

Abstract
The transcription factors that bind to EpRE elements play a key role in the regulation of phase II genes. In this study, we examined whether c-Jun, a partner of Nrf2 in binding to EpRE, requires phosphorylation by JNK for binding and transcriptional activation. We used chromatin immunoprecipitation assays (ChIP) to measure recruitment of transcription factors to EpRE sequences in nqo2, gclc and gclm, western analysis for phosphorylation of JNK, and EpRE driven reporters along with a JNK specific inhibitor peptide to determine the potential importance of c-Jun phosphorylation. Human bronchial epithelial (HBE1) and human hepatoma (HepG2) cells were exposed to 4-hydroxy-2-nonenal (HNE) and differences in regulation of the same EpRE sequences examined. We found binding of c-Jun to EpRE sequences increased subsequent to HNE exposure in HepG2 cells; however, in HNE-exposed HBE1 cells, binding of only phosphorylated c-Jun to the three EpRE sequences increased. Despite the increase in binding of phosphorylated c-Jun, reporter assays for EpREs showed that inhibition of c-Jun phosphorylation had variable effects on basal and HNE-induced transcription of gclc and gclm in HBE1 cells. Thus, in terms of its role in mediating HNE-induction of EpRE-mediated transcription, c-Jun appears to be a partner of Nrf2 and while its phosphorylated form may predominate in one cell type versus another, the effect of phosphorylation of c-Jun on transcription can vary with the gene. This contrasts markedly with the well-established requirement for phosphorylation of c-Jun in the activation of AP-1/TRE mediated transcription.
Keywords: 4-hydroxy-2-nonenal, electrophile responsive elements, human hepatoma cell line, human bronchial epithelial cells, JNK, c-Jun
Introduction
The electrophile responsive element (EpRE), also known as the antioxidant responsive element (ARE) [1], is a cis-acting regulatory element that plays an important role in the gene expression of phase II detoxification enzymes and antioxidant proteins such as glutathione S-transferases (GSTs) [2], NAD(P)H:quinone oxidoreductase-1 (NQO1) [3], NAD(P)H: quinone oxidoreductase 2 (NQO2) [4], and glutamate-cysteine ligase (GCL) [5–7]. Many studies have reported an important role of EpRE in protecting cells from oxidative stress. For example, tert-butylhydroquinone pretreatment, which activates EpRE and subsequently increases the expression of many EpRE-driven genes, protected neuroblastoma cells from oxidative cell death [8]. Regardless, not all EpRE-driven gene expression responds equally to stimulation. Indeed, there are multiple EpRE sequences in the promoters of some genes, such as gclc [7], where only one may be involved in the response to stimulation. Furthermore, there may be differences in responses of the same EpRE sequence in different cells within the same species. Part of these differences may be in the proteins that form the binding complexes to the EpRE sequences and in the signaling for their activation.
The transcription factors that bind to EpRE have been the subject of much debate. It is accepted that Nrf2, and to a lesser extent Nrf1, a member of the NF-E2 related factor family of transcription factors (TFs) is a principal player in activation of the EpRE [9]. Under unstimulated conditions, Nrf2 in the cytoplasm associates with the bricabrac, tramtrack, and broad complex (BTB) domain-containing protein Keap1. In response to endoplasmic reticulum stress or oxidative stress, Keap1 becomes modified, which triggers Nrf2-Keap1 dissociation [10]. Nrf2 then translocates to the nucleus where it forms an active heterodimeric transcription factor inducing the transcription of target genes involved in redox homeostasis [10]. TRE or TRE-like sequence within EpRE suggested the possibility of binding of c-Fos/c-Jun dimers or other members of the Fos/Jun families of transcription factors. Studies by Jaiswal and his collaborators strongly suggest that c-Jun is a binding partner of Nrf2 in activation of EpRE-dependent transcription while members of the Fos family are repressors [11, 12]. It is well established that the phosphorylation of c-Jun is required for transcriptional activation when c-Jun plays a role in TRE-dependent transcription [13, 14]. The important issue that is addressed here is whether phosphorylation of c-Jun is also required when c-Jun plays a role in EpRE-dependent transcription.
4-Hydroxy-2-nonenal (HNE), is a major lipid peroxidation product formed by the reaction of reactive oxygen or nitrogen species with arachidonic acid in cellular membranes during exposure to tobacco smoke, inflammation, and in chronic obstructive pulmonary disease [15, 16]. The physiological concentration of HNE in the plasma is reportedly 0.3–0.7 μM [17, 18] but its concentration can approach millimolar in plasma membranes under conditions of oxidative stress [19–21]. HNE possesses both an electrophilic carbon-carbon double bond that can react in Michael additions with a wide variety of cellular components, including DNA and proteins, and an aldehydic functional group that can reversibly form Schiff bases with amines that equilibrate with free HNE [17].
Differences in the ability of cells to transcriptionally up-regulate genes are expected to contribute to the susceptibility of different types of cells to toxic insults [22]. Therefore, in this study, we compared results of two cell types. Human bronchial epithelial cells (HBE1) and human hepatoma cells (HepG2) were studied in order to verify whether the phosphorylation of c-Jun is general or varies. Our results revealed that HNE increased the induction of phase II gene products through signaling leading to transcription. The effect of c-Jun phosphorylation on these responses to HNE treatment differed between the genes but also between the two cell lines. A significant major difference between the cells was that in HBE1 cells almost all of the c-Jun in the bound to EpRE was phosphorylated, while in HepG2 cells greater than 80% of c-Jun was not phosphorylated.
Materials and Methods
Chemical and reagents
All chemicals and reagents were obtained form Sigma Chemical (St. Louis, MO) unless stated otherwise. HNE was purchased from Cayman Chemical (Ann Arbor, MI). TaqMan reverse transcription reagent and SYBR Green Master Mix were from Applied Biosystems (Foster City, CA). Fu-GENE 6 transfection reagent was from Roche (Indianapolis, IN). Anti-actin antibody was from BD transduction laboratories (San Jose, CA). Anti-p-c-Jun, anti-Jun, anti Nrf2 and anti-lamin were from Santa Cruz Biotechnology (Santa Cruz, CA). Chromatin immunoprecipitation (ChIP) assay kit was from Upstate (Chicago, IL). M-PER mammalian protein extraction reagent and NE-PER nuclear extraction reagent were from Pierce (Rockford, IL). JNK inhibitor was purchased from Calbiochem (La Jolla, CA). All chemicals used were at least analytical grade.
Cell culture
HBE1 cell were a gift from Dr James Yankaskas of the University of North Carolina. The cells were maintained at 37°C in a humidified 5% CO2 incubator and grown in serum-free F12 Ham media supplemented with 6 hormones (5μg/ml Human insulin, 1×10−6 M Hydrocortisone, 3.7 μg/ml endothelial cell growth supplement, 25 ng/ml epidermal growth factor, 3×10−8 M tri-iodothyronine, 5 μg/ml transferrin) 100 units/ml penicillin, 100 μg/ml streptomycin and 40 μg/ml gentamicin on collagen-coated dishes. HNE was dissolved in ethanol, and the final concentration of ethanol in the medium was 0.05%. HBE1 cells were treated at about 85% confluence with 10μM of HNE as indicated in the results session. HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA). The cells were maintained at 37°C in a humidified 5% CO2 incubator and grown in EMEM supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin.
Western blot
Proteins were resolved on a 4–20% Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA) under denaturing conditions before being transferred electrophoretically onto a polyvinylidene difluoride (PVDF) membrane (Immobilon P; Millipore, Bedford, MA). Membranes were blocked with 5% nonfat dry milk (NFDM) at room temperature for 1 h and then incubated overnight at 4°C with primary antibody diluted in 5% NFDM in Tris buffer saline (TBS) as indicated (1:500 anti-p-c-Jun; 1:5000 anti-actin; 1:100 anti phospho-JNK; 1:500 anti Nrf2; 1:500 c-Jun). After being washed with TBS containing 0.05% Tween 20, the membrane was incubated with goat anti-mouse IgG or goat anti-rabbit conjugated to horseradish peroxidase (1:3000) at room temperature for 2 h. The blots were developed by the enhanced chemiluminescence technique (ECL Plus, Amersham, Arlington Heights, IL) according to the manufacturer’s instructions. The bands of interest were imaged with and quantified by photon counting using the charged-coupled device camera of a Kodak Image Station 2000R (Kodak, Rochester, NY, USA) and Kodak 1D 3.6 Image Analysis Software. Photon counting was used for graphing and statistical analysis.
Immunoprecipitation
Nuclear extract contains 500 μg of proteins were precleared with protein A-Sephar-ose beads for 1 h and incubated with 5 μg of antibody to either Nrf2 or c-Jun or p-c-Jun at 4°C overnight. Immunoprecipitated complexes were washed 3 times with lysis buffer and then boiled in SDS sample buffer for 10 minutes. The immunoprecipitation products were run on acrylamide gel and electrophoretically transferred to PVDF as described above.
Transfection procedure and assay of luciferase and β-galactosidase activity
HepG2 and HBE1 (70–80% confluence) cells were plated into each well of 12-well plates and cultured for 24 h. The cells were then transfected with 0.1 μg gclc (EpRE4) or gclm (EpRE) -pGL3 promoter encoding luciferase plasmid by using FuGENE 6 transfection reagent (Roche), and β-galactosidase plasmid (1/10 of total amount of plasmids) was cotransfected as an internal control. Twelve hours after transfection, the medium was replaced; 12 h later, the cells were treated with HNE in 0.05% ethanol, 0.05% ethanol as a control, or JNK inhibitor for 24 h. The cell pellet was lysed with M-PER mammalian protein extraction reagent (Pierce). The supernatant was then used for determination of the activity of luciferase and β-galactosidase. To determine the β-galactosidase activity, 25 μl of supernatant was added to a reaction mixture containing 300 μM 4-methyllumbelliferyl β-D-galactoside. After incubation at room temperature for 20 min with shaking, β-galactosidase activity was determined in a fluorescence microplate reader (Molecular Device Corp., Sunnyvale, CA) at an excitation wavelength of 360 nm and an emission wavelength of 450 nm. For the luciferase assay, a luciferase assay kit (Promega) was used. Briefly, 20 μl of cell lysate was added to the reaction mixture provided in the kit, and luciferase activity was determined in a luminometer (Berthold Detection Systems, Pforzheim, Germany). The final luciferase activity was normalized with the activity of co-transfected β-galactosidase.
The primers sequences used were the following: GCLC (EpRE4), (−3154:−3125) sense 5′-CCGCACAAAGCGCTGAGTCACGGGGAGGCG-3′, antisense 5′-GGCGTGTTTCGCGACTCAGTGCCCCTCCGCG-3′; GCLM (EpRE), (−312:−283) sense 5′-CGCTACGATTTCTGCTTAGTCATTGTCTTCC-3′, antisense 5′-GCGATGCTAAAGACGAATCAGTAACAGAAG-3′.
ChIP Assay
ChIP assays were performed following a protocol provided with the kit from Upstate. Briefly, cells were incubated with formaldehyde by directly adding it into the medium (1% final concentration) at room temperature [23] for 10 min. The cell pellet was then lysed on ice for 10 min and sonicated under conditions that cause DNA to be broken into 200- to 800-bp fragments. Sonicated cell lysate was pre-cleared with 60 μl of salmon sperm DNA/agarose, and the supernatant was used for immunoprecipitation with antibodies to specific transcription factors overnight at 4°C. The protein/DNA complex was eluted from agarose in elution buffer and the DNA/protein complex was reversed by adding 5 M NaCl and incubating the mixture at 65°C for overnight. The DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1). Primers used for PCR in the ChIP assay were h-NQO-2 Sense 5′-CCTTCCTTCTGCTCTATTGTTG-3′; Antisense 5′-GGCGGTGCTGGTGAACAGTT-3′; GCLC (AP-1) Sense 5′-CATTCTGCCGCTCTCACTCTAA-3′ Antisense 5′-CTTTACGCAAACGCGACATATC-3′; GCLM (137 bp) Sense 5′-AAGGGCCAGTCACTTCGG-3′ Antisense 5′-AGCTGTTTCCTGGAAGACAATG-3′; GCLC (EpRE4) (127 bp) Sense 5′-ATCGACTGCGGCAATCCTAG-3′ Antisense 5′-CGTGACTCAGCGCTTTGTG-3′.
Immunoprecipitation analysis was conducted using control rabbit IgG, anti-Nrf2 (Santa Cruz; H-300, sc-13032, however (c-20) was examined as well), anti-c-Jun (Santa Cruz; h79), anti-p-c-Jun (SANTA Cruz), anti-ATF2 (Santa Cruz), anti-Nrf1 (Santa Cruz) and anti-RNA polymerase II (Santa Cruz; sc-899) antibodies.
2% of the chromatin DNA was also subjected to PCR analysis and indicated as input. PCR products were quantified using iQ SYBR Green Supermix (Bio-Rad) with the Chromo 4™ Real-Time PCR Detection System (Bio-Rad). Specific enrichment of DNA by anti-Nrf2, anti-c-Jun anti-p-c-Jun anti-Nrf1 and anti-ATF2 antibodies was normalized by the PCR value of certain antibody to the PCR input value.
Statistical Analysis
Results are reported as mean ± standard error for n=3–6, independent experiments, as indicated in the legends. Statistical significance was accepted when p < 0.05. Comparison of variants between experimental groups was performed with ANOVA and Tukey’s test.
Results
Expression of transcription factors following treatment with HNE
We determined the nuclear content as well as the cytosolic content of Nrf2 and p-c-Jun/c-Jun in both HBE1 and HepG2 cells. As shown in Figure 1, Nrf2 accumulated in the nucleus of HBE1 cells following treatment with 10 μM HNE. In HBE1 cells, the maximal Nrf2 content was achieved after 3 h stimulation, and then decreased. Over the same time period, the amount of Nrf2 in the cytosol decreased (Fig. 1A). In HepG2 cells, however, Nrf2 translocation to the nucleus increased during the first hour of exposure then declined by 4 h (Fig. 1B).
Fig. 1. HNE increases the amount of endogenous nuclear Nrf2 in a time-dependent manner.

A, Translocation of Nrf2 into the nucleus of HBE1 cells. B, Effects of HNE on Nrf2 in HepG2 cells. C, Changes in nuclear Nrf2 of HBE1 cells. D, Changes in nuclear Nrf2 of HepG2 cells. Both cells were treated with 10 μM HNE for the indicated time points. The nuclear and cytosolic extracts containing 30 μg of proteins were prepared and subjected to Western blot analysis. Anti- actin or lamin antibodies were used as markers for the cytosolic and nuclear extracts, respectively.
The ratio between p-c-Jun and c-Jun in the nucleus of HBE1 cells increased after stimulation with HNE and was maximal after 3 h then decreased (Fig. 2A). In HepG2 cells, the ratio between p-c-Jun and c-Jun did not increase but rather decreased following 30 min of HNE exposure (Fig. 2B). No further changes were observed at longer times of exposure. No changes were observed in the nuclear content of either c-Fos or Maf G/F/K (data not shown).
Fig. 2. HNE increases phosphorylation of c-Jun in a time-dependent manner.

HBE1 (A) and HepG2 (B) cells were treated with 10 μM HNE for the indicated time points. The nuclear extract was prepared and subjected to Western blot analysis with indicated antibodies. Anti actin antibody was used as markers for the nuclear extract. Results are reported as mean ± S.E. of three determination.*p<0.05, **p<0.01 significantly different compared with control.
Recruitment of Nrf2, c-Jun and p-c-Jun to EpRE, upon HNE treatment in vivo
To determine the recruitment of transcription factors (Nrf2, c-Jun and p-c-Jun) to EpRE in vivo, we performed ChIP assays. HepG2 cells: Exposure to 10 μM HNE was associated with increased binding of c-Jun to the promoters of nqo2, gclc and gclm by 5.8, 4.8 and 4 fold, respectively (Fig. 3A). Surprisingly, binding of Nrf2 was not increased by HNE despite the increase in nuclear Nrf2 described above.
Fig. 3.


Fig. 3A. Effect of HNE on the recruitment of transcription factors to EpRE of (A) nqo2, (B) gclc and (C) gclm in HepG2 cells. ChIP assays were performed to examined the interaction in vivo of Nrf2, c-Jun and p-c-Jun with EpRE that either treated or untreated with HNE for 3 h. After 3 h stimulation of cells with 10 μM HNE, cells were fixed with1% formaldehyde, lysed, and sonicated to shear chromatin in 0.2–0.8-kb fragments, which were then immunoprecipitated with anti-Nrf2, c-Jun and p-c-Jun antibodies. Pure DNA then analyzed with RT-PCR with specific primers amplifying EpRE elements. Results are reported as mean ± S.E. of at least three determination. *p<0.05, **p<0.01 significantly different compared with control.
Fig. 3B. Effect of HNE on the recruitment of transcription factors to EpRE of (A) nqo2, (B) gclc (EpRE4), (C) gclc (AP-1) and (D) gclm in HBE1 cells. ChIP assays were performed to examined in vivo the interaction of Nrf2, c-Jun and p-c-Jun with EpRE that either treated or untreated with HNE. After 3 h stimulation of cells with 10 μM HNE, cells were fixed with1% formaldehyde, lysed, and sonicated to shear chromatin in 0.2–0.8-kb fragments, which were then immunoprecipitated with anti-Nrf2, c-Jun and p-c-Jun antibodies. Pure DNA then analyzed with RT-PCR with specific primers amplifying EpRE elements. Results are reported as mean ± S.E. of at least three determination. *p<0.05, **p<0.01 significantly different compared with control.
HBE1 cells
Recruitment to the EpRE sequences of nqo2, gclc (EpRE4 and TRE) and gclm of the phos-phorylated form of c-Jun, p-c-Jun, increased dramatically following exposure to HNE (Fig. 3B). The HNE-induced fold increase in p-c-Jun binding was 8 (nqo2 EpRE), 8 (gclc EpRE4), 12 (gclm EpRE), and 24 (gclc TRE), respectively. The only significant increase in the recruitment of Nrf2 to EpRE after stimulation with HNE, appeared in gclm, where a 5-fold increase was observed (Fig. 3B). No changes in recruitment to EpRE were observed for either ATF2 or Nrf1 (data not shown).
Interaction of Nrf2 with c-Jun and p-c-Jun
It was observed that the amount of both Nrf2 and p-c-Jun in the nuclear extracts of HBE1 cells increased following treatment with HNE (Figs. 4 and 5). In order to determine whether c-Jun and/or p-c-Jun bound directly to Nrf2 we performed immunoprecipitation using nuclear extracts from both cell types. In HepG2 cells, immunoprecipitation of either c-Jun or p-c-Jun followed by immunobloting with anti Nrf2 revealed that Nrf2 bound to both c-Jun and p-c-Jun (Fig. 4). Similar results were found with HBE1 cells (Fig. 5). Negative results were obtained using the immunoprecip-itation of Nrf2 followed by immunobloting for p-c-Jun, which was likely due to the very small amount of Nrf2 in cells combined with a low ratio of binding of p-c-Jun compared with other transcription factors so that p-c-Jun protein was below the level of detection.
Fig. 4. Interaction of Nrf2 with either c-Jun or p-c-Jun.

HepG2 cells were treated with or without 10μM HNE for 1 hour. Nuclear proteins were immunoprecipitated with the indicated antibodies: (A) p-c-Jun (B) c-Jun (C) Nrf2, and visualized by Western blot analysis with Nrf2 antibody.
Fig. 5. Interaction of Nrf2 with either c-Jun or p-c-Jun.
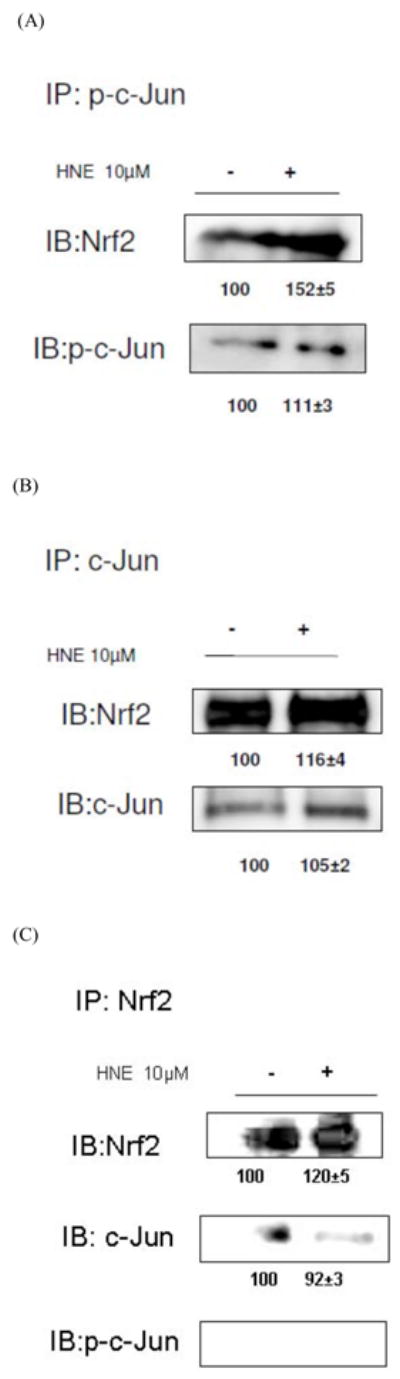
HBE1 cells were treated with or without 10μM HNE for 3 hours. Nuclear proteins were immunoprecipitated with the indicated antibodies; (A) p-c-Jun (B) c-Jun (C) Nrf2, and visualized by Western blot analysis with Nrf2 antibody.
Does HNE activation of the JNK pathway affect GCL-EpRE-driven gene expression?
In whole cell extracts, the increase of p-JNK, following treatment with 10 μM HNE was 1.4 and 1.6 in HepG2 and HBE1 cells, respectively (Fig. 6A and B). In HBE1 cells, HNE increased phosphorylation of c-Jun by 20%, at 30 min. JNKi, not only prevented the HNE-induced increase but decreased p-c-Jun to 60% of the initial amount (data not shown). To examine whether the induction by HNE of GCLM and GCLC through their EpRE cis elements involved the phosphorylation of c-Jun, we performed a promoter activity assay on both genes by transfecting cells with constructs containing the EpRE promoter of the gclm gene or the EpRE4 promoter sequence of the gclc gene using JNKi to prevent c-Jun phosphorylation. HepG2 cells treated with 15 μM HNE increased their EpRE- and EpRE4- promoter activity by 30 % and 19 %, respectively (Fig. 7A and B). In order to study if JNK is involved in the signaling pathway leading to induction through EpRE, we used a cell permeable peptide that inhibits JNK (JNKi) [24]. In HepG2 cells, JNKi did not significantly inhibit the luciferase activity that arose after HNE treatment with gclm EpRE and gclc EpRE4 driven luciferase expression (Fig. 7A and B). In HBE1 cells, HNE increased gclm EpRE-driven and gclc EpRE4-driven promoter activity by 2.5 and 1.45 times fold, respectively, compared to un-stimulated cells (Fig. 7C and D). JNKi inhibited basal expression of the gclc-EpRE4 driven reporter by 44% but did not affect basal gclm EpRE regulated luciferase expression (Fig. 7D and C). Surprisingly, JNKi caused an increase in HNE-induced expression of the gclm EpRE-driven reporter.
Fig. 6. Effect of HNE on the phosphorylation of JNK in HepG2 (A) and HBE1 (B) cells.

Cells were treated with 10 μM HNE for the indicated time points. The whole cell extracts containing 40 μg of proteins were prepared and subjected to Western blot analysis with the indicated antibodies. Anti-actin antibody was used as marker for the cell extracts. Results are reported as mean ± S.E. of three determination. *p<0.05, **p<0.01 significantly different compared with control.
Fig. 7. Signal pathway involved in HNE induced gclc (EpRE4) and gclm (EpRE) promoters in HepG2 cells.

HepG2 (A and B) and HBE1 (C and D) cells were transfected with EpRE- gclm (A and C) or EpRE4- gclc luciferase (B and D) promoter plasmids for 24 h and pretreated with JNK inhibitor (JNKi) for 1 h before treated with or without 15 μM HNE. Luciferase activity was determined 24 h after HNE treatment and transfection efficiency was controlled by normalization with cotransfected β galactosidase activity. Results presented are relative to control (pGL-3 promoter luciferase vector). Results are reported as mean ± S.E. of three determination. *p<0.05, **p<0.01 significantly different compared with control. #p<0.05 significantly different compared with treatment.
Discussion
In the present study we examined whether the c-Jun bound to the functional EpRE sequences of two genes needs to be phosphorylated by JNK in order to increase transcription. In order to know if the results were general or variable, we used two different cell types; human hepatoma (HepG2) and human bronchial epithelial (HBE1) cells. The two cell lines used in this report are homogeneous cultures of transformed, immortalized cells. These cells mimic their normal counterparts in many respects and maintain varying degrees of lineage specific traits. For example, HepG2 cells, derived from a human hepato-cellular carcinoma, display morphological and biochemical features of hepatocytes [25] and have been utilized as an in vitro model of liver cells for decades. The HBE1 cells, a papilloma virus-immortalized human bronchial epithelial cell line, derived from human lung, have also been used for many years [26]. HBE1 cells maintain similar responses to HNE as do normal human bronchial epithelial cells in terms of GCL induction (unpublished observation). While direct extrapolation from the current studies to regulation of phase II genes expression in specific cell types in vivo is not intended, the observed variations in regulation of these genes are indicative of the existence of differential regulatory mechanisms, which may segregate in a cell-type specific manner. We demonstrated that HNE, an α,β-unsaturated aldehyde that is a major product derived from peroxidation of ω-6 polyunsaturated fatty acids, regulates EpRE activation of phase II genes in both cells. We have shown, herein, distinct differences in the regulation of the EpRE elements between cell types in both basal and inducible expression.
In the resting state, most Nrf2 associates with Keap 1, which is anchored to the cytoskeleton [27, 28]. Keap1 however, mediates the ubiquitination and degradation of Nrf2 [29]. Upon exposure to stress, Keap1 is modified so that it does not bind Nrf2, thereby allowing Nrf2 to escape degradation and translocate to the nucleus. This study shows dramatic translocation of Nrf2 from cytoplasm to the nucleus as a result of HNE treatment in HBE1 cells. In HepG2 cells, only a moderate increase was observed in the nucleus; however it appeared during the first 1 h of exposure, then declined by 3 h (Fig. 1) while in HBE1 cells the timing of the increase (maximal at 3 h) differed markedly. Following translocation of Nrf2 to the nucleus, it forms complexes that transactivate gene expression by binding to EpRE in a large number of proteins. Several transcription factors, such as c-Jun, ATF2, and ATF4, have been proposed as potential partners of Nrf2 of EpRE [11, 12, 30–33]. The composition of the transcription factor complexes binding to EpRE sequences has been the subject of controversy but may actually vary depending on cell type, stimuli, treatment and the gene in question. Nrf2/EpRE signaling has been found to be responsible for the induction of many phase II genes in response to electrophiles, including NQO1 [3], NQO2 [4] and GCL [5–7, 34]. In this study, we have shown that the recruitment of c-Jun to EpRE sequences of nqo2, gclc and gclm genes increases following exposure to HNE, in HepG2 cells (Fig. 3A). However, in HBE1 cells, the recruitment of only phosphorylated c-Jun increased following exposure to HNE. This increment was observed for all 3 genes (Fig. 3B).
It has been shown that for AP-1 complexes, the c-Jun protein must be phosphorylated to be transcription-ally active [13]. For EpRE binding complexes, there is a debate about whether c-Jun or other small Mafs are the partner of Nrf2/Nrf1 [11, 35, 36]. Previous studies in our laboratory, using electrophoretic mobility shift assays (EMSA) showed that binding of small Mafs to a consensus EpRE sequence and nuclear extracts from HBE-1 cells stimulated with curcumin (an HNE analogue) actually declined while binding of JunD and phosphorylated c-Jun increased [5]. EMSA, an ex vivo assay, provided evidence for the potential involvement of phosphorylated c-Jun. Thus, here we investigated whether c-Jun needed to be phos-phorylated to bind to and/or activate EpRE for phase II gene expression in living cells using two other approaches. ChIP assays allowed determination of binding in the context of the gene in situ. EpRE-driven reporters in combination with a JNK-specific inhibitor peptide, that we have previously demonstrated to inhibit HNE-activated c-Jun phosphorylation and gene expression [5], allowed the determination of whether of c-Jun phosphorylation was required for EpRE-driven gene expression. Nonetheless, rather than finding an absolute requirement for c-Jun phosphorylation, differences between the human two cell lines in recruiting c-Jun or phosphorylated c-Jun to EpRE were observed.
Another noteworthy point is that even though there was increased translocation of Nrf2 to the nucleus in both cell lines and we and others have shown that silencing of Nrf2 decreases expression of these EpRE-regulated genes, a significant increase in Nrf2 binding to EpRE occurred only in gclm in HBE cells (Fig. 3B). Nrf2 activation and translocation appears to be the most common factors in EpRE-mediated gene expression [11, 12, 31, 32, 37, 38] and our results do not conflict with this. Rather, the results suggest that activation of Nrf2 translocation by HNE maintains but does not increase Nrf2 binding to the EpRE sequences studied here with the exception of gclm in HBE1 cells. These results were in agreement with our previous work [39] using the in vitro EMSA approach, which demonstrated that the total binding of proteins to the gclc EpRE sequence observed as three bands was unchanged by HNE treatment in HBE1 cells although changes in the relative density of three bands occurred. This suggested that changes in composition of the EpRE binding complexes were changed by HNE. In HepG2 cells, where Nrf2 was decreased, it is possible that other transcription factors, such as Nrf1 or c-Fos compete with or inhibits Nrf2 binding to EpRE to a small extent under the conditions used here.
Phase II gene expression in response to HNE varies considerably within, as well as between, the different cell lines, suggesting that additional mechanisms might be involved in the induction of the genes beside EpRE-dependence. In this study we have shown that HNE induced the activity of both gclm and gclc promoter genes, as determined by reporter assays (Fig. 7). Several studies have suggested a differential induction of either the gclc or gclm genes. In cultured rat hepatocytes, for example, only GCLC mRNA was up-regulated by hydrocortisone or insulin, while treatment with buthionine sulfoximine (BSO) or the oxidants tert-butyl hydroquinone (tBHQ) and diethyl maleate (DEM) increased expression of both subunits to a similar extent [40]. Similarly, in cultured rat lung epithelial cells, expression of both subunits is up-regulated by the specific GCL inhibitor BSO [41] and by the redox-cycling quinone, 2,3-dimethoxy-1,4-naphthoquinone (DMNQ) [41, 42]. In HepG2 cells, preferential up-regulation of the gclc gene has been observed in cells incubated under hypoxic conditions or in the presence of the iron chelator desferrioxam-ine [22]. Differential subunit regulation apparently extends to down-regulation of expression as well. In mice constitutively expressing the HIV-1 encoded transactivator protein, Tat, GCLM mRNA levels are lower than in controls in some tissues, while levels of GCLC mRNA are not affected [43]. While many possible mechanisms could be invoked to explain these differences in the regulation of GCL subunit expression, none have yet been verified experimentally.
Our results obtained with ChIP assays (Fig. 3B) in HBE1 cells, suggested a role for JNK in the activation of nqo2, gclc and gclm genes following treatment with HNE. We have shown a 5 fold increase in the phosphorylation of c-Jun (Fig. 2A) as well as 1.6 fold increase in the phosphorylation of JNK 1 (Fig. 6B) following treatment with HNE. Regardless, while gclc (EpRE4) promoter activity inresponse to HNE was slightly inhibited by JNKi, JNKi alone decreased basal expression (Fig. 7D). Surprisingly, the promoter activity of a gclm EpRE-driven reporter was actually increased by JNKi suggesting that phosphorylation by JNK actually had an inhibiting role. This suggests that JNK played a variable role in regulating transcription through EpRE in HBE1 cells even though phosphorylation of c-Jun in the EpRE binding complexes were consistently markedly increased. Several years ago, our lab suggested that the role of JNK in activation of gclc in HNE-treated HBE1 cells was more through TRE than EpRE based upon studies using ex vivo electrophoretic mobility shift studies and JNK inhibition [44]. The relatively greater increase of the p-c-Jun/AP-1/TRE (24 fold) compared to the increase in p-c-Jun/EpRE4 in the gclc promoter (8 fold) in HBE1 cells (Fig. 3B) was consistent with that suggestion. Moreover, the results with EpRE-driven promoters suggests that the major dependence upon activation of JNK, which was essential for HNE induction of GCLC and GCLM in HBE1 cells was, as suggested by our earlier study [44], in activating AP-1/TRE rather than EpRE. In HepG2 cells, however, no increase in phosphorylation of c-Jun and only minor increase in p-JNK was observed following HNE treatment (Fig. 2B and 6A). Additionally, no phosphorylation was required for the recruitment of c-Jun to EpRE following activation of transcription (Fig. 3A). This suggests that JNK has a more dominant role in the response to HNE in HBE1 cells compared with HepG2 cells. The mechanism of how HNE activates the JNK pathway is a related but separate issue. Recently, however, we have shown that HNE activated JNK and its upstream protein kinase kinase, MKK4, in HBE1 cells through the inhibition of the protein tyrosine phosphatase, SHP-1 [45].
The present study addressed the issue of whether or not phosphorylation of the c-Jun that has been reported to be a component of the EpRE binding complex needs to be phosphorylated in order to facilitate transcription. While the results suggest that there is variability in the response among genes and cell types, the broader implication is that regulation of transcription through EpRE is far more complex than currently thought. Any attempt to regulate the transcription of a gene must then take into consideration the observation that the same cis element may be regulated differently in one gene than another and that even the same cis element in the same gene may be differentially regulated in one cell versus another. Thus, for development of therapeutic approaches based on regulation of transcription, this will present an enormous challenge.
Acknowledgments
This work was supported by a grants from the National Institutes of Environmental Health Sciences (ES05511) and the California Tobacco Related Diseases Research Program (14RT-0059). The authors thank Drs. Hongqiao Zhang and Jinah Choi for their useful comments and suggestions.
List of abbreviations
- HNE
4-hydroxy-2-nonenal
- EpRE
electrophile responsive elements
- GST
gluta-thione S-transferase
- AP-1
activator protein-1
- GCLC
catalytic subunit of GCL
- GCLM
modulator subunit of GCL
- NQO2, NAD(P)H
quinone oxidoreductase 2
- NQO1, NAD(P)H
quinone oxidoreductase-1
- HepG2
human hepatoma
- HBE1
human bronchial epithelial
- Nrf2
NF-E2 related factor 2; activation transcription fator-2
- JNK
Jun N-terminal kinase
- EMSA
electrophoretic mobility shift assay
- JNKi
JNK inhibitor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 2.Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- 3.Li Y, Jaiswal AK. Human antioxidant-response-element-mediated regulation of type 1 NAD(P)H:quinone oxidoreductase gene expression. Effect of sulfhydryl modifying agents. Eur J Biochem. 1994;226:31–39. doi: 10.1111/j.1432-1033.1994.tb20023.x. [DOI] [PubMed] [Google Scholar]
- 4.Harada S, Fujii C, Hayashi A, Ohkoshi N. An association between idiopathic Parkinson’s disease and polymorphisms of phase II detoxification enzymes: glutathione S-transferase M1 and quinone oxidoreductase 1 and 2. Biochem Biophys Res Commun. 2001;288:887–892. doi: 10.1006/bbrc.2001.5868. [DOI] [PubMed] [Google Scholar]
- 5.Dickinson DA, Iles KE, Zhang H, Blank V, Forman HJ. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. Faseb J. 2003;17:473–475. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- 6.Mulcahy RT, Gipp JJ. Identification of a putative antioxidant response element in the 5′-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Biophys Res Commun. 1995;209:227–233. doi: 10.1006/bbrc.1995.1493. [DOI] [PubMed] [Google Scholar]
- 7.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272:7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Johnson JA. Time-dependent changes in ARE-driven gene expression by use of a noise-filtering process for microarray data. Physiol Genomics. 2002;9:137–144. doi: 10.1152/physiolgenomics.00003.2002. [DOI] [PubMed] [Google Scholar]
- 9.Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 13.Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 14.Franklin CC, Sanchez V, Wagner F, Woodgett JR, Kraft AS. Phorbol ester-induced amino-terminal phosphorylation of human JUN but not JUNB regulates transcriptional activation. Proc Natl Acad Sci U S A. 1992;89:7247–7251. doi: 10.1073/pnas.89.15.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoshiba K, Koinuma M, Yokohori N, Nagai A. Immunohistochemical evaluation of oxidative stress in murine lungs after cigarette smoke exposure. Inhal Toxicol. 2003;15:1029–1038. doi: 10.1080/08958370390226431. [DOI] [PubMed] [Google Scholar]
- 16.Yang K, Fang JL, Hemminki K. Abundant lipophilic DNA adducts in human tissues. Mutat Res. 1998;422:285–295. doi: 10.1016/s0027-5107(98)00210-3. [DOI] [PubMed] [Google Scholar]
- 17.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 18.Strohmaier H, Hinghofer-Szalkay H, Schaur RJ. Detection of 4-hydroxynonenal (HNE) as a physiological component in human plasma. J Lipid Mediat Cell Signal. 1995;11:51–61. doi: 10.1016/0929-7855(94)00027-a. [DOI] [PubMed] [Google Scholar]
- 19.Schwarzer E, Muller O, Arese P, Siems WG, Grune T. Increased levels of 4-hydroxynonenal in human monocytes fed with malarial pigment hemozoin. A possible clue for hemozoin toxicity. FEBS Lett. 1996;388:119–122. doi: 10.1016/0014-5793(96)00523-6. [DOI] [PubMed] [Google Scholar]
- 20.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 21.Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 2008 doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahl EL, Mulcahy RT. Cell-type specific differences in glutamate cysteine ligase transcriptional regulation demonstrate independent subunit control. Toxicol Sci. 2001;61:265–272. doi: 10.1093/toxsci/61.2.265. [DOI] [PubMed] [Google Scholar]
- 23.Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/s0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- 24.Iles KE, Dickinson DA, Watanabe N, Iwamoto T, Forman HJ. AP-1 activation through endogenous H(2)O(2) generation by alveolar macrophages. Free Radic Biol Med. 2002;32:1304–1313. doi: 10.1016/s0891-5849(02)00840-7. [DOI] [PubMed] [Google Scholar]
- 25.Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science. 1980;209:497–499. doi: 10.1126/science.6248960. [DOI] [PubMed] [Google Scholar]
- 26.Yankaskas JR, Haizlip JE, Conrad M, Koval D, Lazarowski E, Paradiso AM, Rinehart CA, Jr, Sarkadi B, Schlegel R, Boucher RC. Papilloma virus immortalized tracheal epithelial cells retain a well-differentiated phenotype. Am J Physiol. 1993;264:C1219–1230. doi: 10.1152/ajpcell.1993.264.5.C1219. [DOI] [PubMed] [Google Scholar]
- 27.Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224:171–184. doi: 10.1016/j.canlet.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 29.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayes JD, McMahon M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001;174:103–113. doi: 10.1016/s0304-3835(01)00695-4. [DOI] [PubMed] [Google Scholar]
- 31.Itoh K, Ishii T, Wakabayashi N, Yamamoto M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic Res. 1999;31:319–324. doi: 10.1080/10715769900300881. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen T, Huang HC, Pickett CB. Transcriptional regulation of the antioxidant response element. Activation by Nrf2 and repression by MafK. J Biol Chem. 2000;275:15466–15473. doi: 10.1074/jbc.M000361200. [DOI] [PubMed] [Google Scholar]
- 33.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 34.Moinova HR, Mulcahy RT. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun. 1999;261:661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 35.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 36.McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, Wolf CR, Cavin C, Hayes JD. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 37.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 38.Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem. 2000;275:27694–27702. doi: 10.1074/jbc.M004729200. [DOI] [PubMed] [Google Scholar]
- 39.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 40.Cai J, Huang ZZ, Lu SC. Differential regulation of gamma-glutamylcysteine synthetase heavy and light subunit gene expression. Biochem J. 1997;326(Pt 1):167–172. doi: 10.1042/bj3260167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian L, Shi MM, Forman HJ. Increased transcription of the regulatory subunit of gamma-glutamylcysteine synthetase in rat lung epithelial L2 cells exposed to oxidative stress or glutathione depletion. Arch Biochem Biophys. 1997;342:126–133. doi: 10.1006/abbi.1997.9997. [DOI] [PubMed] [Google Scholar]
- 42.Shi MM, Kugelman A, Iwamoto T, Tian L, Forman HJ. Quinone-induced oxidative stress elevates glutathione and induces gamma-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J Biol Chem. 1994;269:26512–6517. [PubMed] [Google Scholar]
- 43.Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ. Molecular mechanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem. 2000;275:3693–698. doi: 10.1074/jbc.275.5.3693. [DOI] [PubMed] [Google Scholar]
- 44.Forman HJ, Dickinson DA, Iles KE. HNE--signaling pathways leading to its elimination. Mol Aspects Med. 2003;24:189–194. doi: 10.1016/s0098-2997(03)00013-x. [DOI] [PubMed] [Google Scholar]
- 45.Rinna A, Forman HJ. SHP-1 inhibition by 4-hydroxynonenal activates Jun N-terminal kinase and glutamate cysteine ligase. Am J Respir Cell Mol Biol. 2008;39:97–104. doi: 10.1165/rcmb.2007-0371OC. [DOI] [PMC free article] [PubMed] [Google Scholar]