

Abstract
Tumour necrosis factor (TNF) blockade has become an important immunomodulatory therapy, particularly in patients refractory to conventional immunosuppression, but responses can be unpredictable. Understanding the complex biology of TNF processing may be key to predicting such responses and reduce unwanted side effects. TNF bioavailability is regulated partly by TNF-α converting enzyme (TACE) cleavage; however, it can also be cleaved by proteinase-3 (PR-3). We have demonstrated this mechanism previously in healthy human alveolar macrophages (AM), leading us to hypothesize that PR-3-mediated TNF processing may be an important mechanism in inflammatory lung disease. Furthermore, this may be more apparent in diseases with a neutrophil component typical of usual interstitial pneumonia (UIP), compared with sarcoidosis, where lymphocytes predominate. We isolated AM from patients with UIP and sarcoidosis and healthy subjects. We found increased TACE expression on AM in sarcoidosis. In contrast, TACE was not increased in UIP; we found increased cleavage of glutathione S-transferase-proTNF) substrate, relative to both sarcoidosis and healthy controls. Furthermore, cleavage was subject to inhibition by serine protease inhibitor, rather than a TACE inhibitor BB-3103. Cleavage was proportional to the number of neutrophils isolated from bronchoalveolar lavage, whereas there was an inverse relationship between neutrophils and BB-3103 inhibition. There was also increased PR-3 on the AM surface in UIP relative to healthy controls. These data provide evidence for PR-3-mediated cleavage in UIP, which may have implications for future therapeutic targeting of TACE.
Keywords: human, inflammation, lung, macrophages, TNF
Introduction
Tumour necrosis factor (TNF)-α is implicated in the pathogenesis of numerous immunologically mediated conditions and its central role has been emphasized recently by the success of anti-TNF therapies in diseases such as rheumatoid arthritis [1], Crohn's disease [2] and steroid refractory asthma [3]. Anti-TNF therapies have also been used in refractory sarcoidosis [4], idiopathic pulmonary fibrosis [5] and moderate to severe chronic obstructive pulmonary disease [6], but with less impressive results. An understanding of the complex biology of TNF processing and activity may be key to predicting responses to TNF blockade. This could potentially minimize unwanted side effects such as increased risk of infection [7].
The TNF-α is produced in a membrane-associated form (mTNF) which is cleaved by TNF-α converting enzyme (TACE) to yield the soluble product. Both forms can initiate cellular responses by binding to receptors on neighbouring cells [8,9]. It is likely that these responses are different because of altered ligation of TNF receptors. Soluble ligation initiates cytokine production, whereas mTNF can induce apoptosis and anergy in neighbouring cells [10]. Membrane-bound TNF is also able to reverse signal, a phenomenon known to exist in the TNF superfamily. This mechanism is thought to be important in down-regulating inflammatory responses, as ligation of mTNF in monocytes has demonstrated inhibition of lipopolysaccharide (LPS) responses [11] and this may be related to enhanced mRNA degradation [12].
Clearly, mechanisms regulating the relative availability of mTNF and soluble TNF are important. It is now recognized that TNF cleavage can be mediated by the serine protease proteinase-3 (PR-3) [13], in addition to TACE. PR-3 is the target antigen for the autoantibody in Wegener's granulomatosis, but whether or not it contributes to the pathogenesis or is an epiphenomenon is not clear [14]. PR-3 is able to degrade extracellular matrix, and its potential contribution to pulmonary inflammatory disease has been demonstrated by the induction of emphysema in hamsters following intratrachael installation [15]. PR-3 is also increased in the sputum of cystic fibrosis patients, and correlates with disease severity [16]. We have described PR-3-mediated cleavage of mTNF on the alveolar macrophage (AM) surface in healthy subjects [17]. We have also demonstrated that PR-3 is expressed on AM and that this is not de novo expression but exogenous PR-3.
Because lung neutrophils are the most likely source of PR-3, we hypothesized that there would be an increased contribution of PR-3 to TNF processing in diseases with abundant alveolar neutrophils. Thus we selected subjects with usual interstitial pneumonia (UIP) as a model of neutrophil-predominant disease and pulmonary sarcoidosis as a model where T cells predominate (thus PR-3 expression should be minimal). We evaluated expression of both TACE and PR-3 on the surface of AM from our study groups. In addition, we determined the relative impact of TACE and PR-3 catalytic activity using specific inhibitors.
Materials and methods
Subjects
Patients with UIP were diagnosed according to American Thoracic Society/European Respiratory Society criteria [18]. The UIP group (n = 20) had a median age of 73 years (range 42–88 years). The normal control population (n = 15), with no previous history of lung disease, had a median age of 54 years (range 23–87 years). Patients with pulmonary sarcoidosis (n = 15) had disease confirmed by histology on lung biopsy and a median age of 43 years (range 27–78 years; see Table 1). No patients were receiving steroids prior to inclusion. The study was approved by North Bristol National Health Service Trust Ethics Committee.
Table 1.
Characteristics of bronchoalveolar lavage (BAL) cell populations, median (range).
| Normal | Sarcoidosis | UIP | |
|---|---|---|---|
| Total cells × 106 | 11·2 (6·6–21·2) | 9·7 (8·1–21·7) | 18·4 (10·6–35·7) |
| Percentage of total BAL cells | |||
| Alveolar macrophages | 96·4 (88·2–100) | 72·7 (46·1–89·1) | 68·4 (41·1–74·9) |
| Lymphocytes | 1·5 (0–4·7) | 25·1 (11·0–53·7) | 4·1 (0·6–7·1) |
| Neutrophils | 1·9 (0–5·4) | 2·1 (0·6–2·7) | 25·2 (7·6–52·3) |
| Eosinophils | 0 | 0·9 (0–3·2) | 3·1 (1·1–4·5) |
UIP, usual interstitial pneumonia.
The AM culture
Bronchoalveolar lavage and AM isolation were performed as described previously [19]. AM were cultured for 24 h with LPS (Sigma, Poole, UK) in the presence or absence of 10 µM BB-3103 metalloproteinase inhibitor (courtesy of British Biotech, Oxford, UK) and serine protease inhibitor Pefabloc (Roche Diagnostics, Mannheim, Germany). For flow cytometry, AM were incubated overnight in sterile Teflon wells to prevent adherence (Savillex Corp., MN, USA). Supernatant was collected and analysed for the presence of soluble TNF.
Generation of cleavage substrate
A recombinant pGEX-2T vector containing a glutathione S-transferase (GST)-proTNF plasmid was donated by British Biotech [20]. The plasmid was transfected into Escherichia coli (TOP10F; Invitrogen, Paisley, UK) and protein expression was controlled by the Taq promoter, induced by isopropylthiogalactosidase. Following induction, GST-proTNF was expressed for 2·5 h before lysis by sonication. The 50 kDa fusion protein was purified from sonicates using affinity chromatography (GST purification module; Amersham Biosciences, Little Chalfont, UK) and resuspended at 1 mg/ml. The fusion protein was stored at −80°C until required.
Cleavage assay
Purified AM were adhered onto 24-well tissue culture plates, 1 × 106 per well (Gibco-Nunc, Paisley, UK). Protein trafficking was arrested by preincubation for 1 h with 5 µg/ml brefeldin A (Sigma) in the presence or absence of BB-3103 and/or Pefabloc (Sigma), before addition of 10 µg GST-proTNF substrate. After 2 h supernatant was removed and spun briefly at 1398 g to remove cell debris. Supernatants were concentrated by centrifugation at 15 339 g for 1·5 h, 4°C in microcon filters (YM3; Millipore, Watford, UK). Samples were run through a 15% polyacrylamide gel and transferred onto nitrocellulose membrane (Immobilon; Millipore) using a semi-dry transfer system (Bio-Rad, Hemel Hempstead, UK). Membranes were stored in 1 × Tris-buffered saline (TBS) prior to Western blotting.
Western blotting
Nitrocellulose membranes were incubated with blocking solution (5% Marvel/TBS/0·05% Tween) for 30 min before addition of biotinylated anti-GST antibody (Autogen-Bioclear, Calne, UK) dilution in blocking solution. After 1 h incubation, membranes were washed in TBS/0·05% Tween. A 1 : 2000 dilution of streptavidin–horseradish peroxidase (Autogen-Bioclear) in blocking solution was then added for a further 1 h incubation. Following 4 × 10 min washes, electrochemiluminescence detection reagent (GE Healthcare, Little Chalfont, UK) was added for 1 min. Membranes were then exposed immediately to X-ray film (Kodak X-Omat; Sigma) and developed after 1 min. Cleavage by TACE results in a 30 kDa band. The density of this band relative to the 50 kDa parent molecule was quantified using Geldoc 1000 with Quantity One software (Bio-Rad).
Soluble TNF enzyme-linked immunosorbent assay
The AM were cultured for 24 h in the presence or absence of 10 µg/ml LPS, 10 µM Pefabloc (Roche Diagnostics) and 10 µM BB-3103. Supernatants were harvested and stored at −70°C prior to analysis. TNF levels were determined using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Endogen, Perbio Science, Cramlington, UK).
Flow cytometry
Unfixed AM were analysed immediately after isolation and were incubated for 10 min in citric acid buffer, pH 4·0, to remove any receptor bound proteins. Cells were then washed once in phosphate-buffered saline (PBS(/0·5% bovine serum albumin/0·1% sodium azide (wash buffer) before addition of 10 µg human IgG to block non-specific binding. Phycoerythin (PE)-labelled anti-human TACE antibody, fluorescein isothiocyanate-labelled membrane-associated TNF antibody or the appropriate isotype control antibodies (0·5 µg antibody/sample; R&D Systems, Abingdon, UK) were added to the cells for 30 min at 4°C in the dark. PR-3 was detected by incubation with PR-3 Pelikine anti-neutrophil cytoplasm antibody (Research Diagnostics, Concord, MA, USA), followed by rabbit anti-mouse-rPE (Dako, Ely, UK). Cells were then washed twice in wash buffer and fixed in PBS/1% paraformaldehyde solution. Labelled cells (1 × 104) were acquired on a picsXL flow cytometer (Beckman Coulter, High Wycombe, UK) and analysed using Expo 32 software (Beckman-Coulter).
Statistical analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). The data were distributed normally. Comparisons between multiple groups were performed using one-way analysis of variance (anova), with Tukey's multiple comparison tests (mct) to compare individual group differences. Paired data were compared using Student's t-test. Relationships between parameters were assessed using Pearson's correlation. A P value of < 0·05 was regarded as significant.
Results
The TACE expression and function is increased on AM from subjects with interstitial lung disease
There was a significant difference in TACE surface expression between the three subject groups (P < 0·005, one-way anova), with the highest expression in sarcoidosis (Fig. 1a). This was significantly different from both healthy controls (P < 0·01 Tukey's mct) and UIP subjects (P < 0·05). Experiments were carried out to determine whether the increased levels of TACE in interstitial lung disease were reflected by increased cleavage. Cleavage of GST-proTNF was significantly different between the groups (P < 0·0005) and the highest level of cleavage was observed in UIP subjects (Fig. 1b). This was significantly higher than cleavage in sarcoidosis (P < 0·05) and healthy controls (P < 0·001).
Fig. 1.
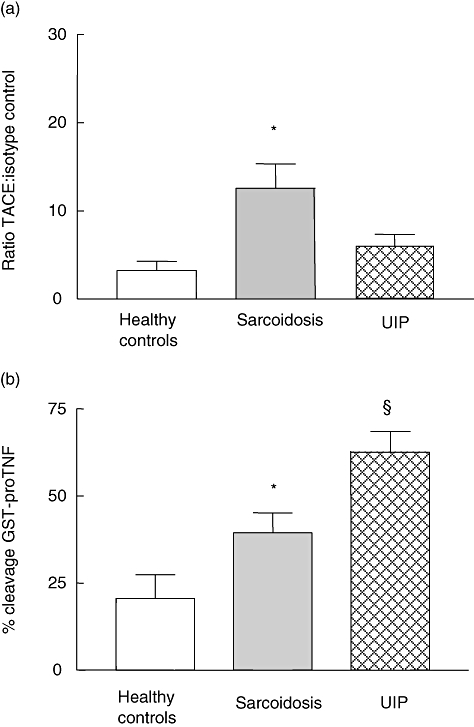
Freshly isolated alveolar macrophages (AM) were incubated with specific phycoerythrin (PE)-labelled anti-tumour necrosis factor (TNF)-α converting enzyme (TACE) antibody and acquired on a flow cytometer (a). Data are expressed relative to PE-labelled isotype control. *P < 0·01 sarcoidosis (n = 15) versus healthy controls (n = 15), P < 0·05 sarcoidosis versus usual interstitial pneumonia (UIP) (n = 20), Tukey's multiple comparison tests (mct). AM were incubated with glutathione S-transferase (GST)-proTNF substrate for 2 h. The cleaved substrate was then detected by Western blotting using a specific GST antibody. *P < 0·05 sarcoidosis (n = 15) versus normal controls (n = 15), §P < 0·001 UIP (n = 20) versus healthy controls, P < 0·05 UIP versus sarcoidosis Tukey's mct (b). (a,b) P = 0·005, one-way analysis of variance.
The relationship between TNF cleavage and TACE demonstrated in healthy subjects is lost in the presence of neutrophilic but not lymphocytic inflammation
We have shown previously that TACE expression on the surface of AM from healthy subjects is proportional to cleavage [17]. We have also found that this relationship is maintained in sarcoidosis (Fig. 2a). When we related cleavage to TACE expression we found that the association seen in healthy controls and in sarcoidosis subjects was lost in patients with UIP, with a median bronchoalvelar lavage fluid (BALF) neutrophil count 25% of cell population (Fig. 2b). This led us to consider the neutrophilic role. Pooling the data from all subjects, we found a significant positive correlation between percentage neutrophils in the BALF and cleavage efficiency (Fig. 2c) and a negative correlation with the inhibitory effect of BB-3103 (Fig. 2d). These data highlighted the possibility that an alternative, non-matrix metalloproteinase (MMP) enzyme, possibly from neutrophils, was responsible for cleaving the GST-proTNF substrate in UIP.
Fig. 2.
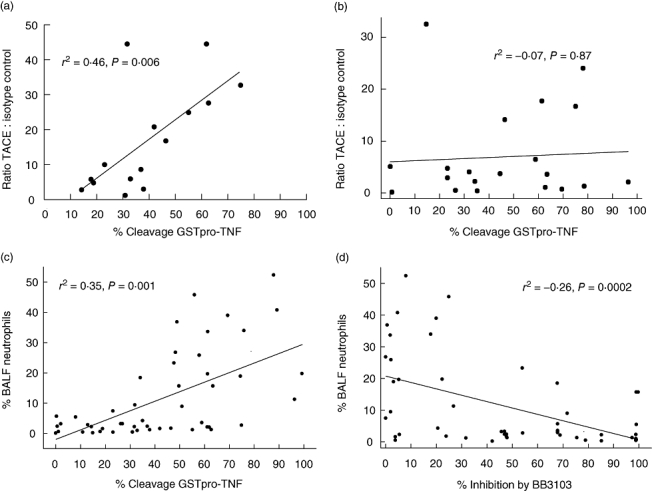
Sarcoidosis alveolar macrophages (AM) (n = 15) (a) and usual interstitial pneumonia (UIP) AM (n = 20) (b) were incubated with glutathione S-transferase (GST)-proTNF) substrate for 2 h. The cleaved substrate was then detected by Western blotting using a specific GST antibody. Cleavage of GST-proTNF by AM (n = 50) is associated with an increased number of neutrophils in the bronchoalveolar lavage fluid (BALF). P = 0·001, Pearson's correlation (c). The ability of BB-3103 to inhibit GST-proTNF cleavage is related inversely to the percentage number of alveolar neutrophils present in the BALF. AM (n = 50) were preincubated for 30 min with 10 µM BB-3103 before addition of GST-proTNF substrate for 2 h. P = 0·006, Pearson's correlation (d).
The TACE inhibition is not sufficient to block TNF cleavage in UIP subjects
There was a significant difference in the ability of BB-3103 to inhibit cleavage between the groups (P < 0·05) (Fig. 3a and b), with increased inhibition in sarcoidosis compared with UIP (P < 0·05). Pefabloc treatment alone had no effect on cleavage in the sarcoidosis group, but reduced cleavage significantly by UIP AM. Furthermore, Pefabloc in addition to BB-3103 increased the level of inhibition further in UIP.
Fig. 3.
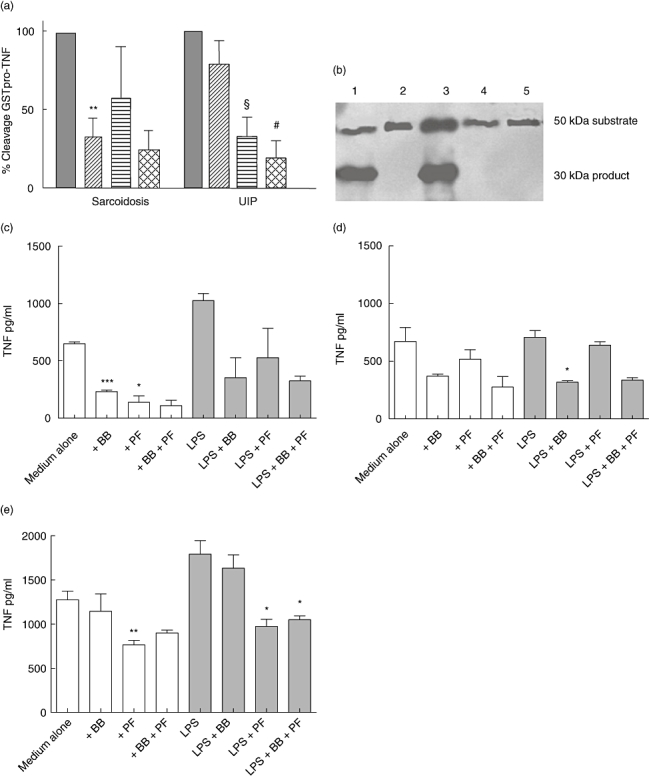
Alveolar macrophages (AM) were cultured in the presence or absence of 10 µM BB-3103 and 1 mM Pefabloc for 30 min before addition of glutathione S-transferase (GST)-proTNF) substrate for a further 2 h. Data are expressed as a percentage of medium alone. Grey bars, medium alone; diagonal bars, 10 µM BB-3103; horizontal bars, Pefabloc; hatched bars, Pefabloc + BB-3103. **P < 0·01 sarcoidosis BB-3103 versus sarcoidosis medium alone; §P < 0·01 usual interstitial pneumonia (UIP) Pefabloc versus UIP medium alone, #P < 0·01 UIP Pefabloc + BB-3103 versus UIP BB-3103 alone, Student's paired t-test (a). In vitro cleavage of GST-proTNF by AM from a subject with UIP. Lane 1, no inhibitors; lane 2, Pefabloc; lane 3, BB-3103; lane 4, BB-3103 + Pefabloc; lane 5, assay in the absence of AM (b). Soluble TNF release by AM from healthy subjects (c), subjects with sarcoidosis (d) and UIP subjects (e) treated with BB-3103 (BB) or Pefabloc (PF), *P < 0·05, **P = 0·01, ***P < 0·001 versus medium alone.
Soluble TNF present in healthy control AM supernatants was reduced in unstimulated AM by the presence of BB-3103 (P < 0·001). Similarly, Pefabloc reduced TNF release (P < 0·05). Pefabloc had no effect on LPS-induced TNF in healthy control AM; BB3103 reduced TNF release, although this failed to reach statistical significance (P = 0·09) (Fig. 3c). In sarcoidosis BB-3103 and Pefabloc had no significant effects on unstimulated TNF release. However, LPS-stimulated TNF was reduced significantly only by BB-3103 (P < 0·05) (Fig. 3d). By contrast, in UIP BB-3103 had no effect on TNF release by AM, but Pefabloc reduced both untreated and LPS-stimulated TNF significantly (P < 0·01 and P < 0·05 respectively) (Fig. 3e). Combined treatment of BB-3103 and Pefabloc had no additive or synergistic effects.
Surface expression of PR-3 on AM
Flow cytometry revealed increased staining for PR-3 on the surface of AM from the UIP group relative to healthy controls and sarcoidosis subjects (ratio to isotype control 3·17 ± 0·56 versus 1·68 ± 0·23 and 1·57 ± 0·3 respectively) (data not shown).
Discussion
It is well recognized that TACE cleavage to generate soluble TNF is a key processing event in host defence, and we and others have demonstrated that TACE is up-regulated in conditions such as sepsis [21], multiple sclerosis [22] and osteoarthritis [23]. We have also shown previously that TACE can be up-regulated in response to LPS and interferon-γ, and conversely down-regulated by the anti-inflammatory cytokine interleukin-10 [17].
We have studied two human diseases in which changes in TNF homeostasis have been implicated, namely UIP [24] and sarcoidosis [19], characterized by neutrophilic and lymphocytic predominance respectively. Surprisingly, this study has demonstrated increased levels of TACE on sarcoidosis AM, but not in UIP. This is despite the fact that AM in UIP are known to secrete relatively high levels of soluble TNF [25]. By contrast, we detected increased cleavage of GST-proTNF in UIP. The measurement of GST-proTNF provides a useful model for the investigation of cleavage activity on the surface of cells. We used GST detection in Western blots to prevent interference from AM-derived TNF-α. However, we cannot be certain that the product we are measuring is being cleaved in an identical fashion to native mTNF. For this reason we have looked at the effect of TACE inhibition on soluble TNF detection in AM supernatants by ELISA. We have also confirmed the specificity of the cleavage by Western blotting and demonstrated a 17·5 kDa band using TNF-α-specific antibodies. Our assumption is also supported by the observed significant correlation between TACE expression and GST-proTNF cleavage in normal subjects and the reduction of cleavage in the presence of the MMP inhibitor BB-3103, which has been demonstrated previously to inhibit TACE catalytic activity in vitro[26].
In UIP, where there are significant numbers of alveolar neutrophils, the association between TACE expression of GST-proTNF cleavage was lost. Furthermore, in contrast to both healthy subjects and patients with sarcoidosis, inhibition of TACE by BB-3103 in UIP subjects did not result in a reduction in GST-proTNF cleavage. Our previous work in healthy subjects suggested that PR-3 might be contributing to cleavage, especially in UIP. When we added the specific serine protease inhibitor Pefabloc to assess any inhibitory effect on cleavage we found that although the effect was minimal in healthy and sarcoidosis subjects, it had increased significance in UIP, both in terms of substrate cleavage and generation of soluble TNF in culture. Although there has been a great deal of interest in the ability of TACE to cleave TNF-α, it was actually demonstrated several years earlier that mTNF could also be cleaved by a serine protease [27], to be identified later as PR-3 [13], and the cleavage site is located at Arg-Val [28], which is immediately adjacent to the Ala-Val cleavage site of TACE, both yielding a 17·5 kDa product. PR-3 is produced by myelomonocytic cells, and the mature enzyme is stored in azurophilic granules of neutrophils. However, it is known to be present on the surface of monocytes [29], endothelial cells [30] and AM [17,31]. Whether this represents production of PR-3 or insertion of exogenous hydrophobic PR-3 into the cell membranes [32] is a matter of contention.
We have reported previously that de novo synthesis of PR-3 did not occur in AM from healthy subjects. Furthermore, AM from patients in the current study did not express mRNA transcripts for PR-3. However, we have demonstrated previously that both purified PR-3 and PR-3 present in activated neutrophil supernatants are able to bind to AM in vitro[17]. This finding is supported by another study which showed that PR-3 was able to bind to the outer plasma membrane of human umbilical vein endothelial cells [30]. We suggest that this is a possible mechanism to account for the increased ability of AM from UIP subjects to process TNF-α, which is supported by an increase in PR-3 on the surface AM in UIP relative to normal controls.
These data provide an explanation for the high levels of soluble TNF found in these lung conditions. While there are anti-TNF therapies available, these target both soluble and membrane forms of the TNF protein. As it is suggested that mTNF may have beneficial effects, such as inducing apoptosis and anergy of immune cells [33,34] which would limit the injurious response, it may be that this approach could be refined. Indeed, in a study by Fujita et al., TNF over-expressing mice were protected from bleomycin and transforming growth factor (TGF)-β-induced lung injury [35]. In addition, local expression of TNF may be important for apoptosis of lung fibroblasts [36] and it has been postulated that fibrosis in UIP may actually be a consequence of low levels of TNF and other proinflammatory cytokines around fibroblastic foci [37]. Interestingly, there have been reported cases of pulmonary fibrosis in patients receiving anti-TNF therapies for rheumatoid arthritis [38,39]. Pharmacological inhibition of the TNF cleavage site has been proposed as a way of preserving functional mTNF while limiting the damaging effects of soluble TNF. These compounds are currently under evaluation [40], but a recent randomized clinical trial has found no efficacy in rheumatoid arthritis [41] and idiopathic pulmonary fibrosis [5]. Our study demonstrates that TACE is not the only means of generating soluble TNF, and it is possible that in diseases where this form of TNF is mediating pathology, TACE inhibition alone may by insufficient.
Acknowledgments
The authors would like to thank Professor Caroline Savage, University of Birmingham, UK for advice regarding the PR-3 investigations. The authors would also like to acknowledge Action Medical Research for supporting Dr Lynne Armstrong and British Biotech for supplying the GST-proTNF vector and the BB-3103 inhibitor.
Disclosure
None.
References
- 1.Maini R, Clair EW, St, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study group. Lancet. 1999;354:1932–9. doi: 10.1016/s0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- 2.Sandborn WJ, Hanauer SB, Katz S, et al. Etanercept for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121:1088–94. doi: 10.1053/gast.2001.28674. [DOI] [PubMed] [Google Scholar]
- 3.Berry MA, Hargadon B, Shelley M, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- 4.Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174:795–802. doi: 10.1164/rccm.200603-402OC. [DOI] [PubMed] [Google Scholar]
- 5.Raghu G, Brown KK, Costabel U, et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am J Respir Crit Care Med. 2008;178:948–55. doi: 10.1164/rccm.200709-1446OC. [DOI] [PubMed] [Google Scholar]
- 6.Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:926–34. doi: 10.1164/rccm.200607-995OC. [DOI] [PubMed] [Google Scholar]
- 7.Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–85. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- 8.Georgopoulos S, Plows D, Kollias G. Transmembrane TNF is sufficient to induce localized tissue toxicity and chronic inflammatory arthritis in transgenic mice. J Inflamm. 1996;46:86–97. [PubMed] [Google Scholar]
- 9.Armstrong L, Thickett DR, Christie SJ, Kendall H, Millar AB. Increased expression of functionally active membrane-associated tumor necrosis factor in acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2000;22:68–74. doi: 10.1165/ajrcmb.22.1.3728. [DOI] [PubMed] [Google Scholar]
- 10.Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004;15:353–66. doi: 10.1016/j.cytogfr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Eissner G, Kirchner S, Lindner H, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages 1. J Immunol. 2000;164:6193–8. doi: 10.4049/jimmunol.164.12.6193. [DOI] [PubMed] [Google Scholar]
- 12.Xin L, Wang J, Zhang H, et al. Dual regulation of soluble tumor necrosis factor-alpha induced activation of human monocytic cells via modulating transmembrane TNF-alpha-mediated ‘reverse signaling’. Int J Mol Med. 2006;18:885–92. [PubMed] [Google Scholar]
- 13.Coeshott C, Ohnemus C, Pilyavskaya A, et al. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA. 1999;96:6261–6. doi: 10.1073/pnas.96.11.6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Geld YM, Limburg PC, Kallenberg CG. Proteinase 3, Wegener's autoantigen: from gene to antigen. J Leukoc Biol. 2001;69:177–90. [PubMed] [Google Scholar]
- 15.Kao RC, Wehner NG, Skubitz KM, Gray BH, Hoidal JR. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J Clin Invest. 1988;82:1963–73. doi: 10.1172/JCI113816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Just J, Moog-Lutz C, Houzel-Charavel A, et al. Proteinase 3 mRNA expression is induced in monocytes but not in neutrophils of patients with cystic fibrosis. FEBS Lett. 1999;457:437–40. doi: 10.1016/s0014-5793(99)01098-4. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong L, Godinho SI, Uppington KM, Whittington HA, Millar AB. Contribution of TNF-alpha converting enzyme and proteinase-3 to TNF-alpha processing in human alveolar macrophages. Am J Respir Cell Mol Biol. 2006;34:219–25. doi: 10.1165/rcmb.2005-0087OC. [DOI] [PubMed] [Google Scholar]
- 18.Demedts M, Costabel U. ATS/ERS international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Eur Respir J. 2002;19:794–6. doi: 10.1183/09031936.02.00492002. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong L, Foley NM, Millar AB. Inter-relationship between tumour necrosis factor-alpha (TNF-alpha) and TNF soluble receptors in pulmonary sarcoidosis. Thorax. 1999;54:524–30. doi: 10.1136/thx.54.6.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gearing AJ, Beckett P, Christodoulou M, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–7. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 21.Robertshaw HJ, Brennan FM. Release of tumour necrosis factor alpha (TNFalpha) by TNFalpha cleaving enzyme (TACE) in response to septic stimuli in vitro. Br J Anaesth. 2005;94:222–8. doi: 10.1093/bja/aei021. [DOI] [PubMed] [Google Scholar]
- 22.Comabella M, Romera C, Camina M, et al. TNF-alpha converting enzyme (TACE) protein expression in different clinical subtypes of multiple sclerosis. J Neurol. 2006;253:701–6. doi: 10.1007/s00415-006-0090-6. [DOI] [PubMed] [Google Scholar]
- 23.Patel IR, Attur MG, Patel RN, et al. F-alpha convertase enzyme from human arthritis-affected cartilage: isolation of cDNA by differential display, expression of the active enzyme, and regulation of TNF-alpha. J Immunol. 1998;160:4570–9. [PubMed] [Google Scholar]
- 24.Freeburn RW, Armstrong L, Millar AB. Cultured alveolar macrophages from patients with idiopathic pulmonary fibrosis (IPF) show dysregulation of lipopolysaccharide-induced tumor necrosis factor-alpha (TNF-alpha) and interleukin-10 (IL-10) inductions. Eur Cytokine Netw. 2005;16:5–16. [PubMed] [Google Scholar]
- 25.Zhang Y, Lee TC, Guillemin B, Yu MC, Rom WN. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J Immunol. 1993;150:4188–96. [PubMed] [Google Scholar]
- 26.Romera C, Hurtado O, Botella SH, et al. In vitro ischemic tolerance involves upregulation of glutamate transport partly mediated by the TACE/ADAM17-tumor necrosis factor-alpha pathway. J Neurosci. 2004;24:1350–7. doi: 10.1523/JNEUROSCI.1596-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niehorster M, Tiegs G, Schade UF, Wendel A. In vivo evidence for protease-catalysed mechanism providing bioactive tumor necrosis factor alpha. Biochem Pharmacol. 1990;40:1601–3. doi: 10.1016/0006-2952(90)90461-s. [DOI] [PubMed] [Google Scholar]
- 28.Bank U, Ansorge S. More than destructive: neutrophil-derived serine proteases in cytokine bioactivity control. J Leukoc Biol. 2001;69:197–206. [PubMed] [Google Scholar]
- 29.Braun MG, Csernok E, Gross WL, Muller-Hermelink HK. Proteinase 3, the target antigen of anticytoplasmic antibodies circulating in Wegener's granulomatosis. Immunolocalization in normal and pathologic tissues. Am J Pathol. 1991;139:831–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Taekema-Roelvink ME, Van Kooten C, Heemskerk E, Schroeijers W, Daha MR. Proteinase 3 interacts with a 111-kD membrane molecule of human umbilical vein endothelial cells. J Am Soc Nephrol. 2000;11:640–8. doi: 10.1681/ASN.V114640. [DOI] [PubMed] [Google Scholar]
- 31.Brockmann H, Schwarting A, Kriegsmann J, et al. Proteinase-3 as the major autoantigen of c-ANCA is strongly expressed in lung tissue of patients with Wegener's granulomatosis. Arthritis Res. 2002;4:220–5. doi: 10.1186/ar410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldmann WH, Niles JL, Arnaout MA. Interaction of purified human proteinase 3 (PR3) with reconstituted lipid bilayers. Eur J Biochem. 1999;261:155–62. doi: 10.1046/j.1432-1327.1999.00259.x. [DOI] [PubMed] [Google Scholar]
- 33.Eissner G, Kohlhuber F, Grell M, et al. Critical involvement of transmembrane tumor necrosis factor-alpha in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin 28. Blood. 1995;86:4184–93. [PubMed] [Google Scholar]
- 34.Vudattu NK, Holler E, Ewing P, et al. Reverse signalling of membrane-integrated tumour necrosis factor differentially regulates alloresponses of CD4+ and CD8+ T cells against human microvascular endothelial cells. Immunology. 2005;115:536–43. doi: 10.1111/j.1365-2567.2005.02190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujita M, Shannon JM, Morikawa O, Gauldie J, Hara N, Mason RJ. Overexpression of tumor necrosis factor-alpha diminishes pulmonary fibrosis induced by bleomycin or transforming growth factor-beta. Am J Respir Cell Mol Biol. 2003;29:669–76. doi: 10.1165/rcmb.2002-0046OC. [DOI] [PubMed] [Google Scholar]
- 36.Frankel SK, Cosgrove GP, Cha SI, et al. F-alpha sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis. Am J Respir Cell Mol Biol. 2006;34:293–304. doi: 10.1165/rcmb.2005-0155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selman M, Thannickal VJ, Pardo A, Zisman DA, Martinez FJ, Lynch JP., III Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs. 2004;64:405–30. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 38.Cairns AP, Taggart AJ. Anti-tumour necrosis factor therapy for severe inflammatory arthritis: two years of experience in Northern Ireland. Ulster Med J. 2002;71:101–5. [PMC free article] [PubMed] [Google Scholar]
- 39.Ostor AJ, Chilvers ER, Somerville MF, et al. Pulmonary complications of infliximab therapy in patients with rheumatoid arthritis. J Rheumatol. 2006;33:622–8. [PubMed] [Google Scholar]
- 40.Le GT, Inhibitors G. Inhibitors of TACE and caspase-1 as anti-inflammatory drugs. Curr Med Chem. 2005;12:2963–77. doi: 10.2174/092986705774462851. [DOI] [PubMed] [Google Scholar]
- 41.Thabet MM, Huizinga TW. Drug evaluation: apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr Opin Investig Drugs. 2006;7:1014–19. [PubMed] [Google Scholar]