

Abstract
The phenanthroindolizidine and phenanthroquinolizidine alkaloids, typified by tylophorine and cryptopleurine, are a family of plant-derived small molecules with significant therapeutic potential. The plant extracts have been used in herbal medicine and the isolated compounds have displayed a range of promising therapeutic activity such as anti-ameobicidal, anti-viral, anti-inflammatory and anti-cancer activity. Despite their therapeutic protential, no compounds in this class have fully passed clinical trials. Drawbacks include low in vivo anti-cancer activity, central nervous system toxicity and low natural availability. A number of biological effects of these compounds, such as protein and nucleic acid synthesis suppression, have been identified, but the specific biomolecular targets have not yet been identified. Significant effort has been expended in the synthesis and structure-activity-relationship (SAR) studies of these compounds with the hope that a new drug will emerge. This review will highlight important contributions to the isolation, synthesis, SAR and mechanism of action of the phenanthroindolizidine and pheanthroquinolizidine alkaloids.
INTRODUCTION
Isolation and Structure
The phenanthroindolizidine and phenanthroquinolizine alkaloids are a family of plant-derived alkaloids composed of over sixty compounds Fig. (1) [1]. They have been isolated from the Asclepiadaceae and Moraceae plant families primarily from India and Japan [2]. One of these plants, Tylophora indica, was included as an official drug in the Bengal pharmacoepia of 1884 [3]. The leaves of this plant have been used for the treatment of asthma as well as bronchitis, rheumatism and dysentery in India.
Fig. 1.


Structures of several phenanthroindolizidines and phenanthroquinolizidines.
The medicinal properties of these plants spurred the isolation and characterization of the individual alkaloid constituents. The isolation of the major alkaloid from Tylophora indica, tylophorine, was reported in 1935 [4] and its structure and stereochemistry were reported in 1960 [5]. Tylophorine was initially reported as having the (S)-absolute stereochemistry, but a total synthesis and optical rotation measurement led to the revision to (R) [6].
The most recent comprehensive review on the isolation, synthesis and structure-activity-relationship studies of the phenanthroindolizidine and phenanthroquinolizidine natural products was published by Huang and co-workers in 2001 [1, 2, 7, 8]. This review will highlight the synthetic and medicinal studies that have appeared subsequently.
Therapeutic Potential
The biological properties of these alkaloids range from cancer cell growth inhibition (in vitro and in vivo) [1, 9–16] and anti-inflammatory [3, 17] activity to anti-ameobicidal [18] and anti-viral [19] activity. A summary of their GI50 concentrations in various cancer cell lines is given in Table 1.
Table 1.
GI50 Values of Some Phenanthroindolizidines and Phenanthroquinolizidines in Various Cancer Cell Lines
| Entry | Compound | Cell Typea | GI50 (M) | Reference |
|---|---|---|---|---|
| 1 | (R)-tylophorine | KB | 17 × 10−8 | [12] |
| 2 | (S)-tylophorine | NCI panelb | 1.9 × 10−8 | c |
| 3 | tylocrebrine | NCI panel | 2.95 × 10−8 | c |
| 4 | (R)-cryptopleurine | NCI panel | 1 × 10−12 | c |
| 5 | (R)-antofine | KB | 1.5 × 10−8 | [12] |
| 6 | (+)-tylophorinidine | HepG2 and PANC-1 | 1 × 10−8 | [14] |
| 7 | 7-desmethyltylophorine | KB | 1.5 × 10−8 | [12] |
| 8 | (S)-isotylocrebrine | KB | 4.5 × 10−8 | [12] |
| 9 | secoantofine | KB | 2.5 × 10−6 | [12] |
| 10 | 6-O-desmethylsecoantofine | KB | 4.0 × 10−7 | [12] |
| 11 | tyloindicine G | NCI panel | 1 × 10−10 | c |
| 12 | tyloindicine F | NCI panel | 1 × 10−10 | c |
| 13 | (S)-tyloindicine I | NCI panel | 4.4 × 10−9 | c |
| 14 | 13(R)-14(R)-hydroxyantofine-N-oxide | KB | 1.4 × 10−7 | [11] |
| 15 | 13(R)-antofine-N-oxide | KB | 1.4 × 10−7 | [11] |
| 16 | tylophoridicine C | HepG2 and PANC-1 | 8 × 10−8 | [14] |
| 17 | tylophoridicine F | HepG2 and PANC-1 | 7 × 10−8 | [14] |
Cell Type: KB = human nasopharyngeal carcinoma (average of one or more cell lines), HepG2 = human hepatocyte carcinoma, PANC-1 = human pancreatic carcinoma.
Average GI50 for NCI 60 cell line panel.
The Ave GI50 values for the NCI cell line panels can be obtained at http://dtp.nci.nih.gov/dtpstandard/dwindex/index.jsp
Although some phenanthroindolizidines are too toxic, several, e.g. both enantiomers of tylophorine, have demonstrated low toxicity in rodents. (R)-Tylophorine was non-toxic in rats up to a dose of 500 mg/kg [3] and non-toxic in hamsters up to 60 mg/kg [18]. Some CNS toxicity, ptosis, sedation, decreased motor activity and staggering gait, has been observed in rats at high doses of (R)-tylophorine [3].
In the early 1960’s tylocrebrine, Fig. (1), was advanced to clinical trials but failed due to CNS toxicity, manifested as disorientation and ataxia [20]. Suffness proposed that more polar analogs, which cannot clear the blood brain barrier, could have less side effects [20].
Medicinal interest in the phenanthroindolizidines was revived in the 1990’s when it was shown that a number of these compounds are highly potent (10−8−10−12 M) growth inhibitors in the NCI’s 60 cell line assay and that their mode of action appears to be different than other known anti-cancer compounds based on their COMPARE [21] analysis [13]. Both drug-sensitive and multidrug-resistant cancer cells demonstrate growth inhibition in the low nanomolar range with this class of compounds (vide infra) [11–13].
MECHANISM OF ACTION
The specific cellular target of the phenanthroindolizidines and quinolizidines is unknown, but a significant amount of cellular activity of these compounds is known. It was determined in the 1970’s that tylocrebrine, tylophorine and cryptopleurine inhibit protein synthesis, and to a lesser extent RNA and DNA synthesis [9, 22]. Inhibition of protein synthesis in Ehrlich ascites-tumor cells by 50% was achieved using 10−6 M tylophorine, 10−7 M tylocrebrine and 10−8 M cryptopleurine [9]. A much higher amount was required for nucleic acid synthesis inhibition. Tylocrebrine inhibits protein and DNA synthesis irreversibly in cells, as determined by lack of incorporation of carbon-14 labeled lysine and thymidine, respectively. Tylocrebrine reversibly inhibited RNA synthesis as measured by the amount of 14C-labeled uridine incorporation [22]. It was observed that tylocrebrine and cryptopleurine prevent the breakdown of polyribosomes and release of nascent peptides and it was inferred that they inhibit peptide chain elongation [22, 23]. It was further determined that tylocrebrine was not an inhibitor of peptide bond formation [22]. The 40S ribosomal subunit has been postulated to house the binding site of these drugs [23, 24].
Other Targets
Besides the ribosome, other targets have been shown to interact with these compounds. Thymidylate synthase, an enzyme important in DNA synthesis, was inhibited by micromolar amounts of tylophorinidine [25]. Dihydrofolate reductase, another enzyme important in nucleic acid biosynthesis, was also inhibited by tylophorinidine [26]. Antofine has been shown to bind bulged DNA [27] and tabacco mosaic virus [19]. Nuclear Factor-kB, a transcription factor, has been suggested to be a mechanism of drug resistance because of its anti-apoptotic role [28, 29]. NF-kB mediated transcription was inhibited by (S)-tylophorine and analogs [13, 15]. Thus, inhibition of NF-kB may account for the cytostatic activity of these drugs in multi-drug-resistant cancer cell lines.
To date, the specific cellular target protein or nucleic acid has not been identified. That the molecules can interact with a number of protein and nucleic acid synthesis cellular machinery is clear. While it is likely that the compounds operate via a number of inhibition mechanisms, which might be to their advantage, to date no protein isolation via pull-down (affinity chromatography) experiments has been reported and no specific nucleic acid targets have been identified. It would be advantageous to drug development to understand how these drugs interact with specific cellular targets.
Many targets may be involved as the compounds inhibit both protein and nucleic acid synthesis. Since protein synthesis is more potently inhibited than nucleic acid synthesis, this is probably the drug’s primary mode of growth inhibition. The nucleic acid synthesis inhibition, including transcription via NF-kB may serve as important supplementary activity that increases potency, especially in MDR cells.
Total Synthesis of Phenanthroindolizidines and Phenanthroquinolizidines
The potent medicinal properties and interesting structures of the phenanthroindolizidines and phenanthroquinolizidines have attracted synthetic organic chemists and medicinal chemists for several decades [1]. The total synthesis of these compounds provides a sustainable source as well as ready access to derivatives for structure-activity-relationship studies. Tylophorine, antofine, cryptopleurine and septicine have been the most popular total synthesis targets to date. Since the target molecules contain a chiral center, their stereoselective synthesis is important to SAR studies and subsequent drug development, where the development of an enantiomerically pure compound would be ideal in order to avoid possible deleterious activity of the other enantiomer as well as use of unnecessarily high doses. Therefore, although a number of racemic routes for the syntheses of these natural products have been reported [1, 2, 16, 30–40] (several of them are quite concise) this review will summarize only the enantioselective syntheses in further detail.
Both the (R)- and (S)-tylophorine enantiomers have been synthesized selectively using the chiron approach, starting with either proline [41, 42], glutamic acid [6] or pyroglutamate [43] as well as the chiral auxiliary approach manifested in diastereoselective Grignard additions [44] and double Michael reactions [45], respectively. Recently, we reported the first catalytic asymmetric route to (S)-tylophorine [46].
(R) and (S)-Antofine have been synthesized enantioselectively starting from proline [42], pyroglutamate [47], and from an allylic alcohol obtained enantioselectively via an enzymatic resolution [48]. Antofine has also been synthesized enantioselectively via enantioselective catalytic phase transfer alkylation [49].
(R)- and (S)-Cryptopleurine has been synthesized enantioselectively starting from (S)-(+)-α-aminoadipate [6] and an allylic alcohol obtained via an enzymatic resolution [48], and by using a proline-derived chiral auxiliary to direct the facial selectivity of an alkylation reaction [50].
The naturally occurring seco analogs of tylophorine and cryptopleurine, septicine and julandine, respectively, have been synthesized enantioselectively as well. (R)-Septicine has been synthesized en route to (R)-tylophorine via an enantioselective alkylation controlled by a chiral ester [44]. (R)-Julandine has been synthesized using the same enantioselective alkylation route as employed in the synthesis of (R)-cryptopleurine, where a proline-derived auxilliary directed the facial selectivity [50].
Asymmetric Synthesis via the Chiral Pool
The first enantioselective synthesis of (S)-(+)-tylophorine, the unnatural enantiomer, was reported by Rapoport and co-workers in 1983 [6]. This synthesis established the absolute configuration of the natural compound, which was opposite the one synthesized by these researchers. The source of chirality was (S)-glutamic acid. The synthesis began with condensation of veratraldehyde (1) with (3,4-dimethoxyphenyl)acetonitrile (2), Scheme 1. The resulting stilbene 3 was oxidized to the phenanthrene with VOF3. Subsequent reduction of the nitrile with diisobutyl-aluminum hydride (DIBAL-H) provided phenanthryl aldehyde 4 in 89% overall yield. Condensation of aldehyde 4 with (S)-diisopropylglutamate provided an initial Schiff base that was rapidly converted to the corresponding aminal upon reaction with an additional equivalent of (S)-diisopropylglutamate (to prevent epimerization). The crude aminal was reduced with NaCNBH3, yielding the desired N-(phenanthrylmethyl)-glutamate 5. Amide formation in warm MeOH/HOAc and hydrolysis of the remaining ester provided acid 6 in 88% yield (four steps). Conversion of the acid to the acid chloride (oxalyl chloride, DMF) and subsequent intramolecular Friedel-Crafts alkylation promoted by SnCl4 provided ketone 7. Depending upon the reducing agent used, ketone 7 provided either the C(14)-R or C(14)-S alcohols, 8 or 9, respectively (tylophorine numbering). Thus, hydrogenation (H2, Pd/C) of 7 provided 8 selectively. Alcohol 8 could be taken on to the pyrrolidine 10. This compound has demonstrated good anti-cancer activity in vitro (vide infra). The C14-(S)-alcohol 9 was obtained by reduction of ketone 7 with L-Selectride. Subsequent reduction of 9 with LiAlH4 provided pyrrolidine 11, which demonstrated very good in vitro and in vivo anti-cancer activity (vide infra). (S)-Alcohol 9 was deoxygenated by conversion to the corresponding chloride and subsequent reduction. The resulting amide was reduced with LiAlH4 to provide (S)-tylophorine. The observed optical rotation of +15° for this compound is somewhat lower than that obtained by other research groups (vide infra). It has been noted that tylophorine decomposes at room temperature, with concomitant decrease in optical rotation, so this may be the source of the relatively lower optical rotation of Rapoport’s synthetic compound [45]. Interestingly, Rapoport and co-workers were unable to perform the de-oxygenation via chlorination/reduction on alcohol 8. Using an approach similar to their tylophorine synthesis, Rapoport and coworkers synthesized (S)-(+)-cryptopleurine starting from (S)-α-aminoadipic acid [6].
Scheme 1.

Glutamic acid approach to the synthesis of (+)-tylophorine
Nordlander and Njoroge subsequently reported a very short, enantioselective synthesis of (S)-tylophorine in 1987 starting from (S)-N-(trifluoroacetyl)prolyl chloride (Scheme 2). 2,3,6,7-Tetramethoxyphenanthrene (14), which can be synthesized in as few as three steps [51, 52] (vide infra) was subjected to Friedel-Crafts acylation with (S)-N-(trifluoroacetyl)prolyl chloride (13) in the presence of AlCl3 in refluxing CH2Cl2. Crystalline ketone 15 was obtained in 51% yield in >98% enantiomeric purity. Deketonization of 15 was accomplished in 64% yield by treatment with Et3SiH in neat BF3•OEt2 at room temperature. Subsequent deacylation in methanolic ammonia provided the amine 17 in 71% yield. Pictet-Spengler annulation afforded (S)-tylophorine in 53% yield in high optical purity.
Scheme 2.

Proline approach to the synthesis of (+)-tylophorine.
Huang and co-workers reported a concise, enantioselective synthesis of (+)-tylophorine using tert-butyl L-pyroglutamate as the chiral pyrrolidine source in 2004 (Scheme 3) [43]. Condensation of veratraldehyde (1) and homoveratric acid (18) in the presence of acetic anhydride and triethylamine afforded an inconsequential mixture of olefin isomers 19 (Scheme 3). Methylation of the carboxylic acid and oxidative cyclization with vanadium oxytrichloride in dichloromethane gave the phenanthryl ester in 99% yield. Reduction with LiAlH4 gave the alcohol that was converted to the phenanthryl chloride 20 in 95% yield. Alkylation of tert-butyl L-pyroglutamate (21) with phenanthryl chloride 20 (NaH, DMSO, rt) provided the phenanthryl amide 22 in 96% yield. Carboxylic acid deprotection, conversion to the acid chloride and subsequent Lewis-acid catalyzed intramolecular Friedel-Crafts cyclization provided the amido ketone 7 in 92% yield. Reduction with LiAlH4 provided a diastereomeric mixture (55:45) of alcohols. Deoxygenation with triethylsilane in trifluoroacetic acid provided (+)-tylophorine in 92% combined yield with high optical purity ([α]D25 = + 74.9°).
Scheme 3.

Pyroglutamate approach to the synthesis of (+)-tylophorine.
Furstner and co-workers reported an enantioselective route to (−)-antofine and (−)-tylophorine using commercially available D-(−)-prolinol as the source of chirality in 2006 (Scheme 4) [42]. Selective N-protection of prolinol (23) [53] was followed by oxidation of the primary alcohol and Wittig olefination of the resulting aldehyde to give 24. Hydroboration then provided primary alcohol 25 that was oxidized and coupled with the Bestmann-Ohira reagent 26 to provide terminal alkyne 27.
Scheme 4.

Cycloisomerization route to (−)-antofine.
Alkyne 27 underwent Sonogashira-type coupling to aryl iodide 28 (synthesized three steps from meta-iodoanisole, see lower part of Scheme 4), providing the biphenylalkyne 29 in 58% yield. Platinum(II)-catalyzed cycloisomerization provided phenanthrene 30 in 72% yield. Removal of the Boc-protecting group and Pictet-Spengler cyclization in one pot afforded (−)-antofine in 91% yield and >98% ee. (−)-Tylophorine was synthesized in the same manner but using a tetramethoxyiodobiphenyl coupling partner in the Sonogoshira-type fragment coupling.
Auxilliary-Directed Asymmetric Synthesis
Fukumoto and co-workers reported the first asymmetric total synthesis of the natural (R)-enantiomer of tylophorine in 1990 (Scheme 5). In this synthesis, a stereoselective intramolecular Michael addition of an amide nucleophile onto a chiral α,β-unsaturated ester provided the stereocenter found in the natural product. The Michael addition was performed in tandem with another Michael addition, thereby synthesizing two ring of the natural product in one pot. (−)-Phenylmenthol was the chiral auxiliary used to obtain the stereocenter in the natural product.
Scheme 5.

Synthesis of (−)-tylophorine via intramolecular double Michael reaction.
The (E)-acid 35 [54] was converted into amide 37 by condensing its acid chloride with 4-aminobutyraldehyde diethyl acetal (36). Conversion of the acetal to its aldehyde and Horner-Wadsworth-Emmons olefination with (−)-phenylmenthyl(trip[henylphosphoranylidene)acetate (38) [55] in refluxing acetonitrile afforded 39. Stereoselective intramolecular double Michael reaction of 39 in the presence of TBSOTf and triethylamine afforded an inconsequential mixture of diastereomers 40. Oxidation with thallium(III) trifluoroacetate afforded phenanthrene 41. Supponification of the ester and decarboxylation followed by reduction of the amide provided the natural product in high enantiopurity.
Kibayashi and co-workers reported the enantioselective syntheses of (R)-(−)-cryptopleurine and (R)-(−)-julandine using a proline derived-auxilliary to direct an amido alkylation in 1995 (Scheme 6). (S)-N-nitrosoprolinol (42) [56] was converted in 5 steps to hemi-aminal 43. The imminium ion formed by treatment of 43 with BF3•OEt2 reacted with enolsilanes 44 and 45 to provide β-aminoketones 46 and 47 in 76% and 70% yield, respectively, with greater than 99% selectivity. The ketone and amide functional groups of 46 were reduced with BH3•THF, along with concomitant removal of the auxiliary. Amide formation with (4-methoxyphenyl)acetyl chloride (50) in the presence of 5% NaOH in CH2Cl2 followed by hydrolysis of the intermediate N,O-bis(phenylacetyl) derivative afforded hydroxy amide 51 in 85% yield. Oxidation of the hydroxyl group with pyridinium dichromate and subsequent aldol condensation in refluxing 5% ethanolic KOH formed quinolizidinone 52 in 53% combined yield. Reduction of the amide with 3:1 LiAlH4/AlCl3 and recrystallization provided (R)-julandine in 58% yield.
Scheme 6.

Synthesis of (R)-(−)-julandine.
(R)-Cryptopleurine was synthesized from the aryl bromide 47 in like manner (Scheme 7). Treatment of the seco-arylbromide intermediate 55 with light or Bu3SnH, AIBN provided the phenyanthryl amide 56. Reduction of 56 with LiAlH4 to provide the natural product.
Scheme 7.

Synthesis of (R)-(−)-cryptopleurine.
(−)-Tylophorine and (−)-Septicine, Chiral Auxiliary Approach
Commins and co-workers reported the development of chiral auxiliaries that could substitute for (−)-8-phenylmenthanol, which is available in only one naturally occurring enantiomer. In particular, trans-2-(α-cumyl)cyclohexanol proved readily available in both antipodes in high enantiomeric purity [57]. Commins’ syntheses of (−)-tylophorine and (−)-septicine used (−)-trans-2-(α-cumyl) cyclohexyl ester as a chiral auxiliary to guide the facial selectivity of a Grignard reagent to a functionalized pyridinium salt (Scheme 8) [44]. Thus, pyridinium salt 57 underwent diasteroselective addition of 3-butenyl magnesium bromide, providing 58 in high diasteremeric purity after recrystallization. Oxidative cleavage of the terminal alkene followed by reduction of the resulting aldehyde provided alcohol 59. Conversion to the chloride 60 and treatment with basic methanol provided the mono-unsaturated indolizidine 61. Electrophilic substitution of bromine for silicon and 1,4-reduction followed by enolate oxygen trapping as the triflate afforded the difunctionalized indolizidine 64. Negishi cross-coupling with 3,4-dimethoxyphenyl zinc bromide (65) provided (−)-septicine in 67% yield. (−)-Tylophorine was obtained in 68% yield by oxidation of (−)-septicine with VOF3 in trifluoroacetic acid and dichloromethane.
Scheme 8.

Synthesis of (−)-tylophorine asymmetric 1,4-addition and cross-coupling.
Asymmetric Synthesis via Chiral Catalysis
In 2003, Kim and co-workers reported an asymmetric total synthesis of (−)-antofine using an enantioselective catalytic phase transfer alkylation (Scheme 9) [49]. The key alkylation of glycine imine 67 with phenanthrylalkyl bromide 66, mediated by the phase transfer catalyst 68, occurred in 97% yield and with 96% enantioselectivity. Subsequent conversion to the unsaturated pyrrolidine 71 was possible via a ring-closing-metathesis route. Reduction of 71 and Pictet-Spengler annulation with formaldehyde generated the natural product.
Scheme 9.

Synthesis of (−)-antofine via enantioselective phase transfer alkylation.
Phenanthryl bromide 66 was synthesized (see lower part of Scheme 9) from alcohol 73 which in turn was synthesized in a four-step sequence starting from condensation of homovaleric acid (18) with p-anisaldehyde (72) as described by Weinreb et al. [58].
Another asymmetric route to antofine was reported by Kim and co-workers in 2004 (Schemes 10–12) [48]. In this route, which was also amenable to the enantioselective synthesis of (−)-cryptopleurine, a common chiral synthon, (R)-(E)-4-(tributylstannanyl)but-3-en-2-ol (74) was the source of chirality. The chiral vinylstannane 74 underwent Pd-catalyzed Stille coupling with phenanthrylbromide 66, providing allylalcohol 75 in 95% yield (Scheme 10). Trichloroacetimidate formation and Overman rearrangement provided the chiral amide 77 in 92% combined yield. Protecting group exchange, N-allylation and ring closing metathesis provided unsaturated pyrrolidine 78 in 85% combined yield. Hydrogenation and Pictet-Spengler annulation provided (−)-antofine in 50% combined yield.
Scheme 10.

Total Synthesis of antofine via Overman rearrangement and ring-closing metathesis.
Scheme 12.

Total synthesis of (−)-cryptopleurine.
(R)-(E)-4-(Tributylstannanyl)but-3-en-2-ol (74) was obtained in three steps as shown in Scheme 11 [59]. Catalytic hydrostannylation of 3-butyne-2-ol (79) with Bu3SnH in THF at −78°C in the presence of Pd(PPh3)2Cl2 (1 mol %) afforded vinylstannanes 80 in 81% yield. Lipozyme-mediated kinetic resolution with vinyl acetate in diisopropylether at 35°C provided 49% of (R)-81 (>99% ee) and 48% of (S)-74 (>99% ee). Alkaline methanolysis of the acetate (R)-81 provided the necessary (R)-74 in 98% yield.
Scheme 11.

Synthesis of chiral vinylalcohol via kinetic resolution.
Intermediate 82 (derived from 77, Scheme 10) was also used en route to a total synthesis of (−)-cryptopleurine by Kim and co-workers (Scheme 12) [48]. Cross-metathesis with homoallyl acetate dimer 83 (5 mol % Grubbs II, refluxing CH2Cl2, 1 d) gave an inseparable E/Z mixture of alkene 84 (82% yield, 8:1 E/Z) along with recovered starting material (14%). Hydrogenation of the alkene and concomitant Cbz deprotection followed by supponification of the acetate provided alcohol 85. Mitsunobu cyclization provided the piperidine in 68% yield. Pictet-Spengler annulation then provided (−)-cryptopleurine in 67% yield.
We have recently accomplished the enantioselective total synthesis of (S)-(+)-tylophorine (Scheme 13) [46]. The synthesis relies on the introduction of the indolizidine ring of tylophorine through a copper(II)-catalyzed enantioselective carboamination reaction developed recently in our lab [60]. Entry into the chiral indolizidine core of these compounds has been referred to as the “limiting synthetic factor” in the synthesis of this class of compounds [61].
Scheme 13.

Enantioselective total synthesis of (S)-(+)-tylophorine.
Conversion of commercially available 3,4-dimethoxy benzylalcohol (86) to the bromide 87 followed by radical-induced dimerization [62] provided 88 (73% yield for two steps). Oxidation [51, 52] with phenyliodine(III)bis(trifluoroacetate) (PIFA) provided phenanthrene 14 (66% yield) which was sulfonylated with chlorosulfonic acid to provide the phenanthrene fragment 89. Convergent fragment coupling of aryl sulfonyl chloride 89 and pentenylamine (90) provided the aryl sulfonamide intermediate 91. Enantioselective copper(II)-catalyzed carboamination with the bis(oxazoline) complex 92, a process recently developed by our research group [60], provided sultam 93 in 64% yield. Reductive removal of the sulfur dioxide group provided the required pyrrolidine 17 in moderate yield (44%). The yield in the conversion of 93 to 17 is moderate because the phenanthrene methoxy groups are labile under the Li/NH3 reaction conditions. Pictet-Spengler cyclization with formaldehyde afforded (S)-tylophorine in 89% yield. The enantiomeric excess of synthetic (S)-tylophorine was 81% as measured by chiral HPLC. Both enantiomers of the chiral bis(oxazoline) ligand, Ph-Box (92), are commercially available and easily synthesized, thus the sulfonamide 91 can in principle provide for the synthesis of either enantiomer of tylophorine. This route should allow for the coupling of diverse arylsulfonyl chlorides with a range of amines for structure-activity-relationship studies. The synthesis of pyrrolidine-substituted analogs of tylophorine is currently underway in our labs.
Structure-Activity-Relationship Studies
The therapeutic properties of the phenanthroindolizidines and phenanthroquinolizidines have attracted the interest of a number of medicinal chemists. An extensive but incomplete understanding of the structure-activity-relationship of these compounds has been achieved. A more full understanding has thus far been limited by a lack of robust and versatile organic synthesis methods (vide infra). As seen above, however, a number of recently reported synthetic routes should allow access to a diversity of analogs for future studies.
The phenanthroindolizidines are composed of a substituted phenanthrene fused to an indolizine Fig. (2). Thus far, researchers have studied the effect of the phenanthrolene substitution pattern, presence or lack of indolizidine ring fusion, presence or lack of C14-hydroxyl groups on the indolizine ring and the oxidation state of the amine (vide infra). Very little information is available on the affect of substituents on the pyrrolidine subunit.
Fig. 2.

Phenanthroindolizidine and phenanthroquinolizidine.
Phenanthrene Substitution
It was found early on that hydroxy and alkoxy substituents on the phenanthrene unit are mandatory for biological activity. The unsubstituted phenanthro[9,10-b]quinolizidine was inactive in in vitro protein synthesis studies and in vivo anti-tumor evaluation, while the unsubstituted phenanthroindolizidine was inactive in in vitro antiamoebic activity assays [9, 18].
The phenanthrene substitution pattern greatly affects the in vitro anti-cancer activity of the phenanthroindolizidines. In an SAR study of antofine (Fig. (3) and Table 2), Kim and co-workers found that changing the C2 substituent from MeO to HO, i-PrO, or OCH2O (shared with C3) all had significantly deleterious effects on the cytotoxicity of the compounds. Thus, the C2 substituent must be a small, H-bond acceptor but not H-bond donor. C3 and C6 could tolerate such changes, and when C6 was changed to HO, the activity increased relative to the parent antofine (Fig. 3 and Table 2).
Fig. 3.
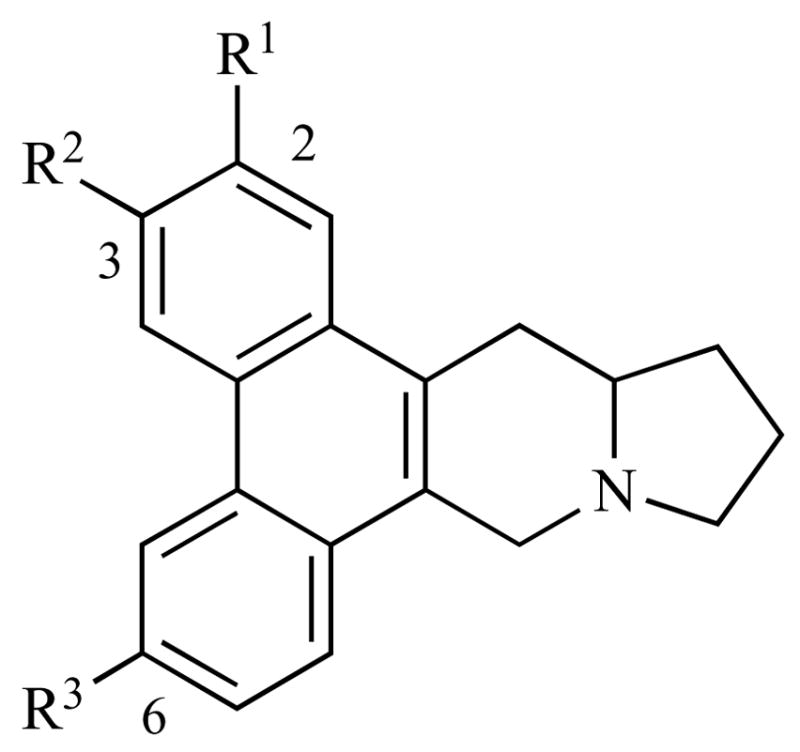
Antofine analogs.
Table 2.
| Entry | R1 | R2 | R3 | IC50 (nM) |
|---|---|---|---|---|
| 1, (R)-antofine | OMe | OMe | OMe | 9.9 |
| 2 (±)-antofine | OMe | OMe | OMe | 29.4 |
| 3 | O-i-Pr | OMe | OMe | 783.2 |
| 4 | OMe | O-i-Pr | OMe | 24.7 |
| 5 | OMe | OMe | O-i-Pr | 19.2 |
| 6 | OH | OMe | OMe | 68.3 |
| 7 | OMe | OH | OMe | 21.4 |
| 8 | OMe | OMe | OH | 1.1 |
| 9 | OMe | OMe | H | 49.3 |
| 10 | -OCH2O- | OMe | 364.1 |
Human colorectal carcinoma.
Phenanthrene Ring Fusion
Seco analogs of antofine, secoantofine, 6-O-desmethylsecoantofine and secoantofine N-oxide are all substantially less active (IC50 400–3000 nM, Fig. 1 and Table 1) in vitro [11, 12]. Tyloindicine I, however, which contains more unsaturation in the indolizidine ring, is highly potent (GI50 10−9 M) in the NCI’s 60 cell line assay. Tyloindicine I is one of the most potent compounds in this family of structures. The greater rotational flexibility of the aryl rings and the decreased basicity of the enamine nitrogen might be beneficial to the activity of this compound.
Nitrogen
The oxidation level of the indolizidine amine affects the in vitro cell growth inhibitory activity of these compounds. Some N-oxides of tylophorine analogs are naturally occurring [e.g. tylophoridicin C and tylophoridicine F and antofine N-oxide, Fig. (1)]. These N-oxides are 5–10 times less active in vitro than their free amine counterparts in KB, HepG2 and PANC-1 cell lines (60–160 nM) [11, 14]. It is interesting to note that while N-oxidation diminishes potentcy, the N-oxides of antofine and tylophoridicine are still active compounds (Table 1).
C9-Keto analogs, amide structures of various phenanthroindolizidines and phenanthroquinolizidines, have also been found to be much less potent in cancer cell growth assays [31, 34, 63].
Interestingly, one study found that the pH of the cell growth medium has a strong effect on the activity of the phenanthroindolizidines and phenanthroquinolizidines. Tylophorine and cryptopleurine were substantially less cytostatic to yeast at pH 5.8 than they were at pH 7.0 [23]. This may indicate that an unprotonated amine (pKa of indolizidines are ca. 10–11) is necessary for transport into the cell or cell membranes. It could also indicate that the unprotonated form of the molecule binds to its receptor.
C(13a) Stereochemistry
Some of the phenanthroindolizidines have C13a(R) stereochemistry while others have C13a(S)-stereochemistry. In human colorectal (HCT 116) and lung (A549) carcinoma cells, the natural (R)-enantiomer of antofine is more cytostatic (IC50 = 9.9 and 10.4 nM, respectively) than the unnatural (S)-enantiomer (IC50 of racemate = 29.4 and 25.1 nM, respectively) [16]. In contrast, as measured in human nasopharyngeal carcinoma (KB) cells, the natural (R)-enantiomer of tylophorine is less cytostatic (IC50 = 173 nM) [12] than the unnatural (S)-enantiomer (IC50 = 12 nM) [13]. The C(13a) stereochemistry of tylocrebrine is (S). It is interesting to note that tylocrebrine and isotylocrebrine are enantiomeric at C(13a) but similar in biological activity and identical in phenanthrene alignment if one allows the pyrrolidine nitrogen to shift, Fig. (4). In the R-series, C-2 methoxylation is very important for activity while in the S-series, C-7 methoxylation is very important for activity.
Fig. 4.

Structural similarities of enantiomers.
C(14) Hydroxylation
In vitro antiproliferative assays (cancer cells) show the same or a slight loss of activity for C(14) hydroxylated analogs of tylophorine, Fig. (5). Gao and co-workers [13, 14] tested the C(14) hydroxylated analogs 10 and 11 of (S)-tylophorine originally synthesized by Rapoport and coworkers [6] (vide supra). The C(14) S, C(13a) S tylophorine analog 11 has an GI50 of 24–40 nM in KB and HepG2 cell lines while the C(14) R, C(13a) S tylophorine analog 10 has GI50 around 100 nM in these cell lines [13]. For comparison, (S)-tylophorine has a GI50 of 9–15 nM in these two cell lines. Naturally occurring tylophorinidine, however, which has C(14) S, C(13a) S stereochemistry and a slightly different phenanthrene substitution pattern, is as active in vitro (GI50 in HepG2 = 11 nM) as (S)-tylophorine [14]. Thus, the C(14) S, C(13a) S stereochemistry may be optimal for these compounds [14].
Fig. 5.

C14-Hydroxylated analogs compared to tylophorine.
Lack of Indolizidine, Quinolizidine
Phenanthrene-based tylophorine derivative (PBT’s) that lack the phenanthroindolizidine ring fusion (fused middle ring) have been studied. The synthesis of the fused indolizidine ring is considered to be the “limiting synthetic factor” of these molecules, thus removal of one of the C-C bonds that make up the indolizidine ring greatly decreases the synthesis burden. A large number of PBTs have been synthesized [61].
In general, the cytotoxicity is in the micromolar to sub-micromolar range and inhibition of tumor growth in vivo is modest [64]. A comparison on the mechanism of action has been made between the PBTs and (S)-tylophorine, and Gao concluded that the PBTs do not operate by the same biological mechanism as (S)-tylophorine [14]. Similar analogs synthesized by other groups confirm that tylophorine and cryptopleurine analogs lacking the indolizidine and quinolizidine rings are much less active in cancer cell growth assays [65]. Interestingly, though, because their structures are similar to the poly-substituted cis-stilbene combretastin A4 [66], a potent anti-mitotic and anti-angiogenic agent, they were tested in anti-angiogenesis assays and found to be quite promising lead compounds [65].
Pyrrolidine Substitution
There is no SAR information on pyrrolidine substitution in the indolizidine class of compounds, likely due to difficulty of their synthesis (vide supra). Most enantioselective syntheses of phenanthroindolizidines start from proline or other naturally occurring compounds that are not easily derivatized. Recently reported syntheses that use chiral auxiliary and chiral catalyst methodology to install the pyrrolidine ring should allow for greater access to pyrrolidine-substituted analogs (vide supra).
In Vivo Activity and Pharmakokinetics
In 1968, Donaldson and co-workers reported that mice infected by Ehrlich ascites-tumor cells (manifested as breast cancer) showed 50% tumor growth inhibition upon administration of 3 micromoles/kg twice daily for ten days with cryptopleurine [9]. Later studies by the NCI found this compound generally toxic. While tylocrebrine showed some promise in vivo, Phase I studies were terminated due to CNS toxicity, manifested as disorientation and ataxia [20]. Decreasing diffusion through the blood brain barrier should minimize the CNS toxicity of the compounds.
Recently, interest has been renewed in the phenanthroindolizidines as anti-cancer compounds. Gao and coworkers recently found that C14 hydroxylated analogs, Fig. (5), are more efficient at reducing tumor growth in vivo. While (S)-tylophorine is equipotent to tylophorinidine in vitro (HepG2, human hepatocarcinoma, cells), it is far less efficacious in vivo (mice) [13, 14]. Likewise, (S)-tylophorine is more active than the synthetic 11 in vitro, Fig. (5), but far less potent in vivo [13]. Both tylophorinidine and 11 share the same C(14) hydroxylation of the indolizidine, which may give them favorable pharmacokinetics in vivo [14]. Antofine, though potent in vitro, is metabolically unstable, resulting in a poor in vivo pharmacokinetic profile (hypothesized to be due to a methoxy moiety on the phenanthrene) [16]. Antofine also lacks a C(14) hydroxyl group. While a couple promising (ca. 60% tumor growth suppression relative to control) [14] compounds have been identified in in vivo studies, a balance between complete inhibition of tumor growth in vivo and mimimization of general toxicity has not been achieved. The factors that affect the pharmacokinetics of these compounds must be more fully understood in order for an optimal compound to be developed into an anti-cancer drug. A better understanding of the structure-activity-relationship of these compounds should facilitate development of a potent anti-cancer compound without undesirable side effects.
Anti-Inflammatory Effects
It should be noted that tylophorine (natural source) is also a promising lead in prevention and treatment of inflammation. The leaves of Tylophora indica have been used for decades in India to treat inflammatory-related conditions such as asthma, bronchitis and rheumatism [3, 17]. In vitro experiments demonstrated that tylophorine prevents NO production in murine macrophage cells (RAW264.7) with an IC50 of 1.8 μM [67]. Tylophorine also inhibits expression of several proinflammatory factors such as iNOS and COX-II. It enhanced Akt activation and decreased c-Jun expression. Akt and c-Jun activity are involved in both apoptosis and stress-induced inflammation [67]. In vivo studies in rats indicated that within hours of ip administered tylophorine (natural source), 25 mg/kg produced significant anti-inflammatory effects. [3] CNS depression was observed at 100 mg/kg or greater. Importantly, tylophorine is very effective in vivo when its effects can be observed shortly after it is administered, when drug metabolism is still at a minimum. 7-Methoxycryptopleurine also exerted potent anti-inflammatory activity in vitro as well as in vivo (rats) [68].
Scheme 14.

Synthesis of phenanthrene-based tylophorine derivatives (PBTs).
Acknowledgments
We are grateful to the National Institutes of Health (NIGMS GM078383) for support of our research program in nitrogen heterocycle synthesis.
References
- 1.Li Z, Jin Z, Huang R. Isolation, total synthesis and biological activity of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Synthesis. 2001;16:2365–2378. [Google Scholar]
- 2.Gellert E. The indolizidine alkaloids. J Nat Prod. 1982;45:50–73. [Google Scholar]
- 3.Gopalakrishnan C, Shankaranarayan D, Kameswaran L, Natarajan S. Pharmacological investigations of tylophorine, the major alkaloid of tylophora indica. Indian J Med Res. 1979;69:513–520. [PubMed] [Google Scholar]
- 4.Rathnagiriswaran AN, Venkatachalam K. The chemical examination of tylophora asthmatica and isolation of the alkaloids tylophorine and tylophorinine. Indian J Med Res. 1935;22:433. [Google Scholar]
- 5.Govindachari TR, Lakshmikantham MV, Pai BR, Rajappa S. Chemical examination of tylophora asthmatica--III. The complete structure of tylophorine. Tetrahedron. 1960;9:53. [Google Scholar]
- 6.Buckley TF, Rapoport H. α-Amino acids as chiral educts for asymmetric products. Chirally specific synthesis of tylophorine and cryptopleurine. J Org Chem. 1983;48:4222–4232. [Google Scholar]
- 7.Bick IRC, Sinchai W. The Alkaloids. 1981;19:193–220. [Google Scholar]
- 8.Govindachari TR, Viswanathan N. Recent progress in the chemistry of phenanthroindolizidine alkaloids. Heterocycles. 1978;11:587–613. [Google Scholar]
- 9.Donaldson GR, Atkinson MR, Murray AW. Inhibition of protein synthesis in Ehrlich Ascites-Tumor cells by the phenanthrene alkaloids tylophorine, tylocrebrine and cryptopleurine. Biochem Biophys Res Comm. 1968;31:104–109. doi: 10.1016/0006-291x(68)90037-5. [DOI] [PubMed] [Google Scholar]
- 10.Ali M, Ansari SH, Grever MR. Cytotoxic alkaloids from Tylophora indica. Pharmazie. 2001;56:188–190. [PubMed] [Google Scholar]
- 11.Staerk D, Christensen J, Lemmich E, Duus JØ, Olsen CE, Jaroszewski JW. Cytotoxic activity of some phenanthroindolizidine N-oxide alkaloids from Cynanchum vincetoxicum. J Nat Prod. 2000;63:1584–1586. doi: 10.1021/np0003443. [DOI] [PubMed] [Google Scholar]
- 12.Staerk D, Lykkeberg AK, Christensen J, Budnik BA, Abe F, Jaroszewski JW. In vitro Cytotoxic activity of phenanthroindolizidine alkaloids from Cynanchum vincetoxicum and Tylophora tanakae against drug-sensitive and multidrug-resistant cancer cells. J Nat Prod. 2002;65:1299–1302. doi: 10.1021/np0106384. [DOI] [PubMed] [Google Scholar]
- 13.Gao W, Lam W, Zhong S, Kaczmarek C, Baker DC, Cheng YC. Novel mode of action of tylophorine analogs as antitumor compounds. Cancer Res. 2004;64:678–688. doi: 10.1158/0008-5472.can-03-1904. [DOI] [PubMed] [Google Scholar]
- 14.Gao W, Busson S, Grill SP, Gullen EA, Hu YC, Huang X, Zhong S, Kaczmarek C, Gutierrez J, Francis S, Baker DC, Yu S, Cheng YC. Structure-activity studies of phenanthroindolizidine alkaloids as potential antitumor agents. Bioorg Med Chem Lett. 2007;17:4338–4342. doi: 10.1016/j.bmcl.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 15.Shiah HS, Gao W, Baker DC, Cheng YC. Inhibition of cell growth and nuclear factor-kb activity in pancreatic cancer cell lines by a tylophorine analogue, DCB-3503. Mol Cancer Ther. 2006;5:2484–2493. doi: 10.1158/1535-7163.MCT-06-0146. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y, Lee SK, Min HY, Lee T, Lee J, Cheng M, Kim S. Synthesis and structure-activity studies of antofine analogues as potential anticancer agents. Bioorg Med Chem Lett. 2007;17:97–100. doi: 10.1016/j.bmcl.2006.09.080. [DOI] [PubMed] [Google Scholar]
- 17.Gopalakrishnan C, Shankaranarayanan D, Nazimudeen SK, Kameswaran L. Effect of tylophorine, a major alkaloid of Tylophora indica, on immunopathological and inflammatory reactions. Indian J Med Res. 1980;71:940–948. [PubMed] [Google Scholar]
- 18.Bhutani KK, Sharma GL, Ali M. Plant Based Antiamoebic Drugs; Part I. Antiamoebic activity of phenanthroindolizidine alkaloids; common structural determinants of activity with emetine. Planta Med. 1987;53:532–536. doi: 10.1055/s-2006-962803. [DOI] [PubMed] [Google Scholar]
- 19.Xi Z, Zhang R, Yu Z, Ouyang D. The interaction between tylophorine B and TMV RNA. Bioorg Med Chem Lett. 2006;16:4300–4304. doi: 10.1016/j.bmcl.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 20.Suffness M, Douros J. Anticancer agents based on natural product models. Academic Press; 1980. pp. 465–487. [Google Scholar]
- 21.Paull KD, Hamel E, Malspeis L. Prediction of biochemical mechanism of action from the in vitro antitumor screen of the national cancer institute. In: Foye WO, editor. Cancer Chemotherapeutic Agents. American Chemical Society; 1995. pp. 4–45. [Google Scholar]
- 22.Huang MT, Grollman AP. Mode of action of tylocrebrine: effects on protein and nucleic acid synthesis. Mol Pharm. 1972;8:538–550. [PubMed] [Google Scholar]
- 23.Grant P, Sanchez L, Jimenez A. Cryptopleurine resistance: genetic locus for a 40s ribosomal component in Saccharomyces cerevisiae. J Bacteriology. 1974;120:1308–1314. doi: 10.1128/jb.120.3.1308-1314.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta RS, Siminovitch L. Mutants of CHO cells resistant to the protein synthesis inhibitors, cryptopleurine and tylocrebrine: genetic and biochemical evidence for common site of action of emetine, cryptopleurine, tylocrebrine and tubulosine. Biochemistry. 1977;16:3209–3214. doi: 10.1021/bi00633a026. [DOI] [PubMed] [Google Scholar]
- 25.Rao KN, Bhattacharya RK, Venkatachalam SR. Thymidylate synthase activity in leukocytes from patients with chronic myelocytic leukemia and acute lymphocytic leukemia and its inhibition by phenanthroindolizidine alkaloids pergularinine and tylophrinidine. Cancer Lett. 1998;128:183–188. doi: 10.1016/s0304-3835(98)00061-5. [DOI] [PubMed] [Google Scholar]
- 26.Rao KN, Venkatachalam SR. Inhibition of dihydrofolate reductase and cell growth activity by the phenanthroindolizidine alkaloids pergularinine and tylophorinidine: the in vitro cytotoxicity of these plant alkaloids and their potential as antimicrobial and anticancer agents. Toxicol in vitro. 2000;14:53–59. doi: 10.1016/s0887-2333(99)00092-2. [DOI] [PubMed] [Google Scholar]
- 27.Xi Z, Zhang R, Yu Z, Ouyang D, Huang R. Selective interaction between tylophorine B and bulged DNA. Bioorg Med Chem Lett. 2005;15:2673–2677. doi: 10.1016/j.bmcl.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 28.Wang CY, Cusack JCJ, Liu RQ, Baldwin ASJ. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kB. Nat Med. 1999;5:412–417. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 29.Cusack JCJ, Liu R, Baldwin ASJ. Inducible chemoresistance to 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxy-campothesin (CPT-11) in colorectal cancer cells and a xenograft model is overcome by inhibition of nuclear factor-kB activation. Cancer Res. 2000;60:2323–2330. [PubMed] [Google Scholar]
- 30.Bhakuni DS. Biosynthesis and synthesis of biologically active alkaloids of indian medicinal plants. J Indian Chem Soc. 2002;79:203–210. [Google Scholar]
- 31.Su CR, Damu AG, Chiang PC, Bastow KF, Morris-Natschke SL, Lee KH, Wu TS. Total synthesis of phenanthroindolizidine alkaloids (±)-antofine, (±)-deoxypergularinine, and their dehydro cogeners and evaluation of their cytotoxic activity. Bioorg Med Chem. 2008;16:6233–6241. doi: 10.1016/j.bmc.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cragg JE, Herbert RB, Jackson FB, Moody CJ, Nicolson IT. Phenanthroindolizidine and related alkaloids: synthesis of tylophorine, septicine, and deoxytylophrinine. J Chem Soc Perkin Trans. 1982;1:2477–2485. [Google Scholar]
- 33.Kim S, Lee YM, Lee J, Lee T, Fu Y, Song Y, Cho J, Kim D. Expedient syntheses of antofine and cryptopleurine via intramolecular 1,3-dipolar cycloaddition. J Org Chem. 2007;72:4886–4891. doi: 10.1021/jo070668x. [DOI] [PubMed] [Google Scholar]
- 34.Chuang TH, Lee SJ, Yang CW, Wu PL. Expedient synthesis and structure-activity relationships of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Org Biomol Chem. 2006;4:860–867. doi: 10.1039/b516152e. [DOI] [PubMed] [Google Scholar]
- 35.Camacho-Davila A, Herndon JW. Total synthesis of antofine using the net [5+5]-cycloaddition of gamma, delta-unsaturated carbene complexes and 2-alkynylphenyl ketones as a key step. J Org Chem. 2006;71:6682–6685. doi: 10.1021/jo061053n. [DOI] [PubMed] [Google Scholar]
- 36.Lebrun S, Couture A, Deniau E, Grandclaudon P. Total synthesis of (±)-cryptopleurine, (±)-antofine and (±)-deoxypergularinine. Tetrahedron. 1999;55:2659–2670. [Google Scholar]
- 37.Pearson WH, Walavalkar R. Synthesis of (±)-tylophorine by the intramolecular cycloaddition of an azide with an omega-chloroalkene. Tetrahedron. 1994;50:12293–12304. [Google Scholar]
- 38.Yerxa BR, Yang K, Moore HW. Synthesis of (±)-septicine. Tetrahedron. 1994;50:6173–6180. [Google Scholar]
- 39.Comins DL, Morgan LA. N-acyldihydropyridones as synthetic intermediates - synthesis of (±)-septicine and (±)-tylophorine. Tetrahedron Lett. 1991;32:5919–5922. doi: 10.1021/jo9711495. [DOI] [PubMed] [Google Scholar]
- 40.Iida H, Watanabe Y, Tanaka M, Kibayashi C. General synthesis of phenanthroindolizidine, phenanthroquinolizidine, and related alkaloids: preparation of (±)-tylophorine, (±)-cryptopleurine, (±)-septicine, and (±)-julandine. J Org Chem. 1984;49:2412–2418. [Google Scholar]
- 41.Nordlander JE, Njoroge FG. A short synthesis of (s)-(+)-tylophorine. J Org Chem. 1987;52:1627–1630. [Google Scholar]
- 42.Furstner A, Kennedy JWJ. Total synthesis of the tylophora alkaloids cryptopleurine, (−)-antofine, (−)-tylophorine, and (−)-ficuseptine c. Chem Eur J. 2006;12:7398–7410. doi: 10.1002/chem.200600592. [DOI] [PubMed] [Google Scholar]
- 43.Jin Z, Li SP, Wang QM, Huang RQ. A concise total synthesis of s-(+)-tylophorine. Chinese Chem Lett. 2004;15:1164–1166. [Google Scholar]
- 44.Comins DL, Chen X, Morgan LA. Enantiopure N-acyldihydropyridones as synthetic intermediates: asymmetric synthesis of (−)-septicine and (−)-tylophorine. J Org Chem. 1997;62:7435–7438. doi: 10.1021/jo9711495. [DOI] [PubMed] [Google Scholar]
- 45.Ihara M, Takino Y, Tomotake M, Fukumoto K. Asymmetric total synthesis of naturally occurring (r)-(−)-enantiomer of tylophorine via intramolecular double michael reaction. J Chem Soc Perkin Trans. 1990;1:2287–2292. [Google Scholar]
- 46.Zeng W, Chemler SR. Total synthesis of (s)-(+)-tylophorine via enantioselective intramolecular alkene carboamination. J Org Chem. 2008;73:6045–6047. doi: 10.1021/jo801024h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faber L, Wiegrebe W. Helv Chim Acta. 1976;59:2201–2212. doi: 10.1002/hlca.19760590633. [DOI] [PubMed] [Google Scholar]
- 48.Kim S, Lee T, Lee E, Lee J, Fan G-j, Lee SK, Kim D. Asymmetric total synthesis of (−)-antofine and (−)-cryptopleurine using (R)-(E)-4-(tributylstannyl)but-3-en-2-ol. J Org Chem. 2004;69:3144–3149. doi: 10.1021/jo049820a. [DOI] [PubMed] [Google Scholar]
- 49.Kim SH, Lee J, Lee T, Park H-g, Kim D. First asymmetric total synthesis of (−)-antofine by using an enantioselective catalytic phase transfer alkylation. Org Lett. 2003;5:2703–2706. doi: 10.1021/ol0349007. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki H, Aoyagi S, Kibayashi C. Asymmetric total synthesis of (R)-(−)-cryptopleurine and (R)-(−)-julandine via highly enantioselective amidoalkylations with N-acylhydrazonium salts. J Org Chem. 1995;60:6114–6122. [Google Scholar]
- 51.Moreno I, Tellitu I, San Martin R, Dominguez E. A new entrance to the preparation of phenanthrene and phenanthrenooid heterocycles. Synthesis. 2001:1161–1163. [Google Scholar]
- 52.Moreno I, Tellitu I, Dominguez E, San Martin R. A simple route to new phenanthro- and phenanthroid-fused thiazoles by a PIFA-mediated (hetero)biaryl coupling reaction. Eur J Org Chem 2002 [Google Scholar]
- 53.Spatola AF, Anwer MK, Rockwell AL, Gierasch LM. Compatibility of beta- and gamma-turn features with a peptide backbone modification: synthesis and conformational analysis of a model cyclic pseudopentapeptide. J Am Chem Soc. 1986;108:825–831. [Google Scholar]
- 54.Russell JH, Hunziker H. Tetrahedron Lett. 1969:4035. [Google Scholar]
- 55.Roush WR, Gillis HR, Ko AI. J Am Chem Soc. 1982;104:2269. [Google Scholar]
- 56.Enders D, Eichenauer H. Chem Ber. 1979;112:2933. [Google Scholar]
- 57.Comins DL, Salvador JM. Efficient synthesis and resolution of trans-2-(1-aryl-1-methylethyl)cyclohexanols: practical alternatives to 8-phenylmenthol. J Org Chem. 1993;58:4656–4661. [Google Scholar]
- 58.Bremmer ML, Khatri NA, Weinreb SM. Quinolizidine alkaloid synthesis via the intramolecular imino Diels-alder reaction. epi-lupinine and cryptopleurine. J Org Chem. 1983;48:3661–3666. [Google Scholar]
- 59.Lee T, Kim S. Efficient preparation of enantiomerically pure (E)-4-(tributylstannanyl)but-3-en-2-ol via lipase-mediated resolution. Tetrahedron: Asymmetry. 2003;14:1951–1954. [Google Scholar]
- 60.Zeng W, Chemler SR. Copper(II)-catalyzed enantioselective intramolecular carboamination of alkenes. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei L, Brossi A, Kendall R, Bastow KF, Morris-Natschke SL, Shi Q, Lee KH. Antitumor agents 251: Synthesis, cytotoxic evaluation, and structure-activity relationship studies of phenanthrene-based tylophorine (PBTs) as a new class of antitumor agents. Bioorg Med Chem. 2006;14:6560–6569. doi: 10.1016/j.bmc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 62.Barrero AF, Herrador MM, Quilez del Moral JF, Arteaga P, Akssira M, Hanbali FE, Arteaga JF, Dieguez HR, Sanchez EM. Couplings of benzylic halides mediated by titanocene chloride: synthesis of bibenzyl derivatives. J Org Chem. 2007;72:2251–2254. doi: 10.1021/jo062492p. [DOI] [PubMed] [Google Scholar]
- 63.Kimball FS, Tunoori AR, Victory SF, Dutta D, White JM, Himes RH, Georg GI. Synthesis, in vitro and in vivo cytotoxicity of 6,7-Diaryl-2,3,8,8a-tetrahydroindolizin-5(1H)-ones. Bioorg Med Chem Lett. 2007;17:4703–4707. doi: 10.1016/j.bmcl.2007.05.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei L, Shi Q, Bastow KF, Brossi A, Morris-Natschke SL, Nakagawa-Goto K, Wu TS, Pan SL, Teng CM, Lee KH. Antitumor agents 253. Design, synthesis, and antitumor evaluation of novel 9-substituted phenanthrene-based tylophorine derivatives as potential anticancer agents. J Med Chem. 2007;50:3674–3680. doi: 10.1021/jm061366a. [DOI] [PubMed] [Google Scholar]
- 65.Banwell MG, Bezos A, Burns C, Kruszelnicki I, Parish CR, Su S, Sydnes MO. C8c-C15 Monoseco-analogs of the phenanthroquinolizidine alkaloids julandine and cryptopleurine exhibiting potent anti-angiogenic properties. Bioorg Med Chem Lett. 2006;16:181–185. doi: 10.1016/j.bmcl.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 66.Cirla A, Mann J. Combretastins: from natural products to drug discovery. Nat Prod Rep. 2003;20:558–564. doi: 10.1039/b306797c. [DOI] [PubMed] [Google Scholar]
- 67.Yang C-W, Chen W-L, Wu P-L, Tseng H-Y, Lee S-J. Anti-inflammatory mechanisms of phenanthroindolizidine alkaloids. Mol Pharm. 2006;69:749–758. doi: 10.1124/mol.105.017764. and references therein. [DOI] [PubMed] [Google Scholar]
- 68.Yang CW, Chuang TH, Wu PL, Huang WH, Lee SJ. Anti-inflammatory effects of 7-methoxycryptopleurine and structure-activity relations of phenanthroindolizidines and phenanthroquinolizidines. Biochem Biophys Res Comm. 2007;354:942–948. doi: 10.1016/j.bbrc.2007.01.065. [DOI] [PubMed] [Google Scholar]