

Abstract
We studied the electronically excited state of the isolated reaction center of photosystem II with high-resolution fluorescence spectroscopy at 5 K and compared the obtained spectral features with those obtained earlier for the primary electron donor. The results show that there is a striking resemblance between the emitting and charge-separating states in the photosystem II reaction center, such as a very similar shape of the phonon wing with characteristic features at 19 and 80 cm−1, almost identical frequencies of a number of vibrational modes, a very similar double-Gaussian shape of the inhomogeneous distribution function, and relatively strong electron-phonon coupling for both states. We suggest that the emission at 5 K originates either from an exciton state delocalized over the inactive branch of the photosystem or from a fraction of the primary electron donor that is long-lived at 5 K. The latter possibility can be explained by a distribution of the free energy difference of the primary charge separation reaction around zero. Both possibilities are in line with the idea that the state that drives primary charge separation in the reaction center of photosystem II is a collective state, with contributions from all chlorophyll molecules in the central part of the complex.
Keywords: fluorescence line narrowing, chlorophyll a
The primary reactions of photosynthetic energy conversion can be divided into three processes. The first is the absorption of (sun)light by one of the antenna pigments, typically a chlorophyll or a carotenoid, specifically bound to a protein embedded in the thylakoid membranes. The second is given by the rapid transfer of the electronically excited state of the irradiated pigment to a nearby chlorophyll. Usually several of these excitation energy transfer reactions are required until a so-called reaction center (RC) is reached. In most types of photosynthetic organisms this process takes about a few hundred ps (1). The third process is the fast transfer of an electron from the electronically excited RC chlorophyll to a nearby acceptor. This primary charge separation subsequently is stabilized by secondary electron transfer reactions and ultimately used for chemical energy fixation.
In green plant photosystem II (PSII) the primary charge separation reaction involves the transfer of an electron within a few tens of ps from a chlorophyll a (Chl-a) species, here referred to as P, to a pheophytin a (Pheo-a) molecule. The mechanism of primary charge separation in PSII is of particular interest, because P+ is able to bring about the oxidation of water to molecular oxygen (2). The smallest PSII unit in which primary charge separation has been observed is the isolated D1D2-cytochrome b559 complex, also called the PSII RC complex (2, 3). In this complex the mechanism of primary charge separation can be studied relatively well because of the small number of pigments bound [in most cases two Pheo-a, one or two β-carotene, and six Chl-a molecules (4), or five Chl-a molecules (5, 6) per complex]. Furthermore, secondary electron transfer reactions do not occur in these isolated complexes. The separated charges ultimately will recombine again in about 100 ns, generating predominantly the triplet state of P (2, 3).
In the absence of detailed structural information on the PSII RC several types of organization of the Chl-a and Pheo-a have been proposed to explain the primary photochemistry (7–12). All recent proposals have in common that they assume a central core part with the two Pheo-a molecules and at least some of the Chl-a molecules in similar positions and orientations as in the related purple bacterial RC of which the structure is well known (13). In addition, one or two other Chl-a molecules are bound at the periphery.
Most models assume that P is a dimer of two weakly coupled “special-pair”-like Chl-a and that excitonic coupling with and between the other Chl-a and Pheo-a is negligible (7–11). In these models all pigments except P operate as separate entities to trap the excitation energy. The excitation energy is first transferred from these monomeric entities to P, after which the singlet-excited P dimer induces the primary charge separation reaction. A key feature of all of these models (7–11) is that the absorption around 680–684 nm originates not only from the P dimer, but also from at least one of the monomeric Chl-a or Pheo-a. The putative red monomeric Chl-a sometimes is referred to as a “linker” (14) of excitation energy between the antenna and P Chl-a. Within the context of this model, the red monomeric pigments will be primarily responsible for the emission at low temperatures, because P will almost exclusively give rise to the fast primary charge separation reaction at low temperatures (15, 16) and therefore will not significantly contribute to the steady-state emission. The emission was shown to be dominated by a lifetime of 4 ns at very low temperatures (17).
A rather different view was proposed by Durrant and coauthors (12), who argued that not only excitonic coupling between the special-pair-like Chl-a should be taken into account, but also the coupling with and between some of the other pigments and the energetic disorder, which all are in the order of 100 cm−1 (12). This view implies that the complete core of the PSII RC (presumably the four central Chl-a and the two Pheo-a) should be regarded as a multimer of weakly coupled pigments, and that a collective excited state drives the primary charge separation reaction. The considerable disorder implies that the lowest collective excited state will be delocalized to very different extents over the central pigments in every individual RC (12). A variety of experiments were successfully explained within the context of this model (18, 19). Key features of the multimer model are that all absorption around 680–684 nm arises from the central core pigments (6), and that only a part of these “red” states include P. The red multimer states that do not include P will be primarily responsible for the emission at low temperatures.
In this study we focus on the emission of the isolated PSII RC complex at 5 K, with the aim to deduce whether the emitting states arise from monomeric pigments or from excitonically coupled states, and thus whether a dimer + monomers model or the multimer model gives the best description of the PSII RC. The emitting states have been studied before by selectively excited fluorescence at nanometer resolution (20, 21) and by spectral hole burning (17). Here we apply fluorescence line narrowing (22) to isolated PSII RC complexes containing six Chl-a per two Pheo-a. This technique recently was applied to the major trimeric Chl-a/b binding light-harvesting complex II of higher plants (23), and enables us to obtain low-temperature emission spectra with much higher resolution than obtained before. The results are fully consistent with the idea that collective excited states play a crucial role in the functioning of the PSII RC.
MATERIALS AND METHODS
Sample Preparation.
PSII RC complexes containing six Chl-a per two Pheo-a molecules were isolated from spinach by using a short Triton X-100 treatment of CP47-RC complexes as described before (4, 15). FPLC gel filtration (24) was used to make sure that the preparation was free of CP47 and other contaminating pigment-protein complexes. Samples were dissolved in a buffer containing 20 mM BisTris (pH 6.5), 20 mM NaCl, 0.06% (wt/vol) N-dodecyl-β,d-maltoside, and 80% (vol/vol) glycerol. All experiments were performed at 5 K by using a helium bath cryostat (Utreks, Maice, Tartu, Estonia).
High-Resolution Fluorescence Measurements.
High-resolution fluorescence emission spectra were recorded as described before (23) with a charge-coupled device camera via a ½-m spectrograph. The bandwidth of detection was 0.25 nm, recording fluorescence every 0.035 nm. Emission spectra were corrected for the sensitivity of the detection system. For nonselective excitation we used the combination of a lamp and a bandpass filter, for selective excitation from 640–710 nm a dye laser was used (spectral bandwidth 1 cm−1). The laser power was kept below 0.2 mW/cm2. The typical illumination time was 1 min per excitation wavelength. Optical densities of ≈0.1 cm−1 (in the Qy maximum) were used (unless stated differently).
RESULTS AND DISCUSSION
Nonselectively Excited Emission Spectra.
To characterize the low-temperature (5 K) emission of the PSII RC in detail we first recorded the nonselectively excited [broadband, full width at half maximum (fwhm) 15 nm, at 487 nm] emission spectrum at high resolution (Fig. 1). The spectrum peaks at 683.7 nm, has a fwhm of 8.2 nm, and is similar to that published before for the same preparation (20). The spectrum does not change on excitation at other wavelengths (up to 610 nm, not shown). Interestingly, the main emission band appears to have a strongly asymmetric shape (Fig. 1, Inset), a feature that has not been noted before. This feature has important implications for the interpretation of the emission spectrum (see below).
Figure 1.

Nonselectively excited emission spectrum of PSII RC at 5 K. Excitation was broad-banded (15 nm fwhm) at 487 nm. The spectral bandwidth of detection was 0.25 nm.
Selectively Excited Emission Spectra.
The nonselective emission spectrum (Fig. 1) is the convolution of the inhomogeneous distribution function (IDF) and the emission spectrum of the individual RCs (single-site spectrum). The inherent glass-like disorder of the protein implies that the optical transitions in the protein are distributed, usually referred to as inhomogeneous broadening (22). Because of this broadening, the fine structure of the single-site spectrum (generally prominent at liquid helium temperature) is lost. The single-site spectrum itself consists of a relatively narrow zero-phonon line (ZPL), caused by the purely electronic transition, and of a broader wing, the so-called phonon wing (PW), at lower energy (in fluorescence) than the ZPL. In a pigment-protein complex phonons can be interpreted as rearrangements of (part of) the protein backbone (25). The relative intensity of the PW with respect to the ZPL is expressed by the Huang-Rhys factor (S), which is minus the natural logarithm of the area fraction of the ZPL in the total spectrum (22). Generally, in a single-site spectrum less intense repeats of the ZPL/PW structure are observed to lower energy than the main ZPL. These repeats are caused by combined electronic, phonon, and vibrational transitions (22, 25). These transitions show up as peaks (vibronic ZPLs, vZPLs) separated with the vibrational frequency from the purely electronic ZPL and provide valuable information on the vibrational modes of the emitting pigments (22).
A technique to overcome inhomogeneous broadening and to obtain high-resolution single-site emission spectra is fluorescence line narrowing (22). In this technique sub-nm resolution emission spectra are recorded on narrow-bandwidth (≈cm−1) continuous-wave laser excitation. When energy transfer reactions can be avoided by exciting the sample at low temperature in the red edge of the absorption spectrum, a narrow distribution of sites is excited, leading to fluorescence line narrowing.
In Fig. 2 some selectively excited emission spectra are shown for PSII RC at 5 K. Note that the x axis in Fig. 2 is the excitation frequency minus the emission frequency (in wavenumbers). The spectra in Fig. 2 are much more structured than those published before (20, 21). This increased line structure is probably because of the better sensitivity, the increased spectral resolution, and the much lower light doses (a factor of about 1,000 less) used for the present experiments.
Figure 2.

Selectively excited emission spectrum of PSII RC at 5 K. The spectral bandwidth of detection was 0.25 nm. Excitation light was provided with a laser (bandwidth 1 cm−1) at 680.1, 681.3, 682.1, 682.9, and 684.0 nm.
The peak at 0 cm−1 is to some extent caused by scattered excitation light, and to some extent by pure electronic emission from the ZPL. On increasing the excitation wavelength, the spectra change from a shape similar to the nonselectively excited spectrum (Fig. 1), via shapes with several peaks in the 0–150 cm−1 region (Fig. 2, upper traces), to a characteristic shape (λexc > 684 nm) with a peak at 19 cm−1 and a large number of sharp peaks up to about 1,700 cm−1 (Fig. 2, lower traces), which are independent of the excitation wavelength. The rich fine structure up to about 1,700 cm−1 is because of a large number of vZPLs. The broader wing with features at 19, 37, and 80 cm−1 represents the PW.
The variation of the shape of the emission spectrum can, in principle, be the result of down-hill energy transfer processes. However, we have shown before by anisotropy measurements (26) that with λexc > 680 nm excitation energy transfer does not occur to a significant extent at 5 K. Absorption by the relatively broad PW also may explain why the shape of the emission spectrum varies as a function of excitation wavelength. At “shorter” excitation wavelengths (λexc < 684 nm) light is not only absorbed in the ZPL, but to an increasing extent also in the PW. We will present simulations of the wavelength-dependence of the emission spectra, which indicate that the variable shape of the emission spectrum is indeed caused by the variable contribution of the PW in the absorption spectrum.
Vibronic Fine Structure.
In Fig. 3 the red-excited line-narrowed emission spectrum of PSII RC at 5 K is shown in detail. The spectrum is an average of five almost identical spectra, excited at different wavelengths (684.0–686.1 nm). These red-excited spectra are very close to the single-site emission spectrum, because the absorption is almost entirely caused by ZPLs in this part of the spectrum. They were recorded by using a relatively high OD (≈0.5 cm−1 in the Qy maximum) to obtain maximal signal quality.
Figure 3.

Line-narrowed emission spectra of PSII RC at 5 K. Measuring conditions are the same as in Fig. 2. The spectrum is the average of five spectra, excited at 684.0–686.1 nm. Also shown is the vibronic region (×5 magnification, upper spectrum). (Insets) Magnification of the PW and the 1,500–1,720 cm−1 region.
The sharp lines in Fig. 3 are caused by vZPLs (0,x ← 1,0 transitions) without contribution of phonons. All fine structure is lost on heating to 60 K (not shown), which proves that the vZPLs are not caused by resonance Raman scattering. Sixty-eight vZPLs can be discerned, the frequencies of which are independent of the excitation wavelength. Fig. 3 (Inset) shows an enlargement of the 1,500–1,720 cm−1 region. This region is of interest, because it comprises the 131 C=O stretch mode of Chl-a (1,650–1,700 cm−1) (27), several chlorin C=C stretch modes (≈1,610, ≈1,550, and ≈1,520–1,530 cm−1) (28) and a characteristic mode of Pheo-a at ≈1,585 cm−1 (29).
In our spectra a contribution at 1,587 cm−1 can be observed. This contribution is, however, small compared with that of isolated Pheo-a (30) and about similar in intensity to that observed before in spectra of light-harvesting complex II (23), which does not contain Pheo-a. We conclude that the contribution of Pheo-a to the selectively excited emission spectrum at 5 K is at most 10%. This observation is in line with earlier, lower-resolution fluorescence measurements (20, 21).
In the inset of Fig. 3 a band at 1,669 cm−1 and a shoulder at 1,660 cm−1 can be discerned, which can be assigned to the 131 C=O stretch mode of Chl-a (27). The fact that we observe two frequencies might indicate that the emitting state is delocalized over several Chl-a, that the emission occurs from more than one state, or that the emitting Chl-a can be present in slightly different environments. Both frequencies (1,660 and 1,669 cm−1) indicate that the keto groups are hydrogen-bonded, otherwise the frequencies would be higher (1,680–1,700 cm−1) (27).
The frequencies of the chlorin C=C stretch modes at 1,536, 1,555, and 1612 cm−1 in Fig. 3 indicate that one axial (protein) ligand is bound to the central magnesium. In case of two axial ligands the frequencies of these modes would be ≈10 cm−1 lower (28).
It has been shown with Raman and Fourier transform infrared spectroscopy (30, 31) that the Pheo-a in the PSII RC have 131 C=O stretch frequencies at ≈1,680 and ≈1,700 cm−1. The 131 C=O stretch modes in our emission spectra are at 1,669 and 1,660 cm−1. The different energies of these stretch modes provide additional proof that the emission at 5 K is not because of Pheo-a. Chl-a-selective Raman spectra of PSII RC showed a broad 131 C=O stretch band centered at 1,674 cm−1 with a shoulder at about 1,660 cm−1 (30). We note that these bands are caused by all of the Chl-a in the complex, whereas our fluorescence experiments only detect the emitting Chl-a. We also note that the relative intensities of the various vibrational bands vary between the resonance Raman and the fluorescence experiments. This difference is caused by the different way of excitation [the Raman experiments have been carried out with Soret excitation (30)], and by the presence of bands caused by β-carotene in the Raman spectra.
Resonance Raman (32) and infrared spectroscopy (10) also have been used to characterize the vibrational differences between the triplet state and the ground state of P, revealing a 131 C=O stretch at ≈1,670 cm−1 and other bands at 1,617 (32), 1,556, and 1,539 cm−1 (10). These frequencies attributed to P are very similar to the ones we observe for the emitting state.
The PW.
The PW of the emission from PSII RC at 5 K (Fig. 3) is quite structured with a sharp main peak at 19 cm−1. A shoulder is present at 37 cm−1, which is most probably caused by the first overtone of the 19 cm−1 mode. At ≈80 cm−1 a relatively broad feature is present, which most likely reflects an extra low-frequency mode. This PW is remarkably similar to the PW used by Kwa and coworkers (26) to simulate site-selected triplet-minus-singlet absorbance difference spectra, which are caused mainly by P. For a good description of the data they had to include a mode at about 80 cm−1. This mode was not observed in similar experiments by Small and coworkers (14, 33), but is clearly present in our spectra. It is remarkable that we have not observed such a mode in similar experiments on the light-harvesting complex II of green plants (23), and on the single Chl-a of the cytochrome b6f complex (34), which indicates that this 80-cm−1 feature is typical for the PSII RC. It might be that it is a frequency of the PSII RC protein phonon bath, at higher frequency than the most intense protein phonon mode at 19 cm−1. Another explanation [as already put forward by Kwa et al. (26)] is that it might be an analogue of the “marker” mode at 115–135 cm−1 observed for the primary donor of bacterial RCs, which is believed to be caused by an intermolecular vibration of the two strongly coupled bacteriochlorophylls forming the primary donor in bacterial RCs (35). The observation that the 80-cm−1 mode of PSII occurs at lower frequency and with less intensity than the marker mode of the bacterial RC then is not unexpected, in view of the smaller coupling strength. If this latter interpretation of the 80-cm−1 mode is correct, it has a very important consequence: both the charge-separating and the emitting state of the PSII RC are caused by two or more excitonically coupled pigments.
Simulation of the Selectively Excited Emission Spectra.
To obtain information on S, characterizing the total electron-phonon coupling strength, and the IDF, we simulated the selectively and nonselectively excited emission spectra of PSII RC at 5 K (see also refs. 14, 25, and 26). The input parameters for these calculations were S, parameters defining the IDF, and the red-excited emission spectrum. We deleted the scatter line/ZPL from the experimental “single-site” emission spectrum (Fig. 3) because of its composite character. The resulting truncated spectrum [Emred(ν)] thus consists of the PW and the vibronic emission. We inserted the ZPL as a delta function (δ(ν)) normalized to unity area and added the PW with relative area e+S−1. The use of a delta function as ZPL is justified within the accuracy of our method.
For the calculation of the relative amount of PW from S we assumed that phonon contributions extend up to 500 cm−1 from the ZPL. We note that a proper discrimination between phonon and vibrational contributions to the single-site emission spectrum cannot be made and that the 500-cm−1 limit is a somewhat arbitrary choice. Consequently, this procedure leads to a summed value of S, including all modes in the 0–500 cm−1 region, and thus to a PW characterized by a relatively broad distribution of modes (25). The effect of the exact choice of this interval on the simulations appeared to be small, well within the error margins.
The single-site emission spectrum [EmSS(ν)] used for the simulations is given by:
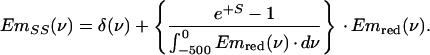 |
1 |
The part between brackets is the scaling of the PW with respect to the ZPL. The selectively excited emission spectra, Em(ν,νexc), were calculated by using (25):
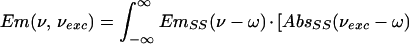 |
2 |
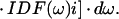 |
Eq. 2 is a convolution integral of the distribution of ZPLs of the excited molecules (the part between brackets) with the single-site emission spectrum.
We numerically calculated the absorption, nonselectively and selectively excited emission spectra with a stepsize of 1 cm−1. The results are shown in Figs. 4 and 5. The best overall agreement of simulated and experimental spectra was obtained with an IDF consisting of two Gaussians, one peaking at 683.6 nm (width 70 cm−1, fwhm), the other at 680.6 nm (width 80 cm−1, relative area 0.91). This form of IDF was required to reproduce the shape of the main peak of the nonselective emission spectrum; a single-Gaussian IDF failed to reproduce the strongly asymmetric shape of the peak at 683.7 nm (Fig. 4, Inset). We emphasize that an IDF with very similar parameters has been used by Kwa and coworkers (26) to simulate their selectively excited triplet-minus-singlet absorption difference spectra, which primarily monitored P. The double-Gaussian IDF might be a reflection of heterogeneity (6, 26). However, a more attractive idea is that the shape of the IDF is influenced by a variation of the dipole strength over the band. Exciton calculations based on the multimer model indeed have predicted such a variation of the dipole strengths and transition energies from RC to RC (12).
Figure 4.

Experimental (solid line, as in Fig. 1) and simulated (dotted line) nonselectively excited emission spectrum of PSII RC. Parameters of the simulation: S = 1.6; IDF, two Gaussians at 683.6, width 70 cm−1 (fwhm) and at 680.6 nm, width 80 cm−1 (fwhm) with relative (area) contribution of 0.91. (Inset) A magnification of the main peak. A simulation (dashed line) is shown with a single Gaussian at 681.7 nm, width 110 cm−1 (fwhm) as IDF and S = 1.6. It should be noted that below 685 nm the dotted and solid curve and above 685 nm the dashed and dotted curves almost coincide. For details see text.
Figure 5.

Experimental (solid line) and simulated (dotted and dashed lines) emission spectra selectively excited at 680.1, 681.3, 682.1, 682.9, and 684.0 nm. The IDF used for the simulations consists of two Gaussians at 683.6, width 70 cm−1 (fwhm) and at 680.6 nm, width 80 cm−1 (fwhm) with a relative (area) contribution of 0.91. (A) S = 1.6. (B) S = 1.2 (dotted line) and 2.0 (dashed line). For details see text.
The best agreement between experimental spectra and simulations was obtained with a value for S of about 1.6 for both nonselective excitation (Fig. 4) and selective excitation (Fig. 5 A and B). In the nonselective emission spectrum (Fig. 4) a lower (higher) value of S leads to relatively too little (much) intensity in the vibronic region (≈710–780 nm). In the selectively excited emission spectra simulations with values for S of 1.2 and 2.0 fail to describe the wavelength dependence of the emission spectra (Fig. 5B).
To conclude: the simulations of the nonselectively excited absorption and emission spectra and of the selectively excited emission spectra show that the S of the total coupling of phonons to the emitting state in PSII RC is 1.6 +/− 0.3. This value of S is significantly higher than the value of 0.7, which was obtained from hole-burning experiments of the emitting state of the same PSII RC preparation (17). In this study a limited part of the PW (up to about 100 cm−1) was used for the determination of S, which explains most of the difference. This value of S is also significantly higher than the value of about 0.8 usually observed for regular antenna chlorophylls (36). Also the single Chl-a in the cytochrome b6f complex was shown to give a low value of S (34). For the primary donor P, however, values for S of about 1.9 have been found from selectively excited triplet-minus-singlet absorption difference spectra (14, 26). The relatively strong electron-phonon coupling of the emitting and charge-separating states of the PSII RC indicates a larger than usual reorganization of the protein environment on (de)excitation of these states.
CONCLUDING REMARKS
The most striking result of our study of PSII RC is the pronounced similarity between the spectroscopic features of the emitting and charge separating states. These similarities include the shape of the PW with the 80-cm−1 feature (Results and Discussion and ref. 26), the frequencies of the vibrational modes (Results and Discussion and refs. 30 and 32), the shape of the IDF (Results and Discussion and ref. 26), and the strength of the electron-phonon coupling (Results and Discussion and ref. 26). We stress that most of these parameters are essentially different for Chl-a in other complexes, such as light-harvesting complex II (23) and cytochrome b6f (34).
The similarity of these features cannot easily be explained in terms of a dimer + monomers model. In such a model, most of the emission is expected to originate from a red monomeric Chl-a or Pheo-a. It is not clear how these monomeric pigments can give rise to almost exactly the same heterogeneity of IDF, shape of PW, vibrational frequencies, and value of S as a dimeric primary electron donor.
On the other hand, the multimer model predicts that in the PSII RC two red-most exciton states may be expected (12, 18), which are very close in energy and are localized on one of the two branches of the RC. The state localized on the active branch pigments is believed to be P, the other is thought to be responsible for the low-temperature “trap” of excitation energy (37). Based on the observed similarities between the trap and P, the excitonic states should be symmetric, especially with respect to the protein environment of the pigments. Based on the delocalization of the exciton states a significant contribution of Pheo-a to the emission would be expected (provided that the site energies of the Chl-a and Pheo-a are similar). Such a contribution is not observed in our spectra, which suggests that the site energies of the Pheo-a are different from those of the Chl-a.
Another possibility is that the emission at 5 K arises from P itself. In other words, the (singlet) excited state of P may be long-lived in part of the complexes at very low temperature. This possibility would in a straightforward way explain why the detailed features of the states responsible for emission and charge separation are spectrally very similar. It would imply that P is unable to perform charge separation in part of the RCs at 5 K, which can be understood in terms of a distribution of the free energy difference of the primary charge separation around zero (15, 38). Then, in part of the complexes charge separation would be activated, and thus impossible at very low temperature, leading to long-lived fluorescence. In other complexes, however, charge separation would not be significantly activated and proceed fast at 5 K, in line with experimental results (15, 33). In the dimer + monomers model P can be responsible only for the emission if all accessory pigments absorb well below 680 nm. This is not in agreement with the absorption spectrum and the spectral models mentioned before (9, 11). In contrast, in the multimer model there is no need to introduce a red monomeric Chl-a to explain the absorption spectrum.
Recent experiments on the purple bacterial RC have shown that in this related RC the primary charge separation may not be driven only by the excited state of the special pair, but also by the excited state of the accessory bacteriochlorophylls of the active branch (39–41). In intact bacteria, the importance of this alternative and very fast charge separation route may be limited because most excitation energy will originate from the bacteriochlorophylls of the LH1 antenna, which absorb much further to the red than the accessory bacteriochlorophyll of the active branch. In PSII, however, the energetic differences between the antenna and accessory Chl-a are negligible at room temperature, and a significant part of the excitation energy will become localized on the multimer with important contributions from the accessory Chl-a. Thus, delocalization of excitation energy in the central core of the reaction center may provide an excellent tool to promote fast and efficient charge separation.
Acknowledgments
We thank Henny van Roon for isolation of PSII RC. This work was supported by the Netherlands Foundation for Scientific Research (NWO) via the Foundation for Life Sciences (SLW) and the European Union (Contract No. 94 0619).
ABBREVIATIONS
- Chl-a
chlorophyll a
- fwhm
full width at half maximum
- IDF
inhomogeneous distribution function
- Pheo-a
pheophytin a
- PSII
photosystem II
- PW
phonon wing
- RC
reaction center
- S
Huang-Rhys factor
- vZPL
vibronic zero-phonon line
- ZPL
zero-phonon line
References
- 1.Van Grondelle R, Dekker J P, Gillbro T, Sundström V. Biochim Biophys Acta. 1994;1187:1–65. [Google Scholar]
- 2.Diner B A, Babcock G T. In: Oxygenic Photosynthesis: The Light Reactions. Ort D R, Yocum C F, editors. Dordrecht, the Netherlands: Kluwer; 1996. pp. 213–247. [Google Scholar]
- 3.Nanba O, Satoh K. Proc Natl Acad Sci USA. 1987;84:109–112. doi: 10.1073/pnas.84.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eijckelhoff C, Dekker J P. Biochim Biophys Acta. 1995;1231:21–28. [Google Scholar]
- 5.Vacha F, Joseph D M, Durrant J R, Telfer A, Klug D R, Porter G, Barber J. Proc Natl Acad Sci USA. 1995;92:2929–2933. doi: 10.1073/pnas.92.7.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eijckelhoff C, Vacha F, van Grondelle R, Dekker J P, Barber J. Biochim Biophys Acta. 1997;1318:266–274. [Google Scholar]
- 7.Bosch M K, Proskuryakov I I, Gast P, Hoff A J. J Phys Chem. 1995;99:15310–15316. [Google Scholar]
- 8.Svensson B, Etchebest C, Tuffery P, van Kan P, Smith J, Styring S. Biochemistry. 1996;35:14486–14502. doi: 10.1021/bi960764k. [DOI] [PubMed] [Google Scholar]
- 9.Mulkidjanian A Y, Cherepanov D A, Haumann M, Junge W. Biochemistry. 1996;35:3093–3107. doi: 10.1021/bi9513057. [DOI] [PubMed] [Google Scholar]
- 10.Noguchi T, Inoue Y, Satoh K. Biochemistry. 1993;32:7186–7195. doi: 10.1021/bi00079a016. [DOI] [PubMed] [Google Scholar]
- 11.Konermann L, Holzwarth A R. Biochemistry. 1996;35:829–842. doi: 10.1021/bi9513158. [DOI] [PubMed] [Google Scholar]
- 12.Durrant J R, Klug D R, Kwa S L S, van Grondelle R, Porter G, Dekker J P. Proc Natl Acad Sci USA. 1995;92:4798–4802. doi: 10.1073/pnas.92.11.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. J Mol Biol. 1984;180:385–398. doi: 10.1016/s0022-2836(84)80011-x. [DOI] [PubMed] [Google Scholar]
- 14.Chang H C, Jankowiak R, Reddy N R S, Yocum C F, Picorel R, Seibert M, Small G J. J Phys Chem. 1994;98:7725–7735. [Google Scholar]
- 15.Groot M L, van Mourik F, Eijckelhoff C, van Stokkum I H M, Dekker J P, van Grondelle R. Proc Natl Acad Sci USA. 1997;94:4389–4394. doi: 10.1073/pnas.94.9.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jankowiak R, Tang D, Small G J, Seibert M. J Phys Chem. 1989;93:1649–1654. [Google Scholar]
- 17.Groot M L, Dekker J P, van Grondelle R, den Hartog F T H, Völker S. J Phys Chem. 1996;100:11488–11495. [Google Scholar]
- 18.Merry S A P, Kumazaki S, Tachibana Y, Joseph D M, Porter G, Yoshihara K, Barber J, Durrant J R, Klug D R. J Phys Chem. 1996;100:10469–10478. [Google Scholar]
- 19.Leegwater J A, Durrant J R, Klug D R. J Phys Chem B. 1997;101:7205–7210. [Google Scholar]
- 20.Kwa S L S, Tilly N T, Eijckelhoff C, van Grondelle R, Dekker J P. J Phys Chem. 1994;98:7712–7716. [Google Scholar]
- 21.Konermann L, Yruela I, Holzwarth A R. Biochemistry. 1997;36:7498–7502. doi: 10.1021/bi9701484. [DOI] [PubMed] [Google Scholar]
- 22.Personov R I. In: Spectroscopy and Excitation Dynamics of Condensed Molecular Systems. Agranovich V M, Hochstrasser R M, editors. Amsterdam: North–Holland; 1983. pp. 555–619. [Google Scholar]
- 23.Peterman E J G, Pullerits T, van Grondelle R, van Amerongen H. J Phys Chem B. 1997;101:4448–4457. [Google Scholar]
- 24.Eijckelhoff C, van Roon H, Groot M L, van Grondelle R, Dekker J P. Biochemistry. 1996;35:12864–12872. doi: 10.1021/bi960991m. [DOI] [PubMed] [Google Scholar]
- 25.Pullerits T, Monshouwer R, van Mourik F, van Grondelle R. Chem Phys. 1995;194:395–408. [Google Scholar]
- 26.Kwa S L S, Eijckelhoff C, van Grondelle R, Dekker J P. J Phys Chem. 1994;98:7702–7711. [Google Scholar]
- 27.Lutz M, Robert B. In: Biological Applications of Raman Spectroscopy. Spiro T G, editor. New York: Wiley; 1988. pp. 347–411. [Google Scholar]
- 28.Fujiwara M, Tasumi M. J Phys Chem. 1986;90:250–255. and 5646–5650. [Google Scholar]
- 29.Lutz M. J Raman Spectrosc. 1974;2:497–516. [Google Scholar]
- 30.Moënne-Loccoz P, Robert B, Lutz M. Biochemistry. 1989;28:3641–3645. [Google Scholar]
- 31.Nabedryk E, Andrianambinintsoa S, Berger G, Leonhard M, Mäntele W, Breton J. Biochim Biophys Acta. 1990;1016:49–54. [Google Scholar]
- 32.Moënne-Loccoz P, Robert B. In: Current Research in Photosynthesis. Baltscheffsky M, editor. Vol. 1. Dordrecht, the Netherlands: Kluwer; 1990. pp. 423–426. [Google Scholar]
- 33.Chang H C, Jankowiak R, Reddy N R S, Small G J. Chem Phys. 1995;197:307–322. [Google Scholar]
- 34.Peterman, E. J. G., Wenk, S. O., Pullerits, T., Pålsson, L. O., van Grondelle, R., Dekker, J. P., Rögner, M. & van Amerongen, H. (1998) Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- 35.Johnson S G, Tang D, Hayes J M, Small G J, Tiede D M. J Phys Chem. 1990;94:5849–5855. [Google Scholar]
- 36.Jankowiak R, Small G J. In: Photosynthetic Reaction Centers. Deisenhofer J, Norris J, editors. II. New York: Academic; 1993. pp. 133–177. [Google Scholar]
- 37.Groot M-L, Peterman E J G, van Kan P J M, van Stokkum I H M, Dekker J P, van Grondelle R. Biophys J. 1994;67:318–330. doi: 10.1016/S0006-3495(94)80483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konermann L, Gatzen G, Holzwarth A R. J Phys Chem B. 1997;101:2933–2944. [Google Scholar]
- 39.Van Brederode M E, Jones M R, van Mourik F, van Stokkum I H M, van Grondelle R. Chem Phys Lett. 1997;268:143–149. [Google Scholar]
- 40.Van Brederode M E, Jones M R, van Grondelle R. Biochemistry. 1997;36:6855–6861. doi: 10.1021/bi9703756. [DOI] [PubMed] [Google Scholar]
- 41.Vos M H, Breton J, Martin J-L. J Phys Chem B. 1997;101:9820–9832. [Google Scholar]