

Abstract
Fractalkine (FKN/CX3CL1) has been detected in synovial fluids from osteoarthritis (OA) patients. Additionally, low-level expression of the FKN receptor, CX3CR1, has been demonstrated in OA synovial lining. This study aimed to determine a biological function for this ligand/receptor pair in OA and to assess a potential signalling mechanism for FKN in this predominant synovial lining cell type, using chemotaxis assays, Western blotting and F-actin staining. Chemotaxis assays demonstrate that the chemokine domain of FKN effectively induces migration of OA fibroblasts. Consistent with this finding, visualization of F-actin demonstrates that 1 or 10 nM FKN induces noticeable reorganization of cytoskeletal structure in OA fibroblasts after 30 min stimulation with a maximal enhancement at approximately 2 h. In addition, Western blotting analysis demonstrates that FKN stimulates phosphorylation of the mitogen-activated protein (MAP) kinases p38, c-Jun N-terminal kinase (JNK) and extracellular-regulated kinase (ERK) 1/2 as well as the serine-threonine kinase Akt at Ser 473 and Thr 308. All these phosphorylation events occur in a time-dependent manner, with little or no activation within 1 min, and maximal activation occurring typically between 5 and 30 min. Moreover, inhibition of ERK 1/2 significantly reduces FKN-induced OA fibroblast migration. These results suggest that FKN is a novel chemoattractant for OA fibroblasts, consistent with FKN-induced alterations in cytoskeletal structure. In addition, FKN induces OA fibroblast signalling via the MAP kinases p38, JNK and ERK 1/2, as well as Akt.
Keywords: fibroblast, Fractalkine/CX3CL1, migration, OA, MAPK
Introduction
Osteoarthritis (OA) is the most common degenerative joint disease and involves the degradation of cartilage, subchondral bone and inflammation of the synovial membrane [1–3]. Cartilage damage can lead to inflammation and ultimately pain. Maintenance of healthy cartilage involves a continuing balance between catabolic and anabolic functions of chondrocytes [4,5]. The disruption of this balance is thought to involve both biochemical factors and mechanical stress [2,4,6]. Biochemical factors such as proinflammatory cytokines and matrix metalloproteinases (MMPs) secreted from a variety of cell types found in the OA joint have been shown to play an important role in maintaining the catabolic processes involved in OA disease progression [1,2,6,7]. In particular, OA synovial fibroblasts have been implicated in sustaining the damage found in the OA joint by producing proinflammatory cytokines as well as MMPs [8–10].
Chemokines are soluble chemotactic cytokines involved in regulating leucocyte trafficking, inflammation and infection [11,12]. The chemokines are designated into four different families based on a conserved cysteine motif. The four families, CXC, CC, C and CX3C, differ in the number and spacing of the first two cysteines near the N terminus. Fractalkine (FKN/CX3CL1), the sole member of the CX3C chemokine family, exists as both a soluble glycoprotein and a membrane-bound molecule [13]. The membrane-bound molecule consists of an intracellular tail, a short membrane-spanning region and a glycosylated mucin-like stalk holding the chemokine domain [13]. These two forms of FKN enable it to function as both an adhesion molecule and a chemoattractant. CX3CR1 is a specific receptor for FKN [14–16]. The expression of both FKN and its receptor can be induced on immune cells [15,17–20]. FKN has been implicated in many inflammatory diseases, such as rheumatoid arthritis (RA) [21–24], inflammatory bowel disease [25], asthma [26] and periodontal disease [27]. FKN has been found in the synovial fluid of OA patients [21]. In addition, the FKN receptor, CX3CR1, has been found in the OA synovium [24,28].
A more thorough understanding of the impact of the inflammatory response on signalling pathways may help to direct future therapies. Signalling pathways involve multiple levels of activation, usually through phosphorylation, leading ultimately to activation of transcription factors which, in turn, activate genes [29,30]. The mitogen-activated protein kinases (MAPK) are involved in mediating the effects of various cytokines associated with OA. Extracellular signal-regulated kinase (ERK) 1/2, c-Jun N-terminal kinase (JNK) and p38 are the terminal components of a three-step kinase cascade [29–31]. Akt, an anti-apoptotic kinase which promotes cell survival, is another kinase relevant to the study of signalling pathways important in OA [32].
In this study, we examined FKN and its receptor in OA fibroblasts to discover the possible biological functions of this ligand/receptor pair. We assessed the ability of FKN to act as a chemoattractant through chemotaxis assays and analysis of cyctoskeleton rearrangement. We further analysed potential signalling pathways activated by FKN stimulation of OA fibroblasts.
Material and methods
Preparation of OA fibroblasts
All synovium were obtained from OA patients undergoing a total knee replacement, with approval by the Midwestern University Institutional Review Board. Patients ranged in age from 48 to 78 years, with a mean age of 62, including two males and eight females. Synovial tissues were minced and digested for 2 h in an enzymatic cocktail at 37°C. OA fibroblasts were cultured in RPMI-1640 + 10% fetal bovine serum (FBS) and antibiotics. Cells were used beyond passage 3, at which time they were considered a homogeneous population of fibroblasts.
F-actin staining
The OA fibroblasts were plated on gelatin-coated glass coverslips in a 24-well culture plate at 50 000 cells/well in RPMI-1640 + 10% FBS. The following day cells appeared 50–60% confluent; they were washed with phosphate-buffered saline (PBS) and media was replaced with FBS-free RPMI-1640. Following 1 h incubation at 37°C, 1 or 10 nM recombinant human chemokine domain of FKN (rhFKN; R&D Systems, Minneapolis, MN, USA) was added to the wells for the times indicated (10 min, 30 min, 1 h, 2 h, 3 h). Fixation took place in 1 ml of 3·7% formaldehyde in PBS for 10 min at room temperature followed by two washes with PBS. Round coverslips were removed from the culture plate and cells were permeabilized with acetone for 3 min at –20°C and washed twice immediately in PBS. Coverslips were blocked in PBS + 1% bovine serum albumin (BSA) for 25 min. Block was replaced with Alexa Fluor® 488 Phalloidin (Molecular Probes, Carlsbad, CA, USA) in PBS + 1% BSA + 4′-6-diamidino-2-phenylindole (10 µg/ml) for 20 min at room temperature. Coverslips were washed two final times, air-dried, mounted on a microscope slide and stored at 4°C in the dark until observed. Representative photographs were taken with a Nikon Eclipse E400 microscope attached to a Spot Digital camera.
Chemotaxis assays
The OA fibroblasts were fed the night before the assay with RPMI-1640 + 10% FBS. Cells (3·9 × 104 cells in 26 µl of RPMI-1640 containing 0·1% FBS) were added to the bottom wells of chemotaxis chambers (Neuroprobe, Gaithersburg, MD, USA) with an 8-µm gelatin-coated polycarbonate membrane on the top. Inverted chambers were incubated for 2 h at 37°C in a 5% CO2 atmosphere, allowing fibroblast attachment to the membrane. Inhibition with pertussis toxin (PTX; Sigma, St Louis, MO, USA), an inhibitor of G protein (Gi/Go) coupled receptors, was accomplished by pretreating OA fibroblasts with 100 ng/ml PTX overnight and chemotaxis was performed the next day, as described previously [33]. To assess the impact of ERK 1/2 activity on fibroblast chemotaxis, OA fibroblasts were pretreated with vehicle (0·1% dimethyl sulphoxide) or with 10 µM U0126 (Cell Signaling, Inc., Beverly, MA, USA) for 2 h while cells adhered to the membrane, prior to introduction of FKN. U0126 inhibits both active and inactive MAP-ERK (MEK) 1/2, the kinase which activates ERK 1/2. PBS served as a negative control, while 3% FBS served as a positive control. Diluted rhFKN was added to the top wells and the chambers were incubated overnight to allow migration at 37°C. The membranes had non-migrated cells removed with a cotton swab; they were fixed in methanol and stained with Diff-Quik (Dade Behring, Deerfield, IL, USA). Samples were assayed in quadruplicate and migrated cells from three randomly selected high power fields were counted. Statistical analysis was performed using a Student's t-test.
Western blot analysis
The OA fibroblasts were cultured in RPMI-1640 + 10% FBS, washed with PBS, and were serum-starved for 1 h in RPMI-1640. Media was replaced with RPMI-1640 + 10 nM FKN prior to lysing for 1 min, 5 min, 15 min or 30 min. Extraction buffer for OA fibroblast lysing included 20 mM Tris pH 7·4, 137 mM NaCl, 1 mM Na4O7P2, 10 µM NaVO4, 100 µM NaF, 1% NP-40 and 10% glycerol. OA fibroblast lysates were combined 1:1 with Laemmlin's sample buffer + 20% sodium dodecyl sulphate and boiled for 5 min prior to being subjected to gel electrophoresis. Transfer of separated proteins onto a nitrocellulose membrane was accomplished using a Trans-Blot® SD Electrophoretic Cell (Bio Rad Laboratories, Inc., Hercules, CA, USA) for 30 min. Non-specific binding was blocked by incubation with 5% milk in Tris-buffered saline + 0·1% Tween-20 (TBST) for 1 h at 4°C. Blots were then incubated overnight at 4°C with primary antibody (Cell Signaling Technology, Inc.). Washing was performed with TBST, followed by blot exposure to horseradish peroxidase-conjugated goat anti-rabbit IgG antibody diluted 1:2000 in TBST + 5% milk for 1 h at 4°C. Detection was accomplished using ECL Plus (Amersham, Piscataway, NJ, USA) and a Storm 860 PhosphorImager (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, USA) together with Image Quant software (Molecular Dynamics, Sunnyvale, CA, USA) to quantitate the bands.
Results
The FKN induces actin rearrangement
To study the reorganization of the actin cytoskeleton in response to FKN, we stained the actin skeleton of fibroblasts from three different OA patients who had been exposed to 1 or 10 nM FKN over a time–course. The non-stimulated cells (Fig. 1a) show thin actin fibres with low levels of staining that could barely be detected. After 10 min stimulation (Fig. 1b) with 1 nM FKN, little change was noted; however, after 30 min some cells began displaying thin bands of actin fibres that stretched the entire length of the cell (Fig. 1c) to 1 h (Fig. 1d). The maximal stimulation of F-actin was observed in all patients after 2–3 h, where many cells showed strong staining of F-actin (Fig. 1e and f) and a completely altered morphology. Stimulation with 10 nM FKN resulted in similar changes to those seen with 1 nM FKN (data not shown).
Fig. 1.
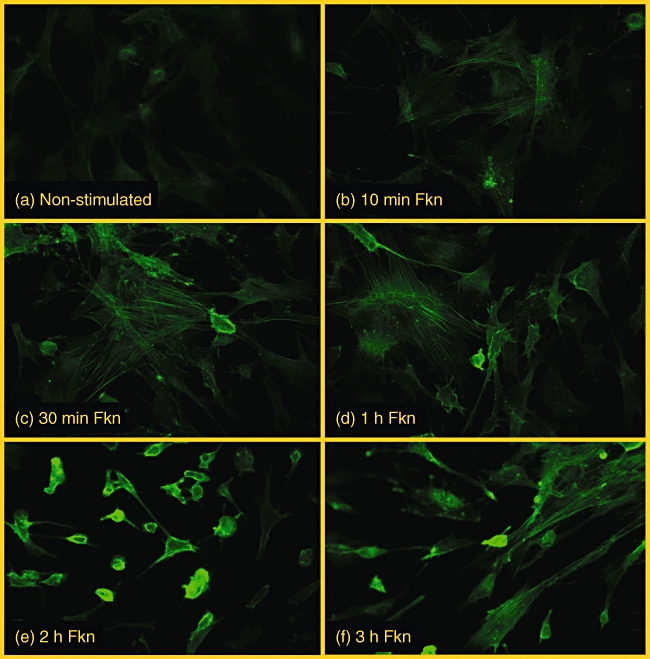
(a) Background staining of F-actin in non-stimulated osteoarthritis (OA) fibroblasts. Cells stimulated with 1 nM fractalkine and stained for F-actin after 10 min (b), 30 min (c), 1 h (d), 2 h (e), and 3 h (f) are shown for comparison.
The FKN induces migration of OA fibroblasts in vitro
Chemotaxis assays were performed to test the effect of FKN on OA fibroblast migration using fibroblasts from 10 different patients. Results of a representative experiment are shown in Fig. 2. The highest migration was seen towards 3% FBS, which was used as a positive control. FKN itself induced migration in a dose-dependent manner. Maximum migration was obtained at 5 nM FKN (Fig. 2), which was significantly higher than background migration shown by PBS (negative control). OA fibroblasts derived from all 10 patients migrated to FKN (n = 10, P < 0·05).
Fig. 2.
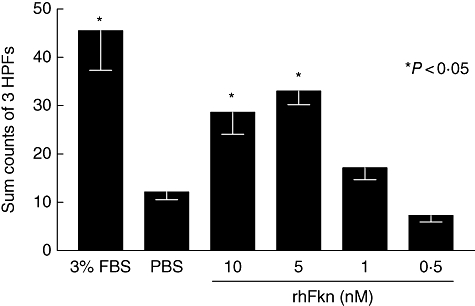
The positive control indicates osteoarthritis (OA) fibroblasts migrating towards 3% fetal bovine serum (a fibroblast chemoattractant). The quantity of background migration is indicated by the negative control, phosphate-buffered saline (PBS). Migration towards several concentrations of fractalkine (FKN) are shown. This graph is representative of assays performed on OA fibroblasts obtained from 10 different patients, where FKN induced significant migration in all cases. Bars and error bars represent the mean ± standard error of the mean of counts summed from three high powered fields (HPFs). An asterisk indicates significant migration above the negative control PBS.
The FKN activates MAPK JNK, p38 and ERK 1/2
Analyses of intracellular signalling revealed that FKN induces the activation of MAPK in OA fibroblasts in a time-dependent manner. In particular, we noted an up-regulation of JNK phosphorylation (Fig. 3a), with the highest levels of activation at 15 min. Bars represent phosphorylated JNK normalized to total JNK in arbitrary units. Blots are representative of similar experiments performed using OA fibroblasts from three different patients (n = 3). Similarly, levels of phosphorylated p38 were also elevated after 1 min stimulation with FKN (Fig. 3b) upon normalization to total p38. Bars represent phosphorylated p38 normalized to total p38 and are representative of three separate experiments using fibroblasts from three different patients.
Fig. 3.
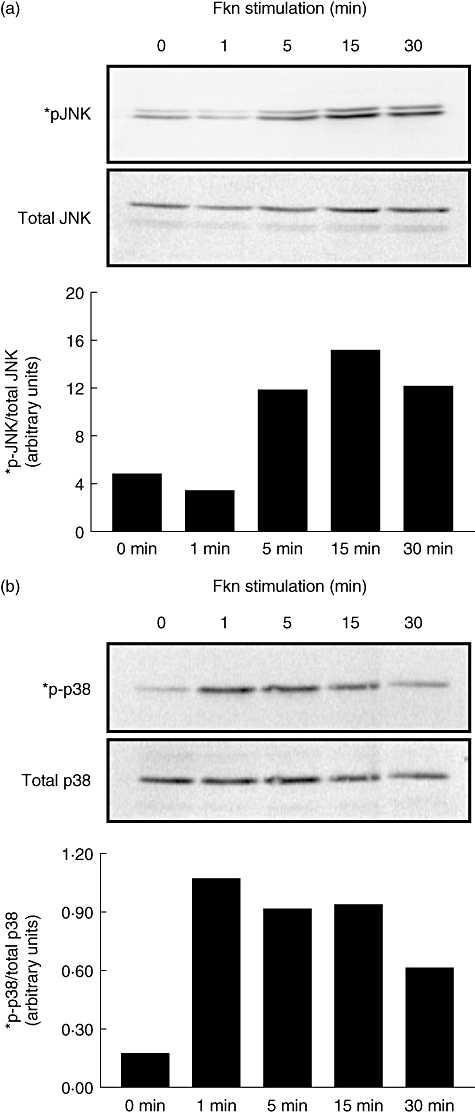
Fractalkine (10 nM) was added to osteoarthritis (OA) fibroblasts for the times indicated and cell lysates were generated. Protein (20 µg/lane) was separated using polyacrylamide gel electrophoresis and blotted using standard Western techniques. Phosphorylated c-Jun N-terminal kinase (a) and p38 (b) were compared with the total amount of their respective proteins to assure equal loading. Bars represent normalized band intensity in arbitrary units. Blots are representative of three different experiments performed using OA fibroblasts from three different patients.
The FKN-mediated phosphorylation of ERK 1/2 was also examined in OA fibroblasts. At 10 nM concentration, FKN induced phosphorylation of ERK 1/2 in a time-dependent manner. Within the 5–30 min time-frame (Fig. 4) of FKN stimulation, phosphorylation was increased, compared with non-stimulated cells. Bars represent phosphorylated ERK 1/2 normalized to total ERK 1/2 and are representative of three separate experiments using fibroblasts from three different patients. The peak of phosphorylation of ERK 1/2 was observed 15 min after stimulation.
Fig. 4.
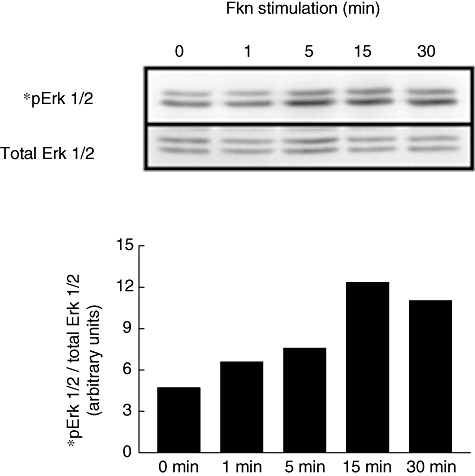
Fractalkine (10 nM) was added to osteoarthritis (OA) fibroblasts for the times indicated and cell lysates were generated. Protein (20 µg/lane) was separated using polyacrylamide gel electrophoresis and blotted using standard Western techniques. Phosphorylated extracellular signal-regulated kinase (ERK) 1/2 is compared with total Erk 1/2 to assure equal loading. Bars represent normalized band intensity in arbitrary units. Blots are representative of three different experiments performed using OA fibroblasts from three different patients.
The FKN fully activates Akt through phosphorylation of Ser 473 and Thr 308
Additional experiments were conducted to establish whether FKN stimulation of fibroblasts can activate downstream signalling pathways. As shown in Fig. 5, Akt was clearly phosphorylated by FKN at the two kinase regulatory sites, Ser 473 (Fig. 5a) and Thr 308 (Fig. 5b), with a maximum effect at 5 min. Bars represent phosphorylated Akt normalized to total Akt and are representative of two separate experiments using fibroblasts from two different patients. The levels of phosphorylation were seen to decline after the peak at 5 min. FKN-stimulated (10 nM; 24 h) and non-stimulated OA fibroblast lysates were also compared for differences in relative amounts of 35 apoptosis-related proteins using Proteome Profiler Human Apoptosis Antibody Array (R&D Systems). While no major differences were noted (n = 2 different patients), there appeared to be a slight decrease in SMAC/Diablo and HSP27 in both experiments (data not shown). In addition, OA fibroblasts were treated with 10 nM FKN prior to inducing apoptosis using 1·5 mM sodium nitroferricyanide(III) dehydrate (SNP), and changes in apoptosis were examined using an In Situ Cell Death Kit (Roche, Indianapolis, IN, USA). Inclusion of 3000 U/ml DNase 1 served as a positive control, whereas a lack of terminal deoxynucleotidyl transferase from calf thymus served as our negative control. FKN-pretreatment did not show any signs of inhibiting apoptosis (n = 3; data not shown).
Fig. 5.
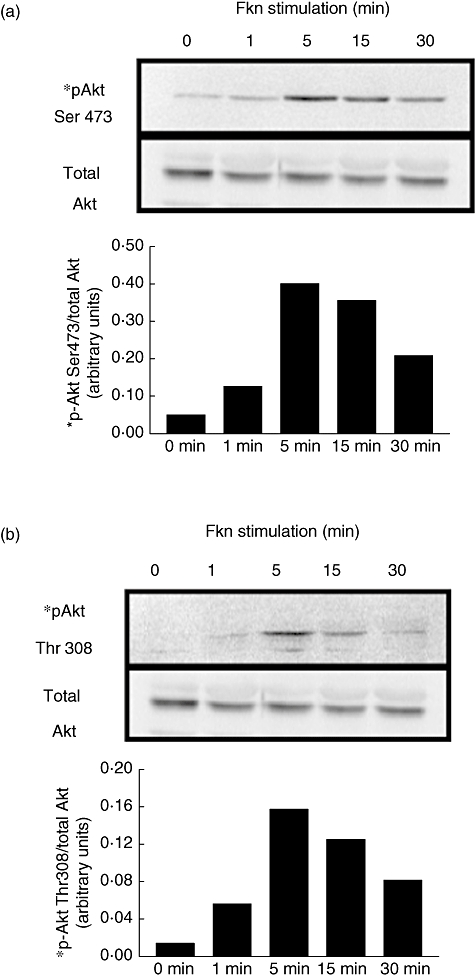
Fractalkine (10 nM) was added to osteoarthritis (OA) fibroblasts for the times indicated and cell lysates were generated. Protein (20 µg/lane) was separated using polyacrylamide gel electrophoresis and then blotted using standard Western blotting techniques. Phosphorylated Akt was compared with total Akt to assure equal loading. Bars represent normalized band intensity in arbitrary units. Blots are representative of two different experiments performed using OA fibroblasts from two different patients.
CX3CR1 and ERK 1/2 probably regulate FKN-induced OA fibroblast migration
To examine whether a G protein-coupled receptor such as CX3CR1 is involved, OA fibroblasts were pretreated with PTX and migration was determined in response to 10 nM FKN. Figure 6a (representative experiment; n = 3; P < 0·05) suggests that PTX reduces significantly any response to FKN. Figure 6b shows results of FKN-induced chemotaxis performed in the presence of a MEK 1/2 inhibitor (10 µM U0126). MEK 1/2 is a kinase upstream from ERK 1/2 and is the only known kinase to date to phosphorylate ERK 1/2. OA fibroblasts pretreated with vehicle control showed significant migration towards FKN at 1, 5 or 10 nM concentrations. OA fibroblasts preincubated with the inhibitor, however, showed significantly reduced migration to FKN. Data in Figure 6b are representative of three separate experiments performed with OA fibroblasts from three separate patients.
Fig. 6.
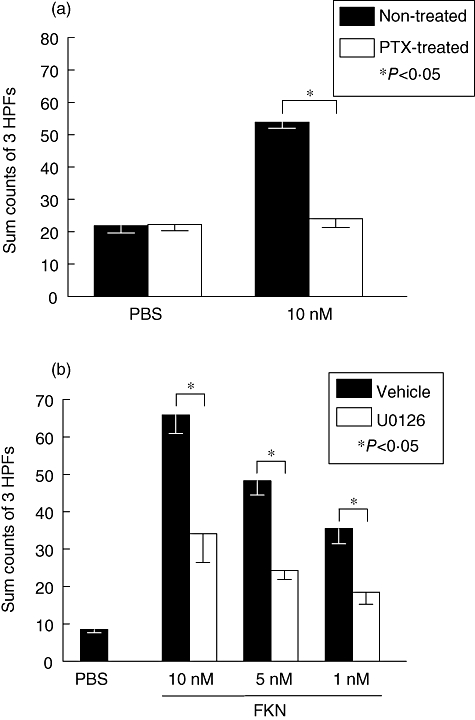
Fractalkine (FKN)-induced osteoarthritis (OA) fibroblast-like synoviocytes (FLS) migration is pertussis toxin (PTX)-sensitive and regulated by extracellular signal-regulated kinase (ERK) 1/2. (a) Migration to FKN is compared in the presence and absence of PTX. This graph is representative of three experiments performed on OA fibroblasts derived from three different patients. The bars represent the mean while error bars represent standard error of the mean of counts summed from three high powered fields (HPFs). A significant decrease in migration with PTX treatment is denoted by an asterisk. (b) OA FLS were pretreated with mitogen-activated protein (MAP)-ERK (MEK) 1/2 inhibitor or a vehicle and used to migrate to different doses of FKN. At 1, 5 and 10 nM FKN concentrations, significant migration was induced that could be reduced significantly by inhibition of ERK 1/2. Bars and error bars represent the mean ± standard error of the mean of counts summed from three HPFs. An asterisk indicates significant migration above the negative control phosphate-buffered saline.
Discussion
Inflammation has long been recognized as a major factor in RA but until recent years has not been the focus of disease progression in OA. Severe inflammation has been demonstrated in synovial tissue samples of OA patients and was associated with the angiogenic vascular endothelial growth factor [7]. It has been demonstrated that fibroblasts may play an important role in the progression of joint destruction in the OA joint. We have demonstrated previously that FKN is present in the synovial fluid of OA patients [21]. Further, the FKN receptor, CX3CR1, has been found in the OA synovium [24,28]. We have also demonstrated previously that RA fibroblast-like synoviocytes (FLS) respond to FKN [22]. In addition, FKN stimulates marked up-regulation of RA fibroblast MMP-2 production [24]. In this study, we investigated the ability of OA fibroblasts to migrate in response to FKN. Our results demonstrate that FKN induces migration of OA fibroblasts effectively in a concentration-dependent manner.
Fibroblasts are an important component of the OA synovium and have been shown to contribute to joint destruction through the release of proinflammatory cytokines, as well as the release of MMPs [6,8,10]. To assess broadly whether FKN-stimulation of OA fibroblasts induces alterations in cytokine expression at the protein level, we performed antibody array experiments to assess changes in 79 different cytokines, chemokines and growth factors (RayBio® Human Cytokine Antibody Array V, Atlanta, GA, USA). An initial experiment exposed OA fibroblasts to 1 nM FKN for 24 h, and compared OA fibroblasts with RA FLS. We noted no differences in any of the 79 proteins when either OA fibroblasts or RA FLS were exposed to FKN (data not shown). Our next experiment extended the time-frame to 48 h stimulation with 1 nM FKN and showed exactly the same result, with no noted differences. A final experiment, using 10 nM FKN stimulation for 48 h, suggested that OA fibroblasts may slightly increase expression of interleukin (IL)-1α, growth-related oncogene and regulated upon activation normal T cell expressed and secreted, but similar changes were not seen in RA FLS (data not shown). Our conclusion from these experiments is that FKN does not appear to induce any major OA fibroblast changes in cytokine expression.
A chemotactic function of FKN is supported by the rearrangement of actin filaments in OA fibroblasts after stimulation with FKN. The effect of FKN on the organization of actin fibres was analysed by Phalloidin staining and followed for several hours after stimulation of OA fibroblasts. The staining demonstrated clear cytoskeleton remodelling within the OA fibroblasts. Taken together with chemotaxis assays, our results suggest that FKN may be directing CX3CR1-expressing OA fibroblasts in the synovium. OA synovial fibroblasts are not unique in this regard, as both normal synovial fibroblasts and RA FLS also migrate to FKN [22]. A key dissimilarity, however, may be at the level of FKN expression, where differences exist between RA, OA and NL synovial tissue and diseased synovial fluids [21,24].
The MAPK activation has been implicated in the induction of many of the mediators thought to be involved in OA. MAPK have also been implicated in the induction of MMPs in chondrocytes as well as apoptosis of chondrocytes [30,34]. The activation of MAPK by proinflammatory cytokines has been well documented [35]. Specifically, JNK and p38 have been activated by tumour necrosis factor and IL-1β[29,36].
The importance of understanding the signalling pathways initiated by cytokines in the progression of OA has been demonstrated by recent studies. Pelletier and colleagues [37], using a rabbit OA model, demonstrated a decrease in the severity of synovial inflammation after treating the OA rabbits with a MEK 1/2 inhibitor. They demonstrated that this effect was associated with a reduction in the level of activated ERK 1/2 in OA chondrocytes [37]. Additionally, the MAPK, p38, was demonstrated to be involved in the regulation of MMP-13 in human OA chondrocytes [38]. Based on these studies, we wanted to determine if MAPK were involved in the response by OA fibroblasts to FKN stimulation. In our study, FKN induced OA fibroblast activation of the three MAPK: p38, JNK and ERK 1/2. This activation of multiple MAPK indicates possible stimulation of multiple mediators from one cell signal initiated by FKN. These findings of JNK and ERK 1/2 phosphorylation in response to FKN are similar to RA FLS [22], while phosphorylation of p38 represents a novel finding not examined previously.
We have demonstrated that RA fibroblasts activate Akt upon FKN-stimulation [22], and investigated a similar mechanism in this study. Cell survival-related pathways were also found to be activated by FKN stimulation of OA fibroblasts. In our study, Akt was found to be phosphorylated at both Ser-473 and Thr-308, demonstrating full activation after stimulation of OA fibroblasts by FKN. Akt is an anti-apoptotic kinase that promotes cell survival through multiple pathways. For example, Akt was shown to play a key role in the survival and proliferation of fibroblasts in the RA synovium [39] and an anti-apoptotic role in NIH 3T3 fibroblasts [35]. The involvement of FKN in cell survival is also documented. Previously, we found Akt to be activated fully by FKN in RA FLS [22]. Furthermore, FKN has been shown to promote cell survival of microglia [40]. However, we demonstrate here that pretreatment of OA fibroblasts with FKN is not sufficient to induce cell survival when apoptosis is induced by SNP. Therefore, it is not clear at this time as to the role of FKN-induced Akt phosphorylation.
In conclusion, we have demonstrated that FKN was able to induce chemotaxis with corresponding actin cytoskeletal changes of OA fibroblasts. These findings indicate a novel role of FKN in regulating OA fibroblast migration. FKN specifically induces OA fibroblast phosphorylation of the MAPK p38, JNK and ERK 1/2, as well as activation of Akt. OA fibroblast migration to FKN, in part, requires ERK 1/2 activation. Taken together with our previous study [22], these results suggest that OA fibroblasts respond to FKN in a similar manner to RA fibroblasts, suggesting that both cell types express functional CX3CR1. However, while both respond to FKN, a major difference between the diseases exists at the level of FKN expression, where FKN is expressed at relatively high levels in RA synovial tissue and fluid compared with comparable samples from OA patients [21,24]. We also note that OA synovium contains shaved cartilage and bone fragments that appear to attach actively to synoviocytes, including OA fibroblasts. Beyond attachment, a limited number of OA synovial fibroblasts are described to deeply invade bone and cartilage fragments, suggesting a behaviour known previously only for RA FLS [41]. Moreover, human osteoblasts express full-length FKN and can release its soluble form in a manner which attracts CX3CR1-expressing cells [42]. Therefore, we speculate that FKN may play a role in OA fibroblast migration to bone fragments, contributing to detritus synovitis. Understanding more fully the mechanisms by which FKN acts upon OA fibroblasts may elucidate a role for FKN in inflammation and the progression of OA.
Acknowledgments
This work was supported by an Arthritis Foundation Arthritis Investigator Award (J. M. W.) and NIH grant R15AR050985 (J. M. W.). The authors are grateful to Ross Sherban DO for his efforts in providing OA synovial tissue.
References
- 1.Sun BH, Wu CW, Kalunian KC. New developments in osteoarthritis. Rheum Dis Clin North Am. 2007;33:135–48. doi: 10.1016/j.rdc.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Haynes MK, Hume EL, Smith JB. Phenotypic characterization of inflammatory cells from osteoarthritic synovium and synovial fluids. Clin Immunol. 2002;105:315–25. doi: 10.1006/clim.2002.5283. [DOI] [PubMed] [Google Scholar]
- 3.Felson DT. An update on the pathogenesis and epidemiology of osteoarthritis. Radiol Clin North Am. 2004;42:1–9. doi: 10.1016/S0033-8389(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 4.Borzì RM, Mazzetti I, Cattini L, Uguccioni M, Baggiolini M, Facchini A. Human chondrocytes express functional chemokine receptors and release matrix-degrading enzymes in response to C-X-C and C-C chemokines. Arthritis Rheum. 2000;43:1734–41. doi: 10.1002/1529-0131(200008)43:8<1734::AID-ANR9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 5.Westacott CI, Sharif M. Cytokines in osteoarthritis: mediators or markers of joint destruction? Semin Arthritis Rheum. 1996;25:254–72. doi: 10.1016/s0049-0172(96)80036-9. [DOI] [PubMed] [Google Scholar]
- 6.Borzì RM, Mazzetti I, Marcu KB, Facchini A. Chemokines in cartilage degradation. Clin Orthop Relat Res. 2004;427:S53–61. doi: 10.1097/01.blo.0000143805.64755.4f. [DOI] [PubMed] [Google Scholar]
- 7.Haywood L, McWilliams DF, Pearson CI, et al. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum. 2003;48:2173–7. doi: 10.1002/art.11094. [DOI] [PubMed] [Google Scholar]
- 8.Lu HT, Liang YC, Sheu MT, et al. Disease-modifying effects of glucosamine HCl involving regulation of metalloproteinases and chemokines activated by interleukin-1beta in human primary synovial fibroblasts. J Cell Biochem. 2008;104:38–50. doi: 10.1002/jcb.21597. [DOI] [PubMed] [Google Scholar]
- 9.Fuchs S, Skwara A, Bloch M, Dankbar B. Differential induction and regulation of matrix metalloproteinases in osteoarthritic tissue and fluid synovial fibroblasts. Osteoarthr Cartil. 2004;12:409–18. doi: 10.1016/j.joca.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Bondeson J, Lauder S, Wainwright S, et al. Adenoviral gene transfer of the endogenous inhibitor IkappaBalpha into human osteoarthritis synovial fibroblasts demonstrates that several matrix metalloproteinases and aggrecanases are nuclear factor-kappaB-dependent. J Rheumatol. 2007;34:523–33. [PubMed] [Google Scholar]
- 11.Bacon K, Baggiolini M, Broxmeyer H, et al. Chemokine/chemokine receptor nomenclature. J Interferon Cytokine Res. 2002;22:1067–8. doi: 10.1089/107999002760624305. [DOI] [PubMed] [Google Scholar]
- 12.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 13.Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–4. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 14.Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–30. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 15.Fong AM, Robinson LA, Steeber DA, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–19. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umehara H, Goda S, Imai T, et al. Fractalkine, a CX3C-chemokine, functions predominantly as an adhesion molecule in monocytic cell line THP-1. Immunol Cell Biol. 2001;79:298–302. doi: 10.1046/j.1440-1711.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- 17.Mizutani N, Sakurai T, Shibata T, et al. Dose-dependent differential regulation of cytokine secretion from macrophages by fractalkine. J Immunol. 2007;179:7478–87. doi: 10.4049/jimmunol.179.11.7478. [DOI] [PubMed] [Google Scholar]
- 18.Fraticelli P, Sironi M, Bianchi G, et al. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001;107:1173–81. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM. T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005;52:1392–401. doi: 10.1002/art.21140. [DOI] [PubMed] [Google Scholar]
- 20.Imaizumi T, Matsumiya T, Tamo W, et al. 15-Deoxy-D12,14 -prostaglandin J2 inhibits CX3CL1/fractalkine expression in human endothelial cells. Immunol Cell Biol. 2002;80:531–6. doi: 10.1046/j.1440-1711.2002.01111.x. [DOI] [PubMed] [Google Scholar]
- 21.Ruth JH, Volin MV, Haines GK, et al. Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvant-induced arthritis. Arthritis Rheum. 2001;4:1568–81. doi: 10.1002/1529-0131(200107)44:7<1568::AID-ART280>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 22.Volin MV, Huynh N, Klosowska K, Chong KK, Woods JM. Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 2007;56:2512–22. doi: 10.1002/art.22806. [DOI] [PubMed] [Google Scholar]
- 23.Volin MV, Woods JM, Amin MA, Connors MA, Harlow LA, Koch AE. Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. Am J Pathol. 2001;159:1521–30. doi: 10.1016/S0002-9440(10)62537-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blaschke S, Koziolek M, Schwarz A, et al. Proinflammatory role of fractalkine (CX3CL1) in rheumatoid arthritis. J Rheumatol. 2003;30:1918–27. [PubMed] [Google Scholar]
- 25.Muehlhoefer A, Saubermann LJ, Gu X, et al. Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol. 2000;164:3368–76. doi: 10.4049/jimmunol.164.6.3368. [DOI] [PubMed] [Google Scholar]
- 26.Tremblay K, Lemire M, Provost V, et al. Association study between the CX3CR1 gene and asthma. Genes Immun. 2006;7:632–9. doi: 10.1038/sj.gene.6364340. [DOI] [PubMed] [Google Scholar]
- 27.Hosokawa Y, Nakanishi T, Yamaguchi D, Nakae H, Matsuo T. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, in periodontal diseased tissue. Clin Exp Immunol. 2005;139:506–12. doi: 10.1111/j.1365-2249.2005.02675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nanki T, Imai T, Nagasaka K, et al. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46:2878–83. doi: 10.1002/art.10622. [DOI] [PubMed] [Google Scholar]
- 29.Firestein GS, Manning AM. Signal transduction and transcription factors in rheumatic disease. Arthritis Rheum. 1999;42:609–21. doi: 10.1002/1529-0131(199904)42:4<609::AID-ANR3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 30.Sweeney SE, Firestein GS. Primer: signal transduction in rheumatic disease – a clinician's guide. Nat Clin Pract Rheumatol. 2007;3:651–60. doi: 10.1038/ncprheum0631. [DOI] [PubMed] [Google Scholar]
- 31.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–15. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 32.Krasilnikov MA. Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation. Biochemistry (Mosc) 2000;65:59–67. [PubMed] [Google Scholar]
- 33.Park KS, Lee HY, Kim MK, et al. Lysophosphatidylserine stimulates L2071 mouse fibroblast chemotactic migration via a process involving pertussis toxin-sensitive trimeric G-proteins. Mol Pharmacol. 2006;69:1066–73. doi: 10.1124/mol.105.018960. [DOI] [PubMed] [Google Scholar]
- 34.Brauchle M, Glück D, Di Padova F, Han J, Gram H. Independent role of p38 and ERK1/2 mitogen-activated kinases in the upregulation of matrix metalloproteinase-1. Exp Cell Res. 2000;258:135–44. doi: 10.1006/excr.2000.4913. [DOI] [PubMed] [Google Scholar]
- 35.Goruppi S, Ruaro E, Varnum B, Schneider C. Requirement of phosphotidylinositol 3kinase dependent pathway and Src for Gas6-Ax1 mitogenic and survival activities in NIH 3T3 fibroblasts. Mol Cell Biol. 1997;17:4442–53. doi: 10.1128/mcb.17.8.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–93. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 37.Pelletier JP, Fernandes JC, Brunet J, et al. In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum. 2003;48:1582–93. doi: 10.1002/art.11014. [DOI] [PubMed] [Google Scholar]
- 38.Boileau C, Pelletier JP, Tardif G, et al. The regulation of human MMP-13 by licofelone, an inhibitor of cyclo-oxygenases and 5-lipoxygenase, in human osteoarthritic chondrocytes is mediated by the inhibition of the p38 MAP kinase signalling pathway. Ann Rheum Dis. 2005;64:891–8. doi: 10.1136/ard.2004.026906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang HG, Wang Y, Xie JF, et al. Regulation of tumor necrosis factor alpha-mediated apoptosis of rheumatoid arthritis synovial fibroblasts by the protein kinase Akt. Arthritis Rheum. 2001;44:1555–67. doi: 10.1002/1529-0131(200107)44:7<1555::AID-ART279>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 40.Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB. Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J Immunol. 1999;163:1628–35. [PubMed] [Google Scholar]
- 41.Schedel J, Wenglén C, Distler O, et al. Differential adherence of osteoarthritis and rheumatoid arthritis synovial fibroblasts to cartilage and bone matrix proteins and its implications for osteoarthritis pathogenesis. Scand J Immunol. 2004;60:514–23. doi: 10.1111/j.0300-9475.2004.01507.x. [DOI] [PubMed] [Google Scholar]
- 42.Shulby SA, Dolloff NG, Stearns ME, Meucci O, Fatatis A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004;64:4693–98. doi: 10.1158/0008-5472.CAN-03-3437. [DOI] [PubMed] [Google Scholar]