

Abstract
Cytochrome P450 2J subfamily (CYP2J) enzymes expressed in mouse hepatocellular carcinoma (HCC) cells were identified as an antigen recognized by specific CD4+ T cells and the structure of its T cell epitope was determined by proteomics-based exploration. The major histocompatibility complex (MHC) class II binding peptides were isolated from I-Ak/peptide complex of dendritic cells (DCs) loaded or unloaded with MIH-2 mouse HCC cells. MHC class II-binding peptides found in MIH-2-loaded DCs but not in unloaded DCs were determined by tandem mass spectrometric analysis. The peptide, consisting of amino acid 276–290 (DFIDAFLKEMTKYPE) of mouse CYP2J enzymes, was identified as an antigenic peptide presented in the context of MHC class II. Preventive treatment of mice with CYP2J peptide stimulated interferon (IFN)-γ production of splenocytes and suppressed the growth of implanted CYP2J-positive MIH-2 cells but not CYP2J-negative murine bladder tumour cells. However, continuous treatment of MIH-2-bearing mice with CYP2J peptide significantly suppressed IFN-γ production of splenocytes and accelerated the growth of implanted MIH-2 tumours in vivo. Increased frequencies of CD4+forkhead box P3 regulatory T cells and CD11b+Gr-1+ myeloid suppressor cells were observed in splenocytes from the continuously immunized mice. These results indicate that antigenecity of CYP2J isoforms expressed in HCC cells activate host anti-tumour immunity at an initial stage of HCC, but suppress host anti-tumour immunity with excessive antigenic stimulation at an advanced stage.
Keywords: dendritic cell, immune regulation, liver cancer, MHC class II, Treg
Introduction
Host anti-tumour immunity could play an important role in the prevention of tumour development, and a tumour-associated antigen (TAA)-specific T cell response is one of the most important mechanisms for tumour rejection [1]. Crosti et al. have reported spontaneous reactivity to carcinoembryonic antigen (CEA) epitopes of peripheral blood CD4+ T cells from patients with early CEA-positive lung cancer [2]. However, the immune system exhibits hyporesponsiveness or unresponsiveness to TAAs when tumours become large, resulting in further tumour development without immunological anti-tumour activity. Analysis of tumour-infiltrating lymphocytes has revealed antigen-specific hyporesponsiveness consistent with a form of T cell anergy [3]. Acquired tolerance to TAAs has been observed even when initial priming of T cells to TAAs was successful [4]. This immunosuppression might be caused by vascular endothelial growth factor (VEGF), interleukin (IL)-10, transforming growth factor (TGF)-beta or prostaglandin E2 produced by tumour cells or tumour-associated immune cells, leading to generation of dysfunctional APCs inducing tolerance for T cell immunity [5,6]. Furthermore, tumour cells form a unique microenvironment to prevent immune attack [7].
Antigen-specific anti-tumour immunity might be inhibited by excessive antigenic stimulation with TAAs by a large tumour burden. In the case of CD8+ T cells, low antigen levels lead to T cell deletion and high antigen levels lead to T cell tolerance [8]. Consequently, in advanced cancer, over-stimulation with immunogenic TAAs on tumour cells might paralyse anti-tumour immunity and favour tumour development. In order to prevent such immune paralysis induced by advanced cancer, it is necessary to identify TAAs generating excessive antigenic stimulation on the host immune system.
CD4+ T cell functions play a pivotal role in the induction of anti-tumour immunity, and T cell activation by stimulation with helper T cell epitopes is probably important for effective anti-cancer immune responses [9–11]. Meanwhile, it has been known that excessive antigenic stimulation with helper epitope peptide induces immune tolerance [12]. Although many TAAs recognized by specific CD8+ cytotoxic T lymphocytes (CTLs) have been reported, fewer helper epitopes of TAAs have been identified than have CTL epitopes [2,13].
Recent progress in mass spectrometric analysis has enabled efficient TAA searches. Known, immunoprecipitated TAAs can be identified clearly with a proteometric approach [14]. Human leucocyte antigen class II presented peptides derived from tissue specimens of primary renal cell carcinoma have been analysed with mass spectrometry, and T helper epitopes from TAAs have been detected [15]. Dendritic cells (DCs), potent APCs, have been analysed with mass spectrometry to identify functional DC-associated molecules. Calreticulin was found with tandem mass spectrometric analysis to be a receptor of NY-ESO-1, a representative TAA, on DCs [16]. Röhn et al. have used nanoelectrospray ionization tandem mass spectrometry to examine DCs upon exposure to necrotic tumour cells and have identified melanotransferrin as a major histocompatibility complex (MHC) class II-restricted tumour antigen [17]. Mass spectrometric analysis could be applied to the detection of animal and human TAAs that would be presented by tumour-cell-captured APCs to T cells.
In this paper, we describe the identification of an immunostimulatory antigen expressed in mouse hepatocellular carcinoma (HCC) cells recognized by CD4+ T cells which, in turn, suppress host anti-tumour immune activity through excessive antigenic stimulation. The antigen was identified with tandem mass analysis of MHC class II binding peptides isolated from DCs loaded with HCC cells.
Materials and methods
Mice, cell lines and reagents
Eight-week-old male C3H/HeNCrj mice were purchased from Charles River Japan (Yokohama, Japan). All animals received humane care in accordance with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals.
A murine HCC cell line, MIH-2, was established in our laboratory from a liver tumour of a 15-month-old C3H/HeNCrj mouse [18]. Another mouse HCC cell line, Hepa 1–6, was purchased from the American Type Culture Collection (Manassas, VA, USA).
Monoclonal antibodies (mAbs) against murine CD11b and Gr-1 were purchased from BD Biosciences (San Jose, CA, USA). A kit for detecting CD4+ forkhead box P3 transcription factor (FoxP3+) regulatory T cells (Tregs) was purchased from eBioscience (San Diego, CA, USA). A mAb against I-Ak for use in affinity chromatography was purchased from BD Biosciences. Rabbit polyclonal anti-CYP2J2pep3 antibody, which recognizes all known mouse cytochrome P450 2J subfamily (CYP2J) isoforms [19], and an anti-CYP2J6pep1 antibody, which is specific for mouse CYP2J6 [20], were kindly provided by Professor Darryl C. Zeldin (National Institute of Environmental Health Sciences, National Institutes of Health, Bethesda, MD, USA).
Preparation of DCs loaded with MIH-2 cells
The preparation of DCs loaded with MIH-2 cells was performed as described previously [18,21]. Briefly, DCs were prepared from bone marrow cells of 8–10-week-old male C3H/HeNCrj mice. Then 2 × 107 DCs were mixed with 107 irradiated MIH-2 cells and centrifuged. After the supernatant had been discarded, 0·5 ml of 50% polyethylene glycol (PEG) (PEG 1450; Sigma-Aldrich, St Louis, MO, USA) was added to the cell pellet, which was left for 1 min at room temperature, after which the PEG solution was diluted gradually. After removal of PEG by centrifugation and overnight incubation, non-adherent or loosely attached cells were collected with gentle pipetting as DCs loaded with MIH-2 cells (DC/MIH-2) and used for isolation of peptides fixed with I-Ak molecules.
Affinity column chromatography
Cell pellets of PEG-treated cells were suspended in TNE buffer (10 mM Tris, 1% Nonidet P-40, 0·15 M NaCl, 1 mM ethylenediamine tetraacetic acid, pH 8·0) and sonicated for 30 s. The supernatant, obtained by centrifugation at 10 000 g for 15 min, was applied to a column containing anti-I-Ak antibody-coated Formyl-Cellulofine beads (Seikagaku Corporation, Tokyo, Japan) equilibrated with phosphate-buffered saline (PBS), after which the column was washed with PBS until A280 absorbance disappeared. The column was prewashed with 0·5 M NaCl and then washed with elution buffer (0·2 M glycine-HCl, pH 2·5). Each 0·5 ml of the eluted samples was collected and examined for A280 absorbance.
Preparation of peptide samples and tandem mass spectrometric analysis
The peptide sample fraction was applied to solid-phase extraction for concentrating peptides and removing detergents. An Octadecyl-Silica SepPack column (Waters Corp., Milford, MA, USA) was activated with 4 ml of methanol and equilibrated with 2 ml of water. After the sample fraction was applied, the SepPack column was washed with water (4 ml) and eluted with an 80% (V/V) acetonitrile aqueous solution containing 0·1% (V/V) formic acid. The organic solvent fraction was frozen and lyophilized. The resulting cloudy precipitate was resolved with 80% (V/V) methanol containing 0·1% (V/V) formic acid. After centrifugation at 16 000 g for 10 min, the 2-µl aliquot was placed in a microcapillary of the nanospray apparatus of the tandem mass spectrometry system.
A Q-Trap tandem mass spectrometer (Applied Biosystems, Foster City, CA, USA) equipped with nanospray systems was used. The BioAnalyst (version 1·3) data-analysis software system (Applied Biosystems) was used for assaying optimization and analysis with tandem mass spectrographs.
Peak reconstruction of mass spectra was recorded under m/z 1500, and the chosen peptide peaks were analysed with tandem mass spectrometric analysis after passage through a nitrogen gas collision chamber. To determine amino acid sequences in tandem mass spectra, the peaks were analysed with automated sequence Tag determination and a database search performed with the data-analysis software.
Reverse transcriptase–polymerase chain reaction
Total RNA was extracted using Isogen (Nippongene, Toyama, Japan), according to the manufacturer's instructions. RNA was reverse-transcribed with Moloney murine leukaemia virus RT (Invitrogen Corp., Carlsbad, CA, USA) and complementary DNA was subjected to the polymerase chain reaction (PCR) with Taq polymerase (Takara Biochemical, Tokyo, Japan) in a thermal cycler. Primers were designed as follows: mouse CYP2J6, forward GTATTTGGAGACAACAATGATTAGTCAGC, reverse TATCAGCAAGTCTTGCTGCCCTCTTC [expected size of the product was 407 base pairs (bp)]; mouse CYP2J9, forward TGTTTGGAGTCATCAATGATGTGC, reverse CGGTCTGCCAGATTCGGCTGTCTTGC (410 bp); and mouse CYP2J11, forward TGTTCTGAGGCATCAATGATGTCAGT, reverse TGGTCATCTAAGGTTGGGTGTCTCCCA (410 bp).
Immunoblot analysis
Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). After blocking in Tris-buffered saline with Tween-20 [25 mM Tris-HCl (pH 8·0), 150 mM Nacl, 0·1% Tween 20] with 10% Difco skimmed milk (BD Biosciences Diagnostic Systems, Sparks, MA, USA), the membranes were incubated with anti-CYP2J2pep3 or anti-CYP2J6pep1 antibodies. Each membrane was then probed for 90 min with peroxidase-conjugated anti-rabbit Ig (Bio-Rad Laboratories) and analysed with an enhanced chemiluminescence detection system (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK).
Vaccination of mice with CYP2J peptide
The CYP2J peptide (NH2-DFIDAFLKEMTKYPE-COOH) and control ovalbumin (OVA) peptide (NH2-GGLEPINFQTAADQA-COOH) were synthesized by Funakoshi Co. Ltd (Tokyo, Japan). The purity of the each peptide was 95·81% and 96·05% respectively. OVA was used as a negative control for MHC class II presented immunogenic antigen. CYP2J peptide or OVA peptide (20 µg/20 µl H2O) was mixed with 0·2 ml of H2O and 0·2 ml of complete Freund's adjuvant (Difco Laboratories, Detroit, MI, USA), agitated vigorously to generate emulsion, and injected subcutaneously once a week.
For preventive treatment of mice with CYP2J peptide, naive mice were immunized twice with the peptide and tumour cells were implanted 1 week after immunization. For continuous treatment of MIH-2-bearing mice with the peptide, immunization started 1 week after the implantation of tumour cells.
Production of cytokines by splenocytes
Splenocytes from untreated control or peptide-vaccinated mice were collected and cultured in 24-well culture plates (5 × 106/well) for 72 h. In some experiments, CYP or OVA peptide (20 µg/ml) was added to the culture to stimulate T cells in vitro. Supernatants were collected and examined for interferon (IFN)-γ, IL-4, IL-10 and TGF-beta using an enzyme-linked immunosorbent assay kit (Biosource International).
Results
Identification of MIH-2-derived MHC class II binding peptide from DCs loaded with MIH-2 cells by mass spectrometric analysis
Tandem mass spectrometry was performed to find peptide peaks of 12–15 amino acids, suitable for binding to MHC class II molecules, from MIH-2-loaded DCs. However, we found no objective ion peaks of 12–15 amino acid peptides. It is probable that isolated MHC class II binding peptides were digested by endogenous peptidases. We then searched for smaller peptide peaks and some ion peaks of peptides were found with total ion chromatography of MIH-2-loaded DCs which were not found in control DCs (Fig. 1). A representative ion peak, indicated by an arrow in Fig. 1 (m/z 821·8), was analysed further using the tandem mass system, and its structural amino acid sequence was determined to be EMTK.
Fig. 1.
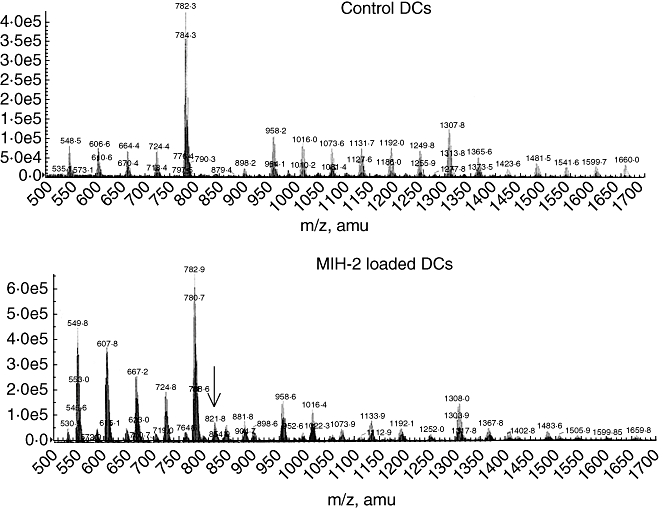
Total ion chromatograph of I-Ak binding peptides derived from control dendritic cells (DCs) (upper) or MIH-2-loaded DCs (lower). Control DCs, treated with 50% polyethylene glycol (PEG) alone, or MIH-2 loaded-DCs, generated by treatment of mixture of DCs and MIH-2 cells with 50% PEG, as described in Materials and methods, were treated with detergent, and I-Ak/peptide complexes were obtained with anti-I-Ak affinity column chromatography. Each binding peptide was analysed with liquid chromatography/mass spectrometry. A representative peptide ion peak that was found only in peptides from MIH-2-loaded DCs and not those from control DCs, indicated by an arrow (821·8 m/z), was analysed further with liquid chromatography/tandem mass spectrometry.
Database analysis revealed that the EMTK polypeptide corresponded to amino acids 284–287 of several enzymes of mouse cytochrome P450 2J (CYP2J) (Fig. 2). Two other candidate molecules, mouse nucleolin (amino acids 285–288) and queuine tRNA-ribosyltransferase (amino acids 200–203), which contain EMTK in their amino acid sequences, were also identified. The I-Ak binding motif structure (DFIDAFLK) was found in amino acids 276–283 of CYP2Js, adjacent to EMTK (Fig. 2), suggesting that a peptide consisting of 15 amino acids, including the I-Ak binding motif and EMTK (DFIDAFLKEMTKYPE) of CYP2Js, would be presented by MIH-2–loaded DCs to CD4+T cells. Mouse nucleolin did not possess a I-Ak binding motif adjacent to EMTK, and mouse queuine tRNA-ribosyltransferase had lower possibility score, indicating that CYP2J is the most possible candidate molecule.
Fig. 2.
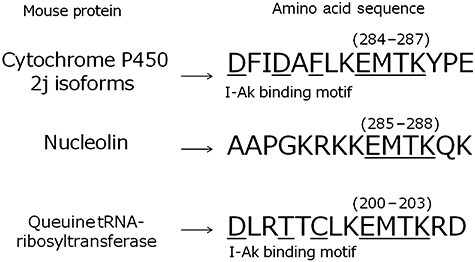
Amino acid sequence of mouse CYP2J6 and a possible epitope recognized by specific CD4+ T cells in the context of major histocompatibility complex (MHC) class II. EMTK (284E–287K) was detected from the peptides binding to I-Ak of MIH-2-loaded dendritic cells (DCs) with tandem mass analysis (TMA). D276–K283, adjacent to EMTK on the N-terminal side, possesses the I-Ak binding motif. A peptide consisting of 15 amino acids containing the I-Ak binding motif and EMTK, as indicated by an underline, was synthesized and examined for further biological activity.
Expression of CYP2Js in HCC cells and normal liver cells
Messenger RNA (mRNA) encoding CYP2J6 was found with reverse transcriptase (RT)–PCR to be expressed in MIH-2 HCC cells as well as Hepa 1–6 HCC cells, B16 melanoma cells and LY-R lymphoma cells (Fig. 3). It was also expressed in C26 cells at a very low level. The enzymes CYP2J6, CYP2J9 and CYP2J11 each contain EMTK, but CYP2J2, CYP2J4, CYP2J5 and CYP2J13 do not [19,20,22]. The mRNA encoding CYP2J9 was expressed in MIH-2 cells at a very low level, and the mRNA encoding CYP2J11 was not expressed in either normal liver or MIH-2 cells (Fig. 3). CYP2J protein (51 kDa) was recognized with an anti-CYP2J2pep3 antibody in MIH-2 cells but not in T3, C26 or MC38 cells on Western blotting analysis (Fig. 3).
Fig. 3.
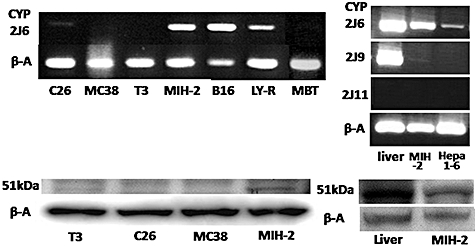
Expression of CYP2J mRNA and protein in various tumour cell lines and normal liver tissue. Expression of mRNA encoding CYP2J6, 2J9 and 2J11 was examined with reverse transcription–polymerase chain reaction, as described in Materials and methods (upper). Expression of CYP2J protein (51 kDa) recognized by anti-CYP2J2pep3 antibody was examined with immunoblot analysis (lower). C26 and MC38, mouse colon cancer; T3, mouse small intestine tumour; MIH-2 and Hepa 1–6, mouse hepatocellular carcinoma; B16, mouse melanoma; LY-R, mouse lymphoma; β-A, beta-actin.
Preventive treatment of naive mice with CYP2J peptide stimulated IFN-γ production of splenocytes and suppressed the growth of implanted MIH-2 cells in vivo
Splenocytes from mice immunized twice with CYP2J peptide (DFIDAFLKEMTKYPE) showed significantly higher IFN-γ production in vitro than did splenocytes from untreated mice or mice immunized with OVA peptide (Fig. 4a). Furthermore, production of IFN-γ by splenocytes from CYP2J peptide-immunized mice was enhanced when CYP2J peptide, but not OVA peptide, was added to the culture (Fig. 4a). In contrast, production of IL-4 by splenocytes was not enhanced (data not shown).
Fig. 4.
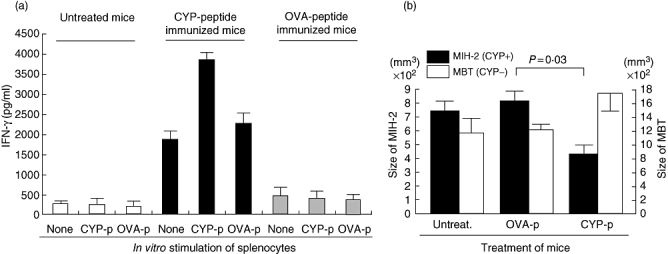
(a) CYP2J peptide was immunogenic in naive C3H/HeN mice. Male C3H/HeN mice received subcutaneous injections of CYP2J peptide (20 µg/mouse) with adjuvant on days 1 and 7. Splenocytes were collected on day 14 and cultured for 72 h with or without CYP peptide (20 µg/ml) or control ovalbumin (OVA) peptide (20 µg/ml). Supernatants were examined for interferon (IFN)-γ with enzyme-linked immunosorbent assay. (b) Immunization with CYP2J peptide suppressed the growth of implanted MIH-2 cells in vivo. Male C3H/HeN mice received subcutaneous injections of CYP2J peptide (20 µg/mouse) or OVA peptide (20 µg/mouse) with adjuvant on days 1 and 7. On day 14, the immunized mice were implanted subcutaneously with MIH-2 cells (106/mouse) or murine bladder tumour (MBT) cells (106/mouse), and the tumour size (long diameter × short diameter) was measured 4 weeks after the tumour cell implantation. CYP2J was detectable in MIH-2 cells but not in MBT cells by immunoblot analysis.
When MIH-2 cells (CYP2J+) were implanted into mice immunized with CYP2J peptide, tumour growth was suppressed significantly (Fig. 4b). However, growth of implanted murine bladder tumour cell (CYP2J−, as shown in Fig. 3) was not suppressed by immunization with CYP2J peptide (Fig. 4b).
Repeated immunization of MIH-2-bearing mice with CYP2J peptide suppressed IFN-γ production and accelerated MIH-2 tumour growth in vivo
The immunostimulatory capacity of CYP2J peptide was studied through repeated immunization of naive mice. Production of IFN-γ by splenocytes increased in mice immunized one to six times (Fig. 5). In contrast, IFN-γ production was suppressed when mice were immunized seven and eight times (Fig. 5). These results suggest that excessive immunization (more than six times) with CYP2J peptide can induce unresponsiveness or anergy to CYP2Js.
Fig. 5.
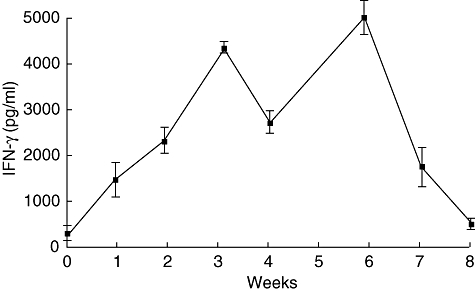
Enhancement of interferon (IFN)-γ production of splenocytes by moderate immunization with CYP2J peptide but suppression by excessive immunization. CYP2J peptide (20 µg/mouse) with adjuvant was injected subcutaneously once a week. Splenocytes were obtained from mice 3 days after each vaccination. Splenocytes (5 × 106/ml) were incubated for 72 h and supernatants were examined for IFN-γ by enzyme-linked immunosorbent assay (n = 3). Splenocytes from control mice injected with phosphate-buffered saline were collected at each time-point and their IFN-γ production was 200–350 pg/ml (n = 3).
Mice bearing MIH-2 tumours and immunized excessively with CYP2J peptide exhibited a significant enhancement of tumour growth compared with mice treated with PBS (Fig. 6a). However, immunization with control OVA peptide did not enhance tumour growth. These results indicate that excessive immunization of MIH-2-bearing mice with CYP2J peptide suppresses host anti-tumour immunity against MIH-2 cells.
Fig. 6.
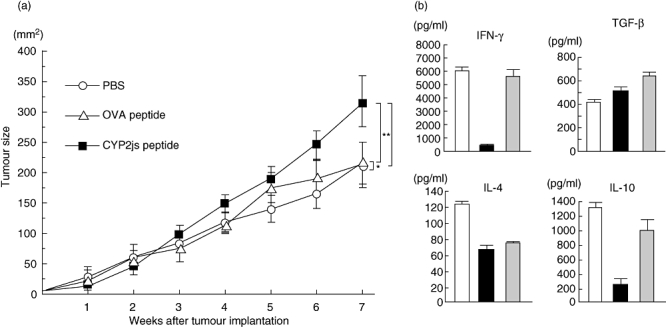
(a) Repeated immunization of MIH-2-bearing mice with CYP2J peptide accelerated MIH-2 tumour growth in vivo. MIH-2 cells (2 × 106/mouse) were implanted into female C3H/HeN mice, which were immunized with CYP2J peptide or control ovalbumin (OVA) peptide (20 µg/mouse) with adjuvant once a week after MIH-2 cell implantation. Tumour size (long diameter × short diameter) was measured every week. Open circle, mice treated with phosphate-buffered saline (PBS); open triangle, mice immunized with OVA peptide; closed square, mice immunized with CYP2J peptide. *P = 0·8692, **P = 0·0261, ***P = 0·0075 determined with repeated-measures analysis of variance (n = 6). This is a representative result of three replicated experiments. (b) Cytokine production of splenocytes from MIH-2-bearing mice immunized excessively (seven times) with CYP2J peptide. Splenocytes were collected and cultured at 5 × 106 cells/ml for 72 h. Supernatants were examined for interferon-γ, interleukin (IL-4), IL-10 and transforming growth factor-β. Open columns, MIH-2-bearing mice treated with PBS; closed columns, MIH-2 bearing mice immunized with CYP2J peptide; shaded columns, MIH-2-bearing mice immunized with OVA peptide.
Splenocytes from MIH-2-bearing mice, even 7 weeks after MIH-2 cell implantation, showed considerable IFN-γ production in vitro by immune stimulation with implanted MIH-2 cells (Fig. 6b). Production of IFN-γ by splenocytes from MIH-2-bearing mice was also suppressed markedly by excessive immunization with CYP2J peptide (Fig. 6b).
Production of IL-4, IL-10 and TGF-beta by splenocytes from mice immunized excessively with CYP2J was examined. Production of IL-10 was suppressed by excessive immunization with CYP2J peptide and IL-4 production was suppressed by CYP2J and OVA peptide, but TGF-beta production was not affected (Fig. 6b). Therefore, suppression of IFN-γ production was not induced by the shift from the T helper type 1 (Th1) to the Th2 immune environment or by enhanced production of immunosuppressive IL-10 or TGF-beta.
Increased frequency of immunosuppressive cells in splenocytes from mice immunized excessively with CYP2J peptide
Splenocytes from mice immunized excessively with CYP2J peptide were examined for the frequency of immunosuppressive cells: CD4+ FoxP3+ Tregs. Splenocytes from MIH-2-bearing mice showed an almost twofold increase in the frequency of CD4+FoxP3+ Tregs (Fig. 7a). Mice that had been immunized excessively with CYP2J peptide showed increased frequency of Tregs in splenocytes even without MIH-2 cell implantation. However, excessive immunization of MIH-2-bearing mice with CYP2J peptide did not increase the Treg frequency further.
Fig. 7.
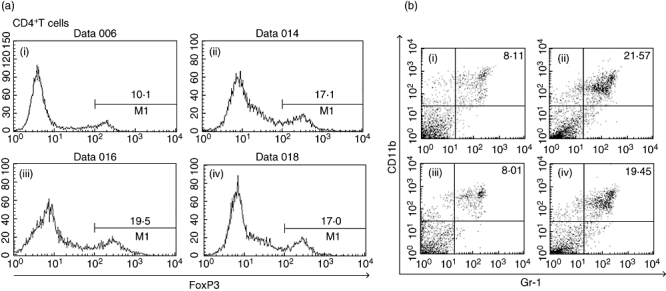
(a) Frequency of forkhead box P3 (FoxP3+) cells in splenic CD4+ T cells from MIH-2-bearing mice or non-tumour-bearing mice immunized excessively with CYP2J peptide. Splenocytes were collected from each group of mice, and CD4+ T cells were obtained by magnetic sorting. The frequency of FoxP3+ cells in CD4+ T cells was examined with flow cytometry. (i) Non-MIH-2-bearing mice treated with phosphate-buffered saline (PBS); (ii) non-MIH-2-bearing mice immunized with CYP2J peptide 7 times; (iii) MIH-2-bearing mice treated with PBS; (iv) MIH-2-bearing mice immunized seven times with CYP2J peptide. (b) Frequency of CD11b+Gr-1+ cells in splenocytes from MIH-2-bearing mice or non-MIH-2-bearing mice immunized excessively with CYP2J peptide. Splenocytes were collected from each group of mice, stained with fluorescein isothiocyanate-conjugated monoclonal antibodies (mAbs) to Gr-1 and with phycoerythrin-conjugated mAbs to CD11b, and analysed with two-colour flow cytometry. (i) Non-MIH-2-bearing mice treated with PBS; (ii) non-MIH-2-bearing mice immunized seven times with CYP2J peptide; (iii) MIH-2-bearing mice treated with PBS; (iv) MIH-2-bearing mice immunized seven times with CYP2J peptide.
The frequency of CD11b+Gr-1+ myeloid suppressor cells (MSCs) was also increased significantly in mice immunized excessively with CYP2J peptide (Fig. 7b). The frequencies of MSCs in each treated group were 8·58 ± 2·11 (non-MIH-2-bearing mice treated with PBS), 23·46 ± 5·56 (non-MIH-2-bearing mice immunized with CYP2J peptide), 7·89 ± 2·89 (MIH-2-bearing mice treated with PBS) and 18·34 ± 4·10 (MIH-2-bearing mice immunized with CYP2J peptide) (n = 3). No increase in the frequencies of Tregs and MSCs was observed in mice bearing or non-bearing MIH-2 tumour immunized with OVA peptide (data not shown).
Discussion
Splenocytes from mice immunized twice with CYP2J peptide showed significantly high IFN-γ production in vitro, and it was enhanced further when CYP2J peptide was added to the culture. These results indicate that CYP2J peptide is immunogenic for naive immunocompetent mice and that adequate immunization with CYP2J peptide can activate the Th1 immune condition. CYP2Js antigenicity providing Th1 might contribute to MIH-2 tumour rejection because MIH-2 cells are susceptible to IFN-γ and to the cytotoxic activity of macrophages activated by IFN-γ[18,23].
The CYP2Js are newly discovered CYP subfamily enzymes that are involved in arachidonic acid metabolism; however, little is known about the role of CYP2Js in immunology. Ma et al.[20] have reported that because the turnover of CYP2J6 in tissue is extremely rapid, the mRNA of only CYP2J6 can be detected with RT–PCR but that CYP2J6 protein cannot be detected with immunoblot analysis. In the present study, we could not detect CYP2J6 protein in MIH-2 cells or normal liver tissue with immunoblot analysis using a CYP2J6-specific anti-CYP2J6pep1 antibody, although these tissues expressed mRNA encoding CYP2J6. It is possible that CYP2Js in normal tissue might have an unusual turnover in vivo which prevents the formation of naturally processed epitopes for immune recognition. Normal hepatocytes under non-pathological conditions would not release CYP2J to the outside of the cells. However, if CYP2J-expressing tumour cells did release CYP2J, it might be recognized as just a ‘non-self’ antigen by the immune system, because the immune system had never encountered the antigenic CYP2J epitope because of its rapid destruction. There is a concern that immunization with CYP2J peptide might induce autoimmune hepatic inflammation by activation of autoreactive T cells responsive to hepatocytes. It was found that about 10% of the vaccinated mice showed mild hepatic inflammation by histological examination of the liver without elevation of liver-derived enzymes such as alanine aminotransferase in sera. This mild liver inflammation might be caused by the action of IFN-γ produced by the activated CD4+ T cells, and antigen-specific activation of CD8+ CTLs would be required for the generation of significant autoimmune liver inflammation.
Considering that splenocytes from MIH-2-bearing mice showed high IFN-γ production, CYP2Js released from MIH-2 tissue might stimulate host anti-tumour immunity to produce IFN-γ. MIH-2 tumours might not develop if tumour cells undergo apoptosis through the direct action of IFN-γ or the cytotoxic activity of IFN-γ-activated immune cells. However, once tumour cells evade IFN-γ-mediated immune attack and continue to proliferate, the large tumour burden would provide excess antigenic stimulation to the host immune system, deleting CYP2J-specific T cells by activation-induced cell death [24], accelerating tumour growth. Using a preclinical mouse model, Sotomayor et al.[25] have shown anergy of CD4+ TAA-specific T cells that correlates with a lack of tumour rejection in vivo. Because anti-tumor activity against MIH-2 tumours is mediated by direct cytotoxicity of IFN-γ or IFN-γ-activated macrophages [18,23], suppression of IFN-γ production of CD4+ T cells must favour the development of MIH-2 tumours.
The Tregs with immunosuppressive activity can be characterized by cell-surface co-expression of CD4 and CD25 [26] and by co-expression of FoxP3 [27]. Tregs are defined functionally as T cells that inhibit immune response by diminishing the activity of other cell types through direct or indirect modes of action [27–29]. Our present results suggest strongly that an increase in Tregs is associated closely with excessive antigenic stimulation with CYP2J. Tregs mediate peripheral tolerance by suppressing self-antigen-reactive T cells [5,30]. Because most TAAs are self-antigens, suppression of TAA-specific lymphocytes by Tregs has been proposed as a mechanism for the failure of anti-tumour immunity [31,32]. Immunization with autoantigens identified through serological identification of antigens by recombinant expression cloning (SEREX) has been reported to activate CD4+CD25+ Tregs[33]. Furthermore, immunization with SEREX-defined autoantigens suppressed anti-tumour immunity and accelerated chemically induced tumour development [34]. Accordingly, it seems likely that excessive antigenic stimulation with some CD4+ T cell epitopes of autoantigens, including TAAs, might induce immune tolerance or unresponsiveness preferentially.
Several studies have shown that the number of MSCs is increased markedly in the peripheral blood of tumour-bearing animals and of patients with cancer, whereas the number of DCs is decreased [35]. Up-regulation of signal transducer and activator of transcription-3 and secretion of tumour-derived cytokines, such as VEGF, TGF-beta, granulocyte–macrophage colony-stimulating factor, IL-10 and prostaglandin, have been shown to arrest differentiation of APCs from their myeloid progenitors and trigger accumulation of MSCs [36,37]. MSCs induce immunosuppression through two enzymes involved in arginine metabolism: inducible nitric oxide (NO) synthetase 2, which generates NO, and arginase 1, which depletes L-arginine, generating amino acid starvation for T cell activation [38]. Tumour-associated MSCs have been reported recently to mediate the development of tumour-induced Tregs and T cell anergy in tumour-bearing hosts [39]. Tregs are differentiated by stimulation with dysfunctional DCs or APCs that are generated in a tumour microenvironment with abundant VEGF, IL-10 or TGF-beta [5,6]. Tumours convert DCs into TGF-beta-expressing immature myeloid DCs that can promote Treg proliferation [40].
Collectively, CYP2J isoforms expressed in HCC cells activate anti-tumour immunity with low or moderate antigenic stimulation but suppress anti-tumour immunity with excessive antigenic stimulation. Immune suppression provided by over-stimulation with CYP2J antigenicity was associated closely with induction of CD4+FoxP3+ Tregs and CD11b+Gr-1+ MSCs. Excessive immune stimulation with CYP2J derived from HCC cells would suppress host anti-tumour immunity, leading to further tumour development in advanced stages of HCC.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (C) and (B), and a Grant-in-Aid for Exploratory Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grants-in-Aid from the Japan Medical Association, the Takeda Science Foundation, the Pancreas Research Foundation of Japan, the Jikei University Research Fund, the Promotion and Mutual Aid Corporation for Private School of Japan and the Science Research Promotion Fund. This work was also supported, in part, with funds from the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
References
- 1.Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–15. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 2.Crosti M, Longhi R, Consogno G, Melloni G, Zannini P, Protti MP. Identification of novel subdominant epitopes on the carcinoembryonic antigen recognized by CD4+ T cells of lung cancer patients. J Immunol. 2006;176:5093–9. doi: 10.4049/jimmunol.176.8.5093. [DOI] [PubMed] [Google Scholar]
- 3.Guilloux Y, Viret C, Gervois N, et al. Defective lymphokine production by most CD8+ and CD4+ tumor-specific T cell clones derived from human melanoma-infiltrating lymphocytes in response to autologous tumor cells in vitro. Eur J Immunol. 1994;24:1966–73. doi: 10.1002/eji.1830240905. [DOI] [PubMed] [Google Scholar]
- 4.Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J Immunol. 2007;178:1268–76. doi: 10.4049/jimmunol.178.3.1268. [DOI] [PubMed] [Google Scholar]
- 5.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 6.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–52. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 7.Gajewski TF. Identifying and overcoming immune resistance mechanisms in the melanoma tumor microenvironment. Clin Cancer Res. 2006;12:2326s–30s. doi: 10.1158/1078-0432.CCR-05-2517. [DOI] [PubMed] [Google Scholar]
- 8.Redmond WL, Marincek BC, Sherman LA. Distinct requirements for deletion versus anergy during CD8 T cell peripheral tolerance in vivo. J Immunol. 2005;174:2046–53. doi: 10.4049/jimmunol.174.4.2046. [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL. MHC class II restricted tumor antigens and the role of CD4+ T cells in cancer immunotherapy. Curr Opin Immunol. 1994;6:741–5. doi: 10.1016/0952-7915(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 10.Wang RF. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 2001;22:269–76. doi: 10.1016/s1471-4906(01)01896-8. [DOI] [PubMed] [Google Scholar]
- 11.Toes RE, Ossendorp F, Offringa R, Melief CJ. CD4 T cells and their role in antitumor immune responses. J Exp Med. 1999;189:753–6. doi: 10.1084/jem.189.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guery JC, Adorini L. Selective immunosuppression of class II-restricted T cells by MHC class II-binding peptide. Crit Rev Immunol. 1993;13:195–206. [PubMed] [Google Scholar]
- 13.Campi G, Crosti M, Consogno G, et al. CD4(+) T cells from healthy subjects and colon cancer patients recognize a carcinoembryonic antigen-specific immunodominant epitope. Cancer Res. 2003;63:8481–6. [PubMed] [Google Scholar]
- 14.Chahal FC, Entwistle J, Glover N, Macdonald GC. A targeted proteomic approach for the identification of tumor-associated membrane antigens using the ProteomeLab PF-2D in tandem with mass spectrometry. Biochem Biophys Res Commun. 2006;348:1055–62. doi: 10.1016/j.bbrc.2006.07.187. [DOI] [PubMed] [Google Scholar]
- 15.Dengjel J, Nastke MD, Gouttefangeas C, et al. Unexpected abundance of HLA class II presented peptides in primary renal cell carcinomas. Clin Cancer Res. 2006;12:4163–70. doi: 10.1158/1078-0432.CCR-05-2470. [DOI] [PubMed] [Google Scholar]
- 16.Zeng G, Aldridge ME, Tian X, et al. Dendritic cell surface calreticulin is a receptor for NY-ESO-1: direct interactions between tumor-associated antigen and the innate immune system. J Immunol. 2006;177:3582–9. doi: 10.4049/jimmunol.177.6.3582. [DOI] [PubMed] [Google Scholar]
- 17.Rohn TA, Reitz A, Paschen A, et al. A novel strategy for the discovery of MHC class II-restricted tumor antigens: identification of a melanotransferrin helper T-cell epitope. Cancer Res. 2005;65:10068–78. doi: 10.1158/0008-5472.CAN-05-1973. [DOI] [PubMed] [Google Scholar]
- 18.Irie M, Homma S, Komita H, et al. Inhibition of spontaneous development of liver tumors by inoculation with dendritic cells loaded with hepatocellular carcinoma cells in C3H/HeNCRJ mice. Int J Cancer. 2004;111:238–45. doi: 10.1002/ijc.20247. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–8. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- 20.Ma J, Bradbury JA, King L, et al. Molecular cloning and characterization of mouse CYP2J6, an unstable cytochrome P450 isoform. Biochem Pharmacol. 2002;64:1447–60. doi: 10.1016/s0006-2952(02)01393-x. [DOI] [PubMed] [Google Scholar]
- 21.Iinuma T, Homma S, Noda T, Kufe D, Ohno T, Toda G. Prevention of gastrointestinal tumors based on adenomatous polyposis coli gene mutation by dendritic cell vaccine. J Clin Invest. 2004;113:1307–17. doi: 10.1172/JCI17323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu W, Bradbury JA, Tsao CC, et al. Cytochrome P450 CYP2J9, a new mouse arachidonic acid omega-1 hydroxylase predominantly expressed in brain. J Biol Chem. 2001;276:25467–79. doi: 10.1074/jbc.M100545200. [DOI] [PubMed] [Google Scholar]
- 23.Komita H, Homma S, Saotome H, Zeniya M, Ohno T, Toda G. Interferon-gamma produced by interleukin-12-activated tumor infiltrating CD8+T cells directly induces apoptosis of mouse hepatocellular carcinoma. J Hepatol. 2006;45:662–72. doi: 10.1016/j.jhep.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 25.Sotomayor EM, Borrello I, Tubb E, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5:780–7. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- 26.Wing K, Ekmark A, Karlsson H, Rudin A, Suri-Payer E. Characterization of human CD25+ CD4+ T cells in thymus, cord and adult blood. Immunology. 2002;106:190–9. doi: 10.1046/j.1365-2567.2002.01412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakaguchi S. The origin of FOXP3-expressing CD4+ regulatory T cells: thymus or periphery. J Clin Invest. 2003;112:1310–2. doi: 10.1172/JCI20274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065x.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- 29.Shevach EM. Certified professionals: CD4(+)CD25(+) suppressor T cells. J Exp Med. 2001;193:F41–6. doi: 10.1084/jem.193.11.f41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Herrath MG, Harrison LC. Antigen-induced regulatory T cells in autoimmunity. Nat Rev Immunol. 2003;3:223–32. doi: 10.1038/nri1029. [DOI] [PubMed] [Google Scholar]
- 31.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of ‘tumor escape’ phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 33.Nishikawa H, Kato T, Tanida K, et al. CD4+ CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. Proc Natl Acad Sci USA. 2003;100:10902–6. doi: 10.1073/pnas.1834479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishikawa H, Kato T, Tawara I, et al. Accelerated chemically induced tumor development mediated by CD4+CD25+ regulatory T cells in wild-type hosts. Proc Natl Acad Sci USA. 2005;102:9253–7. doi: 10.1073/pnas.0503852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gabrilovich D, Ishida T, Oyama T, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66. [PubMed] [Google Scholar]
- 36.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 37.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced immune dysfunctions by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 39.Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 40.Ghiringhelli F, Puig PE, Roux S, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005;202:919–29. doi: 10.1084/jem.20050463. [DOI] [PMC free article] [PubMed] [Google Scholar]