30. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev. 2005;4:189–94. doi: 10.1016/j.autrev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–50. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 35.Sjöholm AG, Jönsson G, Braconier JH, Sturfelt G, Truedsson L. Complement deficiency and disease: an update. Mol Immunol. 2006;43:78–85. doi: 10.1016/j.molimm.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 36.Botto M. Links between complement deficiency and apoptosis. Arthritis Res. 2001;3:207–10. doi: 10.1186/ar301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Botto M, Dell'Agnola C, Bygrave AE, et al. C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 38.Mitchell DA, Pickering MC, Warren J, et al. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol. 2002;168:2538–43. doi: 10.4049/jimmunol.168.5.2538. [DOI] [PubMed] [Google Scholar]
- 39.Lu JH, Teh BK, Wang L, et al. The classical and regulatory functions of C1q in immunity and autoimmunity. Cell Mol Immunol. 2008;5:9–21. doi: 10.1038/cmi.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]