

Abstract
Inflammatory bowel disease (IBD) is characterized by heavy production of proinflammatory cytokines such as tumour necrosis factor (TNF)-α and interleukin (IL)-1β. Interactions of the autonomic nervous system with local immune cells play an important role in the development of IBD, and the balance of autonomic nerve function is broken in IBD patients with sympathetic overactivity. However, the function of catecholamines in the progress of colitis is unclear. In this study, we examined the role of catecholamines via α2-adrenoreceptor in acute murine colitis. The expression of tyrosine hydroxylase (TH) and dopamine b-hydroxylase (DBH), two rate-limiting enzymes in catecholamine synthesis, was detected by immunohistochemistry in murine colitis. Murine colitis was induced by dextran sodium sulphate or trinitrobenzene sulphonic acid (TNBS), and the mice were administered RX821002 or UK14304, α2-adrenoceptor antagonists or agonists. Colitis was evaluated by clinical symptoms, myeloperoxidase assay, TNF-α and IL-1β production and histology. Lamina propria mononuclear cells (LPMCs) from mice with TNBS colitis were cultured in the absence or presence of RX821002 or UK14304, and stimulated further by lipopolysaccharide. TH and DBH are induced in LPMCs of inflamed colon, the evidence of catecholamine synthesis during the process of colitis. RX821002 down-regulates the production of proinflammatory cytokines from LPMCs, while UK14304 leads to exacerbation of colitis. Together, our data show a critical role of catecholamines via α2-adrenoreceptors in the progress of acute colitis, and suggest that use of the α2-adrenoceptor antagonist represents a novel therapeutic approach for the management of colitis.
Keywords: α2-adrenoreceptor, catecholamines, colitis, immune response
Introduction
The aetiology of inflammatory bowel disease (IBD), which contains mainly two forms as Crohn's disease and ulcerative colitis, is still unclear. It is believed that altered immunological function, resulting from an interplay between genetic susceptibility and certain environmental factors, contributes significantly to mucosal inflammation of the gastrointestinal tract [1]. During the process of IBD many proinflammatory cytokines, including tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 produced mainly by macrophages and lymphocytes in inflamed mucosa, contribute to maintenance of the inflammatory response [2]. Inhibiting the production of those proinflammatory cytokines by the treatment such as corticosteroids, aminosalicyclic acids, antibodies of those cytokines and probiotics can lead to down-regulation of inflammatory response and rehabilitation from tissue damage of IBD, as shown by clinical and experimental data [3,4].
Interaction of the nervous system with local immune cells play an important role in keeping the body in a homeostatic state; for example, during inflammation or infection, proinflammatory mediators released by local immune cells activate neuronal responses. In response to stimulation, the autonomic nervous system modulates local immune and inflammatory responses by means of sympathetic and parasympathetic pathways [5,6] and triggers systemic neuroendocrine and regional neural responses that seek to return the system to a homeostatic state. The cholinergic anti-inflammatory pathway, also well studied, regulates inflammatory responses through the cholinergic receptor and has been shown to be helpful in acute pancreatitis [7], endotoxaemia [8] and myocardial ischaemia/reperfusion injury [9]. Conversely, the transmitters of sympathetic nerves, such as catecholamines, modulate the function of immune cells and the production of cytokines through adrenoreceptors [10]. The functional interplay of the adrenergic nervous system with immune cells counterbalances the effects of the parasympathetic nervous system [11,12].
Recent studies suggest that the balance of autonomic nerve function is broken in IBD patients and experimental colitis, with autonomic vagal neuropathy and nerve dysfunction [13–15] and sympathetic overactivity [16], which leads to imbalance of the transmitters such as acetylcholine and catecholamines in inflamed mucosa. Catecholamines can be produced not only by sympathetic nerves, but also by local immune cells such as phagocytes and T cells during the process of inflammation, as shown by Flierl and colleagues [17]. Therefore, via the receptors, endogenous catecholamines are able to activate the releasing cell itself as well as nearby cells in an autocrine/paracrine manner. Blockade of α- and β-adrenoceptors on macrophages and lymphocytes inhibits the production of proinflammatory cytokines significantly [18–20]. From these findings it is tempting to speculate that the inflammatory cytokine network might be one of the important mediator systems controlled tightly by catecholamines via adrenergic receptors [21,22].
However, the role of catecholamines in the pathogenesis of IBD is still unclear. To explore the initial effect of catecholamines, we detected the expression of tyrosine hydroxylase (TH) and dopamine b-hydroxylase (DBH), two rate-limiting enzymes in catecholamine synthesis, in the mucosa of murine colitis, and investigated the effect of catecholamines on experimental colitis by blockade or stimulation of the α2-adrenoreceptor. We report, first, that catecholamines produced by lamina propria mononuclear cells (LPMCs) with heavy TH and DBH expression facilitate the progress of colitis via the α2-adrenoreceptor, and the blockade of α2-adrenoreceptors can down-regulate the inflammatory response and ameliorate acute murine colitis.
Methods
Animals
Male BALB/c mice were obtained from the Experimental Animal Center of Nanchang University, and kept under specific pathogen-free conditions. The mice used in the study were 7–8 weeks old, weighing approximately 22 g. All experiments using mice were reviewed and approved by the Institutional Animal Care Committee of Nanchang University.
Induction of trinitrobenzene sulphonic acid colitis and study design
Colitis was induced in mice on day 1, as described previously [23,24]. In brief, the mice were anaesthetized lightly after overnight food deprivation, and a 3.5 F catheter was inserted intrarectally 4 cm from the anus. To induce colitis, 100 µl of 2·5 mg trinitrobenzene sulphonic acid (TNBS) (Sigma Chemical Co, St Louis, MO, USA) in 50% ethanol was administered slowly into the lumen via the catheter. Control mice received 50% ethanol alone (100 µl). RX821002 (10 mg/kg/mouse body weight; α2-adrenoceptor antagonist) or UK14304 (2 mg/kg; α2-adrenoceptor agonist) was administered intraperitoneally (i.p.) 2 h after TNBS instillation, and repeated daily until the mice were killed.
The mice were killed on day 7 and the colonic segments were frozen immediately in liquid nitrogen for cytokine determination and myeloperoxidase (MPO) activity measurement. For histological studies, a colonic specimen from the middle part was fixed in 10% buffered formalin, and stained with haematoxylin and eosin (H&E) for subsequent histological examination. Histological examinations were performed by the same investigator in a blinded fashion. Histological scores were performed to grade the degree of colonic inflammation from 0 to 4 using previously described scoring systems for hapten-induced colitis [25]: 0, no signs of inflammation; 1, very low level; 2, low level of leucocyte infiltration; 3, high level of leucocyte infiltration, high vascular density, thickening of the colon wall; and 4, transmural leucocyte infiltration, loss of goblet cells, high vascular density, thickening of the colon wall.
Induction of dextran sulphate sodium colitis
Alternatively, acute colitis was induced in BALB/c mice on day 1 by administration of 5% dextran sulphate sodium (DSS) (molecular weight 500, 000; Amersham Pharmacia Biotech AB, Uppsala, Sweden) dissolved in the drinking water. Fresh DSS solution was provided every second day. Control mice drank only distilled water. RX821002 (10 mg/kg/mouse body weight) or UK14304 (2 mg/kg/mouse body weight) was administered i.p. 24 h after induction of colitis, and repeated daily until the mice were killed on day 10.
Development of colitis was assessed daily by measurement of body weight, evaluation of stool consistency and detection of bloody stools. Disease severity was scored using a clinical disease activity index (DAI) ranging from 0 to 4, calculated as described previously [26,27] using the following parameters: stool consistency, presence or absence of faecal blood and weight loss. Mice were killed on day 10, and the middle section of colon was fixed in 10% formaldehyde–saline. H&E-stained sections were graded based on a scoring system modified from a previous study [28]. Histology scoring was performed in a blinded fashion. A combined score of inflammatory cell infiltration and tissue damage was determined as follows: cell infiltration: score 0, occasional inflammatory cells in the lamina propria (LP); 1, increased infiltrate in the LP predominantly at the base of crypts; 2, confluence of inflammatory infiltrate extending into the mucosa; and 3, transmural extension of infiltrate. Tissue damage: score 0, no mucosal damage; 1, partial (up to 50%) loss of crypts in large areas; 2, partial to total 50–100% loss of crypts in large areas, epithelium intact; and 3, total loss of crypts in large areas and epithelium lost.
Measurement of MPO activity
Colonic tissue from mice was frozen in liquid nitrogen and stored at –70°C until assayed. All experiments were performed within 1 week of collection of tissue. MPO activity, the index of polymorphonuclear infiltration into the colon, was measured according to the method described by Bai et al.[29]. In brief, a portion of colonic tissue was homogenized in 0·5% hexadecyltrimethylammonium bromide in 50 mM potassium phosphate buffer (pH 6·0). Aliquots were then added to O-dianisidine hydrochloride solution. Absorbance was read at 460 nm using a microplate reader. MPO was expressed in units/mg of tissue, where 1 unit corresponds to the activity required to degrade 1 mmol of hydrogen peroxide in 1 min at 24°C.
Cytokine determination
Colonic homogenates and culture supernatants were determined by specific sandwich enzyme-linked immunosorbent assay (ELISA). The colonic tissues of each group were homogenized in phosphate-buffered saline (PBS), as the final concentrations were 10% (W/V). ELISA kits were supplied by R&D Systems (Minneapolis, MN, USA). ELISA was performed according to the manufacturer's instructions. In short, polyclonal goat anti-mouse cytokine antibody was used for capturing antibodies; biotinylated polyclonal rabbit anti-mouse cytokine antibody was the detecting antibody. Streptavidin–horseradish peroxidase and tetramethylbenzidine sulphonate were added as colour indicators. Plates were read at 490 nm immediately after the colour reaction was stopped with acid. All steps were performed at room temperature.
Isolation and culture of LPMCs
The LPMCs were isolated from freshly obtained colonic specimens of the mice with TNBS-induced colitis at the peak of the disease (day 4), using a modification of the method described previously [30]. In brief, the colonic specimens were washed in Hanks's balanced salt solution (HBSS)–calcium–magnesium-free solution, then cut into 5-mm pieces and incubated in HBSS containing 0·75 mmol/l ethylenediamine tetraacetic acid (Sigma) and 1 mmol/l dithiothreitol (Sigma) at 37°C for 30 min to remove epithelium. The tissues were digested further in RPMI-1640 (HyClone, Logan, UT, USA) containing 400 U/ml colleagenase IV (Sigma) and 0·01 mg/ml Dnase I (Sigma) in a shaking incubator at 37°C; the step was repeated two to three times. The cells released from the tissues were layered on a 40–100% Percoll gradient (Pharmacia Biotech, Piscataway, NJ, USA) and spun at 58·06 g for 5 min to collect the LPMCs at the 40–100% Percoll interface.
The LPMCs were incubated in complete medium (RPMI-1640 supplemented with 100 U/ml penicillin/streptomycin, 2 mmol/l L-glutamine and 10% heat-inactivated fetal calf serum) at a concentration of 5 × 105 cells/ml, in the absence (unstimulated) or presence of RX821002 (2 nM, 10 nM) or UK14304 (1 nM, 5 nM). After 15 min, the cells were stimulated further in the presence or absence of lipopolysaccharide (LPS, 50 ng/ml), and culture supernatants were collected and stored at –70°C after 8 h.
Immunohistochemistry
Colonic tissues of mice were fixed in 10% buffered formalin, embedded in paraffin, and 4-µm sections were cut. After blocking inner peroxidase, sections were incubated sequentially with the first antibody solution including rabbit anti-TH or rabbit anti-DBH antibody [Santa Cruz Biotechnology, Santa Cruz, CA, USA; 4 µg/ml in 2% bovine serum albumin in PBS solution respectively]. After three washes in pH 7·4 PBS, the sections were then incubated in goat anti-rabbit immunoglobulin (Ig)G conjugated with peroxidase labelled polymer, coloured using diaminobenzidine reaction and counterstained with haematoxylin. Negative controls were established using rabbit IgG instead of the first antibodies.
Statistical analysis
All data in the text and figures are expressed as mean ± standard deviation. Comparisons of more than two groups were made with a one-way analysis of variance usin Tukey's post hoc test. When appropriate, comparison with two groups was made using Student's t-test for unpaired data. Differences were considered statistically significant if P < 0·05.
Results
The TH and DBH expressions are induced in the progress of experimental colitis
To investigate biosynthesis of catecholamines in the immune cells of colonic tissues, we detected the expression of TH and DBH, two key enzymes in catecholamine synthesis, in colonic tissues of the mice with TNBS- or DSS-induced colitis. There was no appearance of TH and DBH staining in normal colonic tissue of control mice, whereas intestinal inflammation of TNBS or DSS colitis induced TH and DBH expressions strongly in cytoplasm of LPMCs (Fig. 1), implying that catecholamines can be produced strongly by LPMCs in the progress of colitis.
Fig. 1.

Tyrosine hydroxylase (TH) and dopamine b-hydroxylase (DBH) expressions were induced in colonic mucosa during the process of trinitrobenzene sulphonic acid (TNBS)- (day 4) and dextran sulphate sodium (DSS)-induced colitis (day 10). TH and DBH expressions were assessed by immunochemical staining in colonic mucosa of mice with experimental colitis and normal mice (n = 5 in each group). All images are shown at the same magnification (×200).
Blockade of α2-adrenoceptor ameliorates TNBS-induced colitis
To investigate the role of catecholamines in the pathogenesis of TNBS-induced colitis, we induced TNBS colitis and treated the mice daily with RX821002, the α2-adrenoceptor antagonist, and UK14304, the α2-adrenoceptor agonist. Mice subjected to intrarectal administration of 2·5 mg TNBS in 50% ethanol developed severe colitis characterized by bloody diarrhoea, rectal prolapse accompanied by extensive wasting syndrome and sustained weight loss resulting in a mortality of 29·4% (five of 17) on day 7. Mice treated with RX821002 (10 mg/kg) 2 h after TNBS colitis induction and repeated daily showed a survival rate of 84·6% (11 of 13) on day 7 and recovered the lost body weight rapidly, whereas those mice administered UK14304 (2 mg/kg) had the highest mortality of 42·1% (eight of 19) and extensive body weight loss. Histological examination of the distal colon of mice with TNBS colitis showed patchy ulceration, epithelial cell loss, reduction of the density of the tubular glands, focal loss of crypts, infiltration of inflammatory cells consisting of macrophages, lymphocytes and neutrophils, named as LPMCs in the LP, and transmural inflammation involving all layers of the bowel wall. When the mice received RX821002 treatment these histological signs were greatly improved, with significant reduction of inflammatory activity, neutrophil infiltration and MPO activity. However, the mice administered UK14034 showed the highest histological score, with severe mucosal tissue damage, extensive immune cells infiltration and the highest MPO activity (Figs 2 and 3a).
Fig. 2.
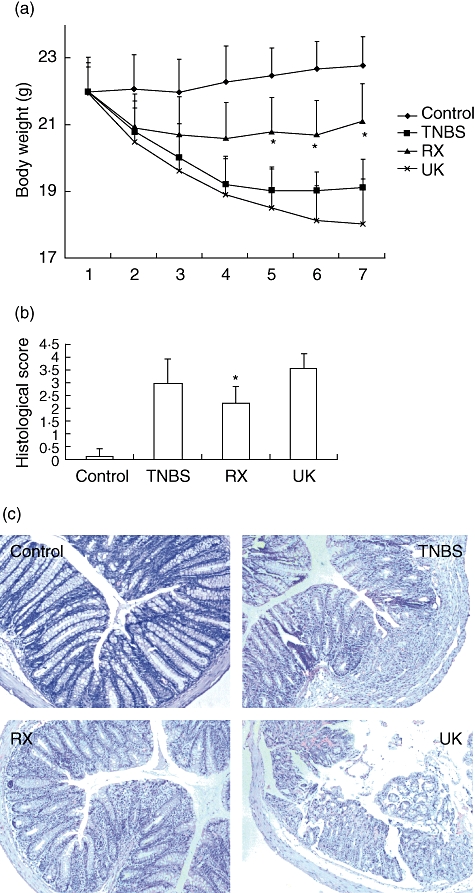
Blocking α2-adrenoceptor treatment after initiation of colitis inhibits the progress of trinitrobenzene sulphonic acid (TNBS)-induced disease. Colitis was induced by intracolonic administration of TNBS. RX821002 (10 mg/kg) or UK14304 (2 mg/kg) were administered intraperitoneally 2 h after TNBS instillation and repeated daily. Mice treated with ethanol alone were used as control. (a) Body weight changes in the mice in each group. *P < 0·05 between RX821002-treated mice and the UK14304 group or TNBS mice. (b) Colonic inflammation was scored by histological analysis at the end of the experiment. *P < 0·05; RX821002 mice versus the UK14304 group or TNBS mice. (c) Haematoxylin and eosin staining of colonic tissues of four groups of mice. All images are shown at the same magnification (×40).
Fig. 3.
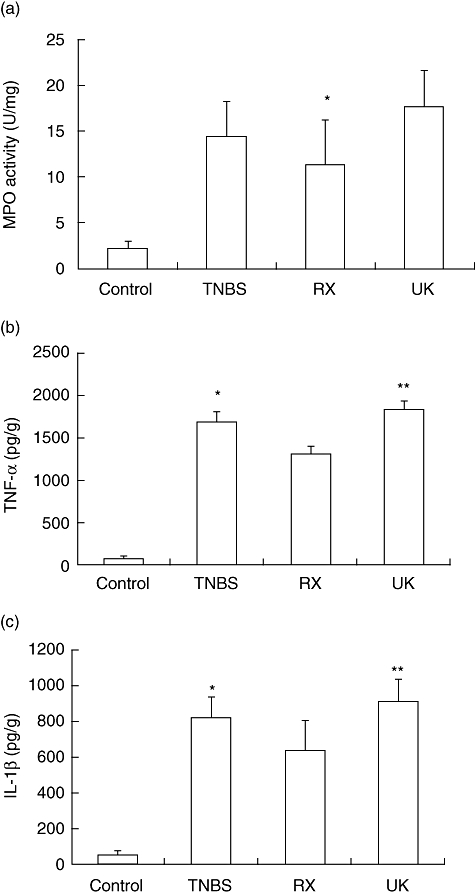
Blocking α2-adrenoceptor treatment inhibits the inflammatory response of trinitrobenzene sulphonic acid (TNBS)-induced colitis. Myeloperoxidase (MPO) activities in colonic tissues and concentrations of tumour necrosis factor (TNF)-α and interleukin (IL)-1β in colonic homogenates of each group of mice were determined by MPO activity assay and specific sandwich enzyme-linked immunosorbent assay (ELISA) kit, respectively, n = 8 in each group. (a) MPO activities in colonic tissues of each group of mice. *P < 0·05; RX821002-treated mice versus treatment-free mice with TNBS colitis. (b) Concentration of TNF-α in colonic homogenates of each group of mice was determined by ELISA. *P < 0·05 compared with the RX821002 group; **P < 0·01, with RX821002. (c) Concentration of IL-1β in colonic homogenates of each group of mice. *P < 0·05 compared with the RX821002 group; **P < 0·01; RX821002.
We next evaluated the effect of α2-adrenoceptor antagonist or agonist treatment on the production of inflammatory mediators such as TNF-α and IL-1β, which are linked mechanistically to TNBS-induced colitis. RX821002 management reduced the production of inflammatory cytokines strikingly in colonic homogenates such as TNF-α (1441·4 pg/g) and IL-1β (663·5 pg/g), compared with those with TNBS-induced colitis (1700·1 pg/g, 818·4 pg/g respectively), whereas UK14304 administration elevated the the production of TNF-α (1840·4 pg/g) and IL-1β (909·3 pg/g), as shown in Fig. 3b and c. These results suggest that blockade of the α2-adrenoceptor is able to turn off an established in vivo inflammatory response.
Blockade of α2-adrenoceptor inhibits cytokine release from LPMCs of TNBS-induced colitis
The TNBS-induced colitis is characterized with a high production of proinflammatory cytokines such as TNF-α and IL-1β, expressed by LPMCs. To evaluate the effect of α2-adrenoceptor blockade on cytokine release from LPMCs during the process of TNBS-induced colitis, we isolated LPMCs from colonic specimens of the mice with TNBS-induced colitis at the peak of the disease (day 4) and cultured the cells in vitro in the presence of RX821002 (2 nM, 10 nM) or UK14304 (1 nM, 5 nM). Ten nM RX821002 could down-regulate slightly TNF-α and IL-1β production of LPMCs from the mice with TNBS-induced colitis, whereas 5 nM UK14304 induced TNF-α and IL-1β secretion slightly from LPMCs, with a significant difference of IL-1β production between the cells treated with 10 nM RX821002 and 5 nM UK14304 (P < 0·05), shown in Fig. 4a.
Fig. 4.
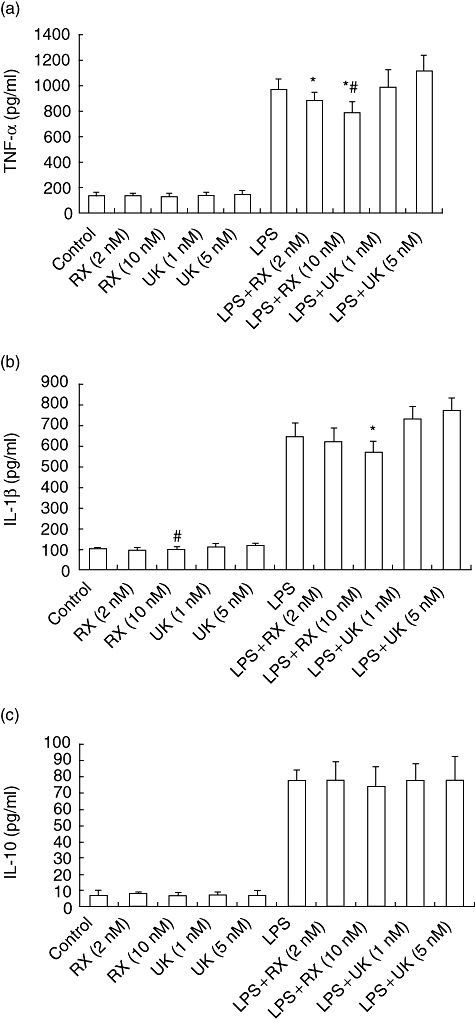
Blocking α2-adrenoceptor treatment down-regulates immune response in trinitrobenzene sulphonic acid (TNBS)-induced colitis. Lamina propria mononuclear cells (LPMCs) were isolated from colonic tissues of mice with TNBS-induced colitis at the peak of the disease (day 4) and cultured with medium in the absence or presence of RX821002 (2 nM, 10 nM) or UK14304 (1 nM, 5 nM). After 15 min, the cells were stimulated further in the presence or absence of lipopolysaccharide (LPS) (50 ng/ml) for 8 h, then culture supernatants were collected and determined by enzyme-linked immunosorbent assay (ELISA), n = 5 in each group. (a) Concentration of tumour necrosis factor (TNF)-α in culture supernatants was measured by ELISA. *P < 0·01 compared with LPS + UK14304 (1 nM) or LPS + UK14304 (5nM); #P < 0·05 compared with LPS. (b) Concentration of interleukin (IL)-1β was measured by ELISA. *P < 0·01 compared with LPS, LPS + UK14304 (1nM) or LPS + UK14304 (5nM); #P < 0·05 compared with UK14304 (5 nM). (c) Concentration of IL-10 in culture supernatants was measured; no significant difference was found among those groups with or without stimulation of LPS.
We stimulated LPMCs further with 50 ng/ml LPS for 8 h in the presence of RX821002 (2 nM, 10 nM) or UK14304 (1 nM, 5 nM). LPS induced proinflammatory cytokine release from LPMCs such as TNF-α and IL-1β, but pretreatment with RX821002 (2 nM, 10 nM) decreased TNF-α and IL-1β production of LPMCs initiated with LPS, with significant inhibition of TNF-α production by 10 nM RX821002 (P < 0·05). Conversely, UK14304, the α2-adrenoceptor agonist, boosted proinflammatory cytokine secretion from LPMCs induced by LPS. Proinflammatory cytokine production of LPMCs treated with 50 ng/ml LPS and 10 nM RX821002 was significantly lower than those cells treated with 50 ng/ml LPS and 1 nM or 5 nM UK14304 (P < 0·01). The results show that blockade of the α2-adrenoceptor by RX821002 treatment abrogates the responsiveness of LPMCs to LPS stimulation, in contrast to the excitatory effect of the α2-adrenoceptor using UK14304 treatment.
We next detected the effect of blockade or stimulation of the α2-adrenoceptor in LPMCs on the production of regulatory cytokines such as IL-10 in the presence or absence of LPS. There was no significant difference in IL-10 production among those groups with or without LPS stimulation (Fig. 4c), which implied that an inhibiting inflammatory response of TNBS-induced colitis by α2-adrenoceptor antagonist was initiated mainly by down-regulating the production of proinflammatory cytokines such as TNF-α and IL-1β, instead of inducing regulatory cytokines such as IL-10.
Blockade of α2-adrenoceptor ameliorates DSS-induced colitis
To investigate further the role of catecholamines in the pathogenesis of DSS-induced colitis via the α2-adrenoceptor, we induced colitis by administering 5% DSS in drinking water and treated the mice with RX821002 or UK14304. DSS administration was associated with significant clinical changes, inculding body weight loss (starting on day 5), appearance of occult faecal blood (on day 7) and diarrhoea (on day 8). Treatment with RX821002 resulted in significant amelioration of colitis, as shown by a decrease in DAI, improvement in stool consistency and reduced rectal bleeding and colonic MPO activity. Conversely, treatment with UK14304 worsened body weight loss and increased DAI and MPO acitvity. Histological examination of the distal colon of mice with DSS colitis showed multi-focal dropouts of crypts, and infiltration of inflammatory cells consisted of macrophages, lymphocytes and neutrophils. When the mice received RX821002 treatment these histological signs were much improved, with significant reduction of inflammatory activity and immune cell infiltration compared with those DSS coitic mice (P < 0·05) or UK14304-treated mice (P < 0·01). However, mice that received UK14034 administration showed the highest histological score, with severe mucosal tissue damage and extensive immune cell infiltration (Fig. 5c and d).
Fig. 5.
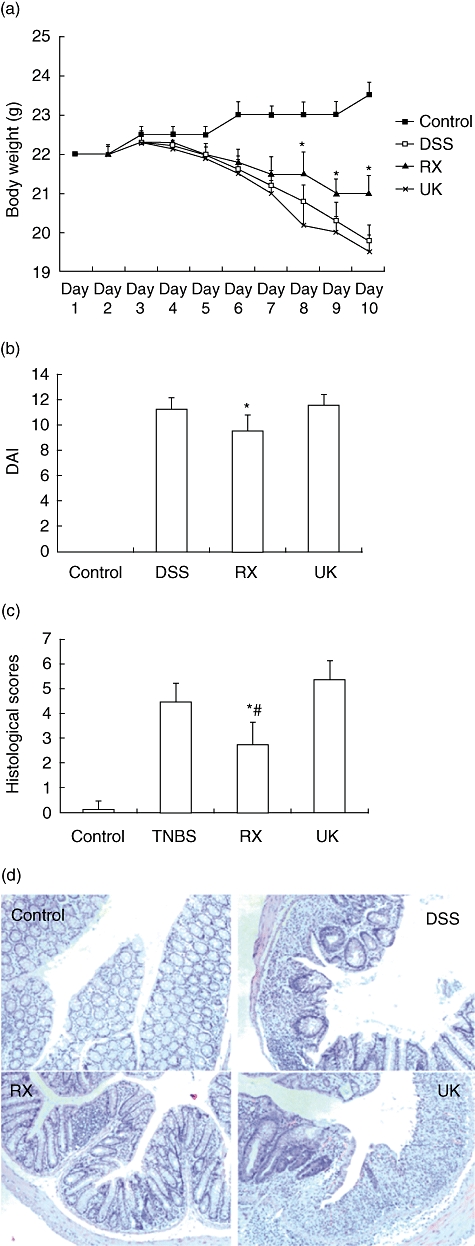
Blocking α2-adrenoceptor management decreases systemic and colonic inflammatory responses in the dextran sulphate sodium (DSS) model of colitis. Colitis was induced in mice by administration of 5% DSS dissolved in drinking water. RX821002 (10 mg/kg) or UK14304 (2 mg/kg) was administered intraperitoneally 24 h after induction of colitis, and repeated daily until the mice were killed on day 10. Control mice drank only distilled water. (a) Body weight changes in the mice in each group, nine mice in each group. *P < 0·05 compared with DSS group mice or UK14304 group mice. (b) Disease activity index (DAI) was scored on day 10 using the following parameters: stool consistency, presence or absence of faecal blood and weight loss. *P < 0·05 compared with DSS group mice or UK14304 group mice. (c) Colonic inflammation was scored by histological analysis at the end of the experiment. *P < 0·05 compared with DSS mice; #P < 0·01 compared with UK14304 mice. (d) Haematoxylin and eosin staining of colonic tissues of four groups of mice. All images are at shown the same magnification (×40).
We next evaluated the effect of the antagonist or agonist of α2-adrenoceptor treatment on the production of inflammatory mediators such as TNF-α and IL-1β in colonic tissues, which are linked mechanistically to DSS-induced colitis. RX821002 strikingly reduced the production of inflammatory cytokine management in colonic homogenates such as TNF-α (1079·4 pg/g) and IL-1β (765·9 pg/g), compared with DSS-induced colitis (1276·7 pg/g, P < 0·05; 881·5 pg/g, P < 0·05 respectively), while UK14304 administration elevated the the production of TNF-α (1412·6 pg/g) or IL-1β (961·5 pg/g), P < 0·01 compared with that of RX821002-treated mice, as shown in Fig. 6. These results suggest that blockade of the α2-adrenoceptor is able to inhibit inflammatory response of DSS-induced colitis.
Fig. 6.
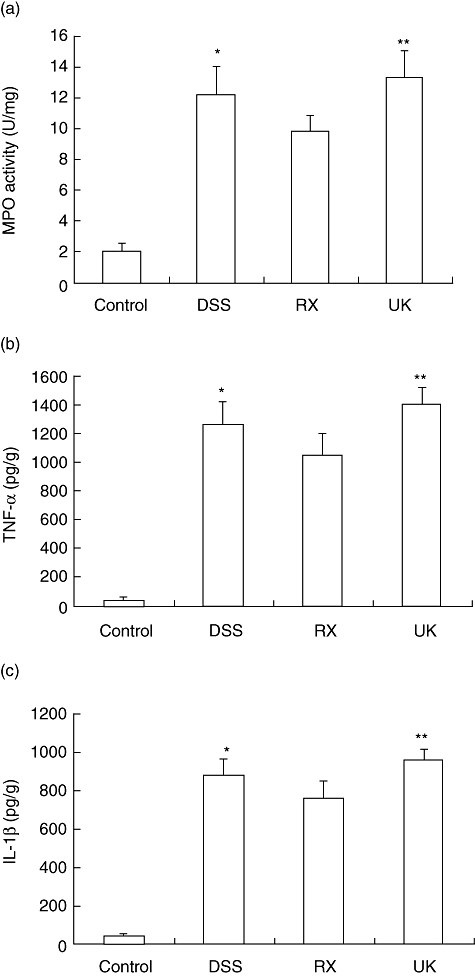
Blocking α2-adrenoceptor treatment inhibits the inflammatory response of DSS-induced colitis. Myeloperoxidase (MPO) activities in colonic tissues and concentrations of tumour necrosis factor (TNF)-α and by interleukin (IL)-1β in colonic homogenates of each group of mice were determined by MPO activity assay and specific sandwich enzyme-linked immunosorbent assay (ELISA) kit respectively. *P < 0·05 compared with the RX821002 group; **P < 0·01, with RX821002. (a) MPO activities in colonic tissues of each group of mice. (b) Concentration of TNF-α in colonic homogenates of each group of mice was determined by ELISA. (e) Concentration of IL-1β in colonic homogenates of each group of mice.
Discussion
Recently, the function of autonomic nerves of IBD patients has been emphasized according to the following: autonomic nerves have close contact with LPMCs and modulate intestinal inflammation by secreting transmitters such as acetylcholine or catecholamines [31,32]; histological evaluation of colon samples reveals alteration of nerve fibres in ulcerative colitis [33]; and psychological stress may exacerbate IBD [34,35], which implies important interactions among brain, autonomic nerve system and gastrointestinal tract. The balance of autonomic nerve function is broken in IBD patients and experimental colitis, with domination of the sympathetic nerve [16] and autonomic vagal nerve dysfunction [13], thus resulting in a maladjusted interaction between the nerve fibres and immune cells in intestinal mucosa, and contributes to the process of IBD and experimental colitis. Furthermore, the transmitters such as acetylcholine or catecholamines are secreted not only by local autonomic nerve fibres, but also by immune cells under inflammatory conditions [17,36]. In this study, we detected the expressions of TH and DBH, two key enzymes in catecholamine synthesis, in the colonic mucosa of two colitis models, including TNBS- and DSS-induced murine colitis, and the results suggest that in the development of colitis local immune cells regulate catecholamine release in an autocrine or paracrine manner.
Neurotransmitters have received considerable attention with regard to their effect on inflammatory response. Recently, it has been reported that choline acetyltransferase, the key enzyme of acetylcholine synthesis, is expressed in LPMCs, endocrine and epithelial cells of normal colonic mucosa or non-IBD mucosa, but is down-regulated in those cells during the process of IBD [37], which implies a deficiency of acetylcholine synthesis, with an imbalance of acetylcholine and catecholamines in the progress of colitis. The cholinergic transmitter can modulate the inflammatory response of experimental colitis [30,38] through thecholinergic anti-inflammatory pathway; the imbalance in transmitter production may then lead to further inflammation in colonic mucosa in the progress of IBD. Catecholamines can be produced not only by the sympathetic nerve, also by local immune cells such as phagocytes and T cells during the process of inflammation [17,36]. Therefore, secreted endogenous catecholamines are able to activate the releasing cell itself as well as nearby cells in an autocrine/paracrine manner, ultimately regulating cell function. Blockade of α- and β-adrenoceptors on macrophages, neutrophils and lymphocytes inhibited significantly the cytokine/chemokine production of these cells [18,19,20]. These findings make it tempting to speculate that the inflammatory cytokine/chemokine network might be one of the important mediator systems controlled tightly by catecholamines via adrenergic receptors [39–41]. However, catecholamines also mediate the anti-inflammatory effect in human T lymphocytes via the β2-adrenoceptor, as shown by Borger et al.[22], in contrast to the proinflammatory response of catecholamines via α2-adrenoreceptors. Thus, sympathetic transmitters show different functions depending upon receptor affinity to different receptor subtypes and the expression of adrenoceptors [42]. It has now been well established that immune/inflammatory cells express multiple receptors for catecholamines, and α2-adrenoreceptors have been identified on human and rodent peripheral blood mononuclear cells, macrophages, neutrophils and lymphocytes [43–47]. Using α2-adrenoreceptor antagonists and agonists, we explored the role of catecholamines in the pathogenesis of experimental colitis, as the results showed that blockade of α2-adrenoreceptors ameliorated acute TNBS-induced colitis and DSS colitis. The results also provide important information: first, via α2-adrenoreceptors, sympathetic transmitters promote the inflammatory response and contribute to the development of experiment colitis in contrast to the effect of cholinergic transmitters. Secondly, the sympathetic transmitters may show different biological functions depending upon the interaction with different receptors involved, and exert a proinflammatory effect via α2-adrenoreceptors in the progress of experimental colitis.
Proinflammatory cytokines such as TNF-α and IL-1β contribute to the progression of IBD and colitis in animal models of IBD [48–51]. We detected the effects of α2-adrenoceptor agonists and antagonists on the production of TNF-α and IL-1β in the development of TNBS- or DSS-induced colitis. RX821002, the α2-adrenoceptor antagonist, down-regulated expression of TNF-α and IL-1β in inflamed colon in accordance with a significant reduction in DAI, decreased weight loss and improvement of the colitis histological score. However, UK14304, the α2-adrenoceptor agonist, up-regulated TNF-α and IL-1β expression, with the highest histological score, severe mucosal tissue damage, intensive immune cell infiltration and the highest MPO activity. The results provide evidence that stimulating the α2-adrenoceptor by catecholamines can induce overproduction of proinflammatory cytokines, while blocking the α2-adrenoceptor can lead to down-regulation of proinflammatory cytokines and turn off the immune response of active experimental colitis.
To study the effect of catecholamines on regulating inflammatory cells function via the α2-adrenoceptor we isolated LPMCs from TNBS-induced colitis and treated the cells with α2-adrenoceptor antagonists and agonists respectively, and detected the effect of blocking or stimulating the α2-adrenoceptor on cytokine secretion. The α2-adrenoceptor antagonist such as RX821002 can down-regulate slightly TNF-α and IL-1β production of LPMCs from mice with TNBS-induced colitis, but the α2-adrenoceptor agonist, such as UK14304, induces TNF-α and IL-1β secretion slightly from LPMCs, whereas neither reagent has any effect on the production of regulatory cytokines such as IL-10; this result differs from the work of others, who have reported the important role of catecholamines in up-regulating IL-1β and IL-10 [52]. Further, we detected LPS-induced production of proinflammatory cytokines such as TNF-α and IL-1β and regulatory cytokine IL-10 from LPMCs in vitro by managing the α2-adrenoceptor. Blockade of the α2-adrenoceptor inhibits LPS-stimulated TNF-α and IL-1β production, compared with the effect of α2-adrenoceptor stimulation on inducing TNF-α and IL-1β production. However, neither the α2-adrenoceptor antagonist nor the α2-adrenoceptor agonist showed a direct effect on the production of IL-10. Our results suggest that, via the α2-adrenoceptor, catecholamines can trigger LPMCs producing proinflammatory cytokines such as TNF-α and IL-1β, but have no effect on IL-10 production.
In summary, to our knowledge we report, for the first time, that intestinal inflammation can trigger catecholamine synthesis by inducing TH and DBH expression in LPMCs and that catecholamines contribute to the development of experimental colitis; moreover, blockade of α2-adrenoreceptors can ameliorate acute TNBS-induced colitis and DSS colitis. Consequently, the use of α2-adrenoreceptor antagonists or blocking its signalling pathway represents a novel therapeutic approach for the management of IBD.
Acknowledgments
This work was supported by grants from the National Natural Scientific Foundation of China (30860108 to Aiping Bai). The guarantor of the article was Aiping Bai MD.
Disclosure
Potential competing interests: none.
References
- 1.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Irving PM, Shanahan F, Rampton DS. Drug interactions in inflammatory bowel disease. Am J Gastroenterol. 2008;103:207–19. doi: 10.1111/j.1572-0241.2007.01559.x. [DOI] [PubMed] [Google Scholar]
- 4.de Boer NK, van Bodegraven AA, Jharap B, de Graaf P, Mulder CJ. Drug insight: pharmacology and toxicity of thiopurine therapy in patients with IBD. Nat Clin Pract Gastroenterol Hepatol. 2007;4:686–94. doi: 10.1038/ncpgasthep1000. [DOI] [PubMed] [Google Scholar]
- 5.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–9. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nature Rev Immunol. 2006;6:318–28. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Westerloo DJ, Giebelen IA, Florquin S, et al. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology. 2006;130:1822–30. doi: 10.1053/j.gastro.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 8.Huston JM, Gallowitsch-Puerta M, Ochani M, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med. 2007;35:2762–8. doi: 10.1097/01.CCM.0000288102.15975.BA. [DOI] [PubMed] [Google Scholar]
- 9.Mioni C, Bazzani C, Giuliani D, et al. Activation of an efferent cholinergic pathway produces strong protection against myocardial ischemia/reperfusion injury in rats. Crit Care Med. 2005;33:2621–8. doi: 10.1097/01.ccm.0000186762.05301.13. [DOI] [PubMed] [Google Scholar]
- 10.Rouppe van der Voort C, Kavelaars A, van de Pol M, Heijnen CJ. Noradrenaline induces phosphorylation of ERK-2 in human peripheral blood mononuclear cells after induction of alpha(1)-adrenergic receptors. J Neuroimmunol. 2000;108:82–91. doi: 10.1016/s0165-5728(00)00253-8. [DOI] [PubMed] [Google Scholar]
- 11.Miksa M, Wu R, Zhou M, Wang P. Sympathetic excitotoxicity in sepsis: pro-inflammatory priming of macrophages by norepinephrine. Front Biosci. 2005;1:2217–29. doi: 10.2741/1691. [DOI] [PubMed] [Google Scholar]
- 12.Hori T, Katafuchi T, Take S, Shimizu N, Niijima A. The autonomic nervous system as a communication channel between the brain and the immune system. Neuroimmunomodulation. 1995;2:203–15. doi: 10.1159/000097198. [DOI] [PubMed] [Google Scholar]
- 13.Lindgren S, Stewenius J, Sjölund K, Lilja B, Sundkvist G. Autonomic vagal nerve dysfunction in patients with ulcerative colitis. Scand J Gastroenterol. 1993;28:638–42. doi: 10.3109/00365529309096103. [DOI] [PubMed] [Google Scholar]
- 14.Jönsson M, Norrgård O, Forsgren S. Presence of a marked nonneuronal cholinergic system in human colon: study of normal colon and colon in ulcerative colitis. Inflamm Bowel Dis. 2007;13:1347–56. doi: 10.1002/ibd.20224. [DOI] [PubMed] [Google Scholar]
- 15.Fujii T, Takada-Takatori Y, Kawashima K. Basic and clinical aspects of non-neuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy. J Pharmacol Sci. 2008;106:186–92. doi: 10.1254/jphs.fm0070109. [DOI] [PubMed] [Google Scholar]
- 16.Furlan R, Ardizzone S, Palazzolo L, et al. Sympathetic overactivity in active ulcerative colitis: effects of clonidine. Am J Physiol Regul Integr Comp Physiol. 2006;290:R224–32. doi: 10.1152/ajpregu.00442.2005. [DOI] [PubMed] [Google Scholar]
- 17.Flierl MA, Rittirsch D, Nadeau BA, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449:721–5. doi: 10.1038/nature06185. [DOI] [PubMed] [Google Scholar]
- 18.Spengler RN, Allen RM, Remick DG, Strieter RM, Kunkel SL. Stimulation of alpha-adrenergic receptor augments the production of macrophage derived tumor necrosis factor. J Immunol. 1990;145:1430–4. [PubMed] [Google Scholar]
- 19.Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL. Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J Immunol. 1994;152:3024–31. [PubMed] [Google Scholar]
- 20.Starkie RL, Rolland J, Febbraio MA. Effect of adrenergic blockade on lymphocyte cytokine production at rest and during exercise. Am J Physiol Cell Physiol. 2001;281:C1233–40. doi: 10.1152/ajpcell.2001.281.4.C1233. [DOI] [PubMed] [Google Scholar]
- 21.Flierl MA, Rittirsch D, Huber-Lang M, Sarma JV, Ward PA. Catecholamines – crafty weapons in the inflammatory arsenal of immune/inflammatory cells or opening Pandora's box? Mol Med. 2008;14:195–204. doi: 10.2119/2007-00105.Flierl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borger P, Hoekstra Y, Esselink MT, et al. Beta-adrenoceptor-mediated inhibition of IFN-gamma, IL-3, and GM-CSF mRNA accumulation in activated human T lymphocytes is solely mediated by the beta2-adrenoceptor subtype. Am J Respir Cell Mol Biol. 1998;19:400–7. doi: 10.1165/ajrcmb.19.3.2765. [DOI] [PubMed] [Google Scholar]
- 23.Kolachala VL, Vijay-Kumar M, Dalmasso G, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology. 2008;135:861–70. doi: 10.1053/j.gastro.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai A, Lu N, Guo Y, Fan X. Tanshinone IIA ameliorates trinitrobenzene sulfonic acid (TNBS)-induced murine colitis. Dig Dis Sci. 2008;53:421–8. doi: 10.1007/s10620-007-9863-8. [DOI] [PubMed] [Google Scholar]
- 25.Santucci L, Fiorucci S, Rubinstein N, et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology. 2003;124:1381–94. doi: 10.1016/s0016-5085(03)00267-1. [DOI] [PubMed] [Google Scholar]
- 26.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 27.Murano M, Maemura K, Hirata I, et al. Therapeutic effect of intracolonically administered nuclear factor-κB (p65) anti-sense oligonucleotide on mouse dextran sulphate sodium (DSS)-induced colitis. Clin Exp Immunol. 2000;120:51–8. doi: 10.1046/j.1365-2249.2000.01183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith P, Mangan NE, Walsh CM, et al. Infection with a helminth parasite prevents experimental colitis via a macrophage-mediated mechanism. J Immunol. 2007;178:4557–66. doi: 10.4049/jimmunol.178.7.4557. [DOI] [PubMed] [Google Scholar]
- 29.Bai A, Hu P, Chen J, et al. Blockade of STAT3 by antisense oligonucleotide in TNBS-induced murine colitis. Int J Colorect Dis. 2007;22:625–35. doi: 10.1007/s00384-006-0229-z. [DOI] [PubMed] [Google Scholar]
- 30.Bai A, Guo Y, Lu N. The effect of the cholinergic anti-inflammatory pathway on experimental colitis. Scand J Immunol. 2007;66:538–45. doi: 10.1111/j.1365-3083.2007.02011.x. [DOI] [PubMed] [Google Scholar]
- 31.Kiba T. Relationships between the autonomic nervous system, humoral factors and immune functions in the intestine. Digestion. 2006;74:215–27. doi: 10.1159/000100512. [DOI] [PubMed] [Google Scholar]
- 32.Di Comite G, Grazia Sabbadini M, Corti A, Rovere-Querini P, Manfredi AA. Conversation galante: how the immune and the neuroendocrine systems talk to each other. Autoimmun Rev. 2007;7:23–9. doi: 10.1016/j.autrev.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Sanovic S, Lamb DP, Blennerhassett MG. Damage to the enteric nervous system in experimental colitis. Am J Pathol. 1999;155:1051–7. doi: 10.1016/S0002-9440(10)65207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerebours E, Gower-Rousseau C, Merle V, et al. Stressful life events as a risk factor for inflammatory bowel disease onset: a population-based case–control study. Am J Gastroenterol. 2007;102:122–31. doi: 10.1111/j.1572-0241.2006.00931.x. [DOI] [PubMed] [Google Scholar]
- 35.Graff LA, Walker JR, Lix L, et al. The relationship of inflammatory bowel disease type and activity to psychological functioning and quality of life. Clin Gastroenterol Hepatol. 2006;4:1491–501. doi: 10.1016/j.cgh.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 36.Bergquist J, Tarkowski A, Ekman R, Ewing A. Discovery of endogenous catecholamines in lymphocytes and evidence for catecholamine regulation of lymphocyte function via an autocrine loop. Proc Natl Acad Sci USA. 1994;91:12912–16. doi: 10.1073/pnas.91.26.12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jönsson M, Norrgård O, Forsgren S. Presence of a marked nonneuronal cholinergic system in human colon: study of normal colon and colon in ulcerative colitis. Inflamm Bowel Dis. 2007;13:1347–56. doi: 10.1002/ibd.20224. [DOI] [PubMed] [Google Scholar]
- 38.Ghia JE, Blennerhassett P, Kumar-Ondiveeran H, Verdu EF, Collins SM. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology. 2006;131:1122–30. doi: 10.1053/j.gastro.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 39.Romero-Sandoval EA, McCall C, Eisenach JC. Alpha2-adrenoceptor stimulation transforms immune responses in neuritis and blocks neuritis-induced pain. J Neurosci. 2005;25:8988–94. doi: 10.1523/JNEUROSCI.2995-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kin NW, Sanders VM. It takes nerve to tell T and B cells what to do. J Leukoc Biol. 2006;79:1093–104. doi: 10.1189/jlb.1105625. [DOI] [PubMed] [Google Scholar]
- 41.Haskó G, Elenkov IJ, Kvetan V, Vizi ES. Differential effect of selective block of alpha 2-adrenoreceptors on plasma levels of tumour necrosis factor-alpha, interleukin-6 and corticosterone induced by bacterial lipopolysaccharide in mice. J Endocrinol. 1995;144:457–62. doi: 10.1677/joe.0.1440457. [DOI] [PubMed] [Google Scholar]
- 42.Straub RH, Wiest R, Strauch UG, Härle P, Schölmerich J. The role of the sympathetic nervous system in intestinal inflammation. Gut. 2006;55:1640–9. doi: 10.1136/gut.2006.091322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang BY, Lee SW, Kim TS. Stimulation of interleukin-12 production in mouse macrophages via activation of p38 mitogen-activated protein kinase by alpha2-adrenoceptor agonists. Eur J Pharmacol. 2003;467:223–31. doi: 10.1016/s0014-2999(03)01628-5. [DOI] [PubMed] [Google Scholar]
- 44.Szelényi J, Kiss JP, Puskás E, Szelényi M, Vizi ES. Contribution of differently localized alpha 2- and beta-adrenoceptors in the modulation of TNF-alpha and IL-10 production in endotoxemic mice. Ann NY Acad Sci. 2000;917:145–53. doi: 10.1111/j.1749-6632.2000.tb05378.x. [DOI] [PubMed] [Google Scholar]
- 45.Ignatowski TA, Spengler RN. Regulation of macrophage-derived tumor necrosis factor production by modification of adrenergic receptor sensitivity. J Neuroimmunol. 1995;61:61–70. doi: 10.1016/0165-5728(95)00074-c. [DOI] [PubMed] [Google Scholar]
- 46.Cupic V, Colic M, Pavicic L, Vucevic D, Varagic VM. Immunomodulatory effect of xylazine, an alpha(2) adrenergic agonist, on rat spleen cells in culture. J Neuroimmunol. 2001;113:19–29. doi: 10.1016/s0165-5728(00)00370-2. [DOI] [PubMed] [Google Scholar]
- 47.Felsner P, Hofer D, Rinner I, Porta S, Korsatko W, Schauenstein K. Adrenergic suppression of peripheral blood T cell reactivity in the rat is due to activation of peripheral alpha 2-receptors. J Neuroimmunol. 1995;57:27–34. doi: 10.1016/0165-5728(94)00158-k. [DOI] [PubMed] [Google Scholar]
- 48.Bode H, Schmitz H, Fromm M, Scholz P, Riecken EO, Schulzke JD. IL-1beta and TNF-alpha, but not IFN-alpha, IFN-gamma, IL-6 or IL-8, are secretory mediators in human distal colon. Cytokine. 1998;10:457–65. doi: 10.1006/cyto.1997.0307. [DOI] [PubMed] [Google Scholar]
- 49.Kucharzik T, Lügering N, Adolf M, Domschke W, Stoll R. Synergistic effect of immunoregulatory cytokines on peripheral blood monocytes from patients with inflammatory bowel disease. Dig Dis Sci. 1997;42:805–12. doi: 10.1023/a:1018872332387. [DOI] [PubMed] [Google Scholar]
- 50.Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- 51.Meijer MJ, Mieremet-Ooms MA, van Duijn W, et al. Effect of the anti-tumor necrosis factor-alpha antibody infliximab on the ex vivo mucosal matrix metalloproteinase–proteolytic phenotype in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:200–10. doi: 10.1002/ibd.20051. [DOI] [PubMed] [Google Scholar]
- 52.Zhou M, Das P, Simms HH, Wang P. Gut derived norepinephrine plays an important role in up-regulating IL-1beta and IL-10. Biochim Biophys Acta. 2005;1740:446–52. doi: 10.1016/j.bbadis.2004.11.005. [DOI] [PubMed] [Google Scholar]