

Abstract
Macrophages ingesting apoptotic cells attenuate inflammatory responses, such as reactive oxygen species (ROS) generation. In atherosclerosis, ongoing inflammation and accumulation of apoptotic/necrotic material are observed, suggesting defects of phagocytes in recognizing or responding to dying cells. Modified lipoproteins such as oxidized LDL (oxLDL) are known to promote inflammation and to interfere with apoptotic cell clearance. Here, we studied the impact of cells exposed to oxLDL on their ability to interfere with the oxidative burst in phagocytes. In contrast to apoptotic cells, cells dying in response to or in the presence of oxLDL failed to suppress ROS generation despite efficiently being taken up by phagocytes. In addition, apoptotic cells, but not oxLDL-treated cells, inhibited phosphorylation of extracellular signal-regulated kinase, which is important for NADPH oxidase activation. oxLDL treatment did not interfere with activation of the antiinflammatory transcriptional regulator peroxisome proliferator-activated receptor γ by apoptotic cells. Moreover, cells exposed to oxLDL failed to suppress lipopolysaccharide- induced proinflammatory cytokine expression, whereas apoptotic cells attenuated these phagocyte responses. Thus, the presence of oxLDL during cell death impaired the ability of apoptotic cells to act antiinflammatory with regard to oxidative burst inhibition and cytokine expression in phagocytes.
Keywords: apoptosis, macrophages, NADPH oxidase, lipoproteins, phagocytosis, reactive oxygen species
An immunologically silent removal of dying cells by professional phagocytes and bystander cells is a prominent feature of programmed cell death. Phagocytes ingesting apoptotic material show diminished inflammatory responses toward pathogen stimulation, thus displaying a desensitized phenotype (1, 2). A major characteristic of this phenotype shift is the attenuated production of reactive oxygen species (ROS), known as the oxidative burst, upon phagocytosis of pathogens or stimulation with phorbol esters (3, 4). Mechanistically, inhibition of the oxidative burst by apoptotic cells was suggested to involve peroxisome proliferator-activated receptor (PPAR)γ, which prevented translocation of protein kinase Cα to the membrane (3). A reduced ROS-producing capacity of macrophages is accompanied by attenuated NO production via enhanced arginase II expression (5) and a switch from pro- to antiinflammatory cytokine production (6–8). These responses are supposed to suppress inflammation following the contact with apoptotic cells.
Cell death accompanies atherosclerosis at all stages of disease progression but is particularly pronounced in advanced plaques (9). These plaques are characterized by the formation of a necrotic core, which is mainly composed of free cholesterol and material derived from dying macrophages and smooth muscle cells. The size of the necrotic core correlates with plaque instability and the risk of plaque rupture (10). Defects in apoptotic cell clearance by phagocytes are now recognized as a fundamental process in the formation of the necrotic core (9, 11). Among the factors attenuating phagocytosis in atherosclerotic plaques, oxidized LDL (oxLDL) has gained considerable importance. Several mechanisms may account for the detrimental effects of oxLDL on apoptotic cell clearance. Specifically, oxLDL interferes with the recognition of apoptotic cells by competing with epitopes on apoptotic cells for the interaction with the CD36 receptor on macrophages (12–16). Alternatively, oxLDL induces oxidative stress, and oxidative stress may avert phagocytosis of apoptotic cells (17). In addition, oxLDL-derived material may be trapped in macrophage lysosome structures following oxLDL uptake (11). This may cause lysosomal dysfunction and account for impaired phagocytosis in atherosclerosis (17).
Although the nature of cell death may affect phagocytosis of dying cells, little is known about phagocytosis of cells dying in response to oxLDL. Similarly, the impact of oxLDL-treated cells on macrophage desensitization remains unclear. It was reported that recognition of murine macrophages undergoing apoptosis in response to oxLDL is normal and suppresses proinflammatory cytokine production, while the uptake of macrophages dying after free cholesterol overload provokes a proinflammatory phagocyte phenotype (18).
We compared the oxidative burst in phagocytes following their interaction with apoptotic cells generated by the treatment with staurosporine versus cells dying in response to or in the presence of oxLDL. We observed that oxLDL-treated cells were incapable of blocking the oxidative burst, although their uptake by phagocytosis was normal. Cells exposed to oxLDL retained their ability to activate PPARγ in macrophages but inefficiently suppressed proinflammatory cytokine formation. Taken together, these data indicate that proinflammatory signals dominate the phenotype shift in macrophages upon contact with cells dying in response to oxLDL.
MATERIALS AND METHODS
Cell culture
The mouse macrophage cell line RAW264.7, human THP-1 monocytic cells, and Jurkat T-cells were maintained in RPMI 1640 containing 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% heat-inactivated fetal calf serum. Human peripheral blood mononuclear cells were isolated from buffy coats using Ficoll density centrifugation and plated in serum-free medium for 1 h followed by washing of nonadherent cells. Monocytes were then differentiated into macrophages with RPMI 1640 containing 10% AB-positive human serum (PAA Laboratories) for 7 days. The investigation conforms to the principles outlined in the Declaration of Helsinki.
LDL isolation and treatment
Human LDL (d = 1.02–1.06 g/ml) was isolated from plasma of healthy volunteers (DRK-Blutspendedienst Baden-Württemberg-Hessen, Institut für Transfusionsmedizin und Immunhämatologie Frankfurt am Main, Frankfurt, Germany) by sequential ultracentrifugation. oxLDL was prepared by incubating LDL with 5 µM CuSO4 at room temperature for 24 h followed by dialysis against PBS with 100 µM EDTA. Oxidation was monitored by measuring relative electrophoretic mobility of LDL and oxLDL in agarose gels (relative electrophoretic mobility of 3–4) and by spectrophotometric detection of thiobarbituric acid reactive substances (6–8 nmol/mg). Endotoxin content of the preparations was <1 ng/mg oxLDL (Pyrotell assay, Associates of Cape Cod, Falmouth, MA).
Apoptosis induction
Apoptosis was induced in THP-1 and Jurkat cells by treatment with 0.5 µg/ml staurosporine for 3 h in serum-free medium. Secondary necrosis was induced by treatment with 0.5 µg/ml staurosporine for 36 h. For oxLDL-induced cell death, Jurkat and THP-1 cells were treated for 8 h with 25 µg/ml and 75 µg/ml oxLDL, respectively. Coincubation of oxLDL and staurosporine was carried out for 3 h. In all cases, cells were washed two times in serum-free medium prior to their exposure to macrophages.
Apoptosis analysis
Apoptotic cells were analyzed by flow cytometry (FACS Canto, BD Biosciences, Heidelberg, Germany) using the Annexin V-FITC apoptosis detection kit (Beckman Coulter, Krefeld, Germany) according to the manufacturer's instructions. Caspase-3/7 activity was quantified as described previously (19).
Oxidative burst
Superoxide production by RAW264.7 cells was analyzed by flow cytometry measuring hydroethidine (HE) oxidation as described (3). Briefly, cells were plated on 3 cm dishes at 1.5 × 105 cells/dish 24 h before the experiment. Human macrophages were plated at 2 × 105/well in 24-well plates. Following their preincubation with apoptotic cells at a ratio of 1:5 (macrophages:AC), macrophages were washed two times with PBS and subsequently resuspended in PBS and treated for 20 min with 1 µM phorbol 12-myristate 13-acetate (PMA). Thereafter, 2 µM HE was added and incubations continued for 20 min before analysis. Macrophages were gated according to their forward scatter/side scatter characteristics. Data from 10,000 gated cells were collected. Quantitative analysis was performed using PE median fluorescence intensities. Data were normalized to median fluorescence intensity values of PMA-treated cells.
Phagocytosis
Phagocytosis of apoptotic cells by human primary macrophages was assessed by fluorescent microscopy. Jurkat cells were labeled with 0.5 µM 5-chloromethylfluorescein diacetate (CMF-DA, Invitrogen, Karlsruhe, Germany) before apoptosis induction. Apoptotic cells were washed two times and incubated with macrophages prelabeled for 30 min with 5 µM CellTracker Orange CMRA (Invitrogen) at a ratio of 1:5 (macrophages:AC). Following 1 h incubations, macrophages were washed three times with PBS and analyzed by microscopy. At least 300 macrophages were counted and the data are expressed as phagocytic index, i.e., percentage of macrophages engulfing one or more apoptotic bodies multiplied by the number of engulfed particles per macrophage. Phagocytosis of apoptotic cells by RAW264.7 macrophages was performed according to Schrijvers et al. (20) with minor modifications. Jurkat cells were labeled with 0.5 µM CMF-DA before apoptosis induction. Apoptotic cells were washed two times and incubated with RAW264.7 cells preincubated for 30 min with 5 µM CellTracker Orange CMRA. Following 1 h incubations, macrophages were washed three times in PBS and analyzed by flow cytometry. Red fluorescent macrophages were measured in the PE channel and green fluorescent apoptotic cells in the FITC channel. Phagocytosis was quantified as the increase of median FITC fluorescence intensity of the macrophage-gated population, which excluded adherent apoptotic cells.
Western blotting
2 × 106 RAW264.7 cells were incubated with 10 × 106 apoptotic or oxLDL-treated Jurkat cells followed by stimulation with 100 nM PMA for 5 min. Then 80 µg of cell lysates were resolved by SDS-polyacrylamide gel electrophoresis. Proteins were blotted onto nitrocellulose. Membranes were incubated overnight with mouse anti-phospho-extracellular signal-regulated kinase (ERK) and rabbit anti-ERK antibodies (#9106 and #4695, Cell Signaling Technology, Danvers, MA), washed three times with Tris-buffered solution containing 0.1% Tween 20, incubated 1 h with a mixture of IRDye680-coupled anti-rabbit and IRDye800CW-coupled anti-mouse antibodies (LI-COR Biosciences, Bad Homburg, Germany), washed four times with Tris-buffered solution containing 0.1% Tween 20, and developed and quantified using Odyssey infrared imaging system (LI-COR Biosciences, Bad Homburg, Germany). Fluorescence intensities of phospho-ERK bands were normalized to total ERK and expressed as fold of PMA-treated cells.
Luciferase reporter assays
106 RAW264.7 cells were seeded on 6 cm dishes. For PPAR response element (PPRE) reporter assays cells were transfected using JetPEI transfection reagent (PolyPlus Transfection) with 4.75 µg p(ACOx)3-TK-luc coding for luciferase under the control of acyl-CoA oxidase PPRE (provided by Christopher Glass, University of California, San Diego) and 0.25 µg pRL-CMV to normalize for transfection efficiency. At 6 h posttransfection, the medium was changed and cells were seeded at 200,000 per well in a 12-well plate. On the next day, cells were treated with 2 × 106 apoptotic cells for 15 h prior to analysis of firefly and Renilla luciferase activities (dual luciferase reporter assay system, Promega).
Quantitative PCR
A total of 106 macrophages were cultured on 6 cm dishes. Cells were treated for 90 min with 10 × 106 apoptotic Jurkat cells generated in the absence or presence of 25 µg/ml oxLDL followed by stimulation with 1 µg/ml LPS for 3 h. Total RNA was isolated using PeqGold RNAPure kit (PeqLab, Erlangen, Germany). Reverse transcription was done with 1 µg of RNA using iScript™ cDNA Synthesis kit (BioRad, Munich, Germany). Quantitative PCR was performed with Absolute QPCR SYBRGreen Fluorescein mix (Abgene, Hamburg, Germany) using MyiQ iCycler system from BioRad. The following primer pairs were selected for quantitative PCR: mouse monocyte chemotactic protein (MCP)-1 forward: 5′-GAT GAT CCC AAT GAG TAG GC-3′, MCP-1 reverse: 5′-GGT TGT GGA AAA GGT AGT GG-3′, mouse tumor necrosis factor (TNF)α forward: 5′-CCA TTC CTG AGT TCT GCA AAG G-3′, TNFα reverse: 5′-AAG TAG GAA GGC CTG AGA TCT TAT C-3′; mouse interleukin (IL)-6 forward: 5′-GAA CAA CGA TGA TGC ACT TGC-3′, IL-6 reverse: 5′-TCT CTG AAG GAC TCT GGC TTT G-3′; mouse 16S ribosomal protein forward: 5′-AGA TGA TCG AGC CGC GC -3′, 16S ribosomal protein reverse: 5′-GCT ACC AGG GCC TTT GAG ATG GA-3′; human β-microglobulin forward: 5′-TCC AAA GAT TCA GGT TTA CTC A-3′, β-microglobulin reverse: 5′-ATA TTA AAA AGC AAG CAA CGA G-3′. For determination of human TNFα and MCP-1 mRNA levels, we used Quanti-Tect® Primer assays (Qiagen, Hilden, Germany). Values were normalized to 16S ribosomal protein (mouse) or β-microglobulin (human) expression.
Statistical analysis
Data are presented as mean ± SE. Data were analyzed by one-way ANOVA test with Bonferroni post hoc means comparison using OriginPro 7.5 (OriginLab Northampton, MA). Differences were considered statistically significant for P < 0.05.
RESULTS
Impact of oxLDL-exposed cells on the oxidative burst in phagocytes
We monitored oxLDL-induced cell death in THP-1 monocytes and Jurkat T-lymphoma cells by flow cytometry following Annexin V/propidium iodide staining (Table 1). Both apoptosis and necrosis were observed. While apoptosis reached a maximum at 8 h, necrosis gradually increased thereafter (Supplementary Fig. I). Jurkat cells were more sensitive toward oxLDL-induced toxicity compared with THP-1 cells. To achieve a similar extent of cell death, i.e., apoptosis plus necrosis, we incubated Jurkat with 25 µg/ml and THP-1 cells with 75 µg/ml oxLDL, respectively. In addition, oxLDL-induced cell death was accompanied by caspase activation (Table 1). For comparison, we used staurosporine to initiate cell death. Incubations with staurosporine for 3 h provoked an exclusive apoptotic mode of cell death. Prolonged exposures to staurosporine initiated secondary necrosis as well (Table 1).
TABLE 1.
Characterization of cell death induced by staurosporine and/or oxLDL.
| Treatment | Apoptosis, % | Necrosis, % | Caspase Activity, Arbitrary Units |
|---|---|---|---|
| YYNTHP-1 untreated | 5.0 ± 0.4 (6) | 5.9 ± 0.9 (6) | 490 ± 40 (4) |
| AC THP-1 | 83.3 ± 1.2* (6) | 7.7 ± 1.1 (6) | 20470 ± 2800* (4) |
| SN THP-1 | 9.1 ± 3.5 (8) | 86.2 ± 4.6* (8) | ND |
| oxLDL THP-1 | 26.5 ± 3.3* (10) | 42.7 ± 5.8 (10) | 11080 ± 835* (7) |
| oxLDL-AC THP-1 | 30.7 ± 2.7* (9) | 51.3 ± 3.4* (9) | 15960 ± 2520* (3) |
| Jurkat untreated | 7.4 ± 0.4 (5) | 7.1 ± 2.0 (5) | 499 ± 187 (4) |
| AC Jurkat | 81.0 ± 2.2* (12) | 11.8 ± 1.5 (12) | 13830 ± 1090* (3) |
| oxLDL Jurkat | 31.3 ± 2.2* (5) | 28.8 ± 5.3* (5) | 1250 ± 210* (3) |
| oxLDL-AC Jurkat | 28.3 ± 4.3* (12) | 62.5 ± 4.9* (12) | 6320 ± 610* (3) |
Apoptotic cells (AC) were generated by treatment with 0.5 µg/ml staurosporine for 3 h. Secondary necrotic cells (SN) were generated by incubating cells for 36 h with staurosporine. oxLDL was added at 25 µg/ml (Jurkat) or 75 µg/ml (THP-1) for 8 h. Cotreatment with staurosporine and oxLDL (oxLDL-AC) was for 3 h. Apoptosis/necrosis was determined by staining cells with Annexin V and propidium iodide. Apoptosis was defined as the percentage of Annexin V positive, propidium iodide negative cells and necrosis as the percentage of Annexin V, propidium iodide double-positive cells. *, P < 0.05 versus untreated cells. The number of experiments for each group is given in parentheses.
In a second step, we compared the ability of apoptotic, secondary necrotic, and oxLDL-treated THP-1 cells to modulate the oxidative burst in RAW264.7 macrophages (Fig. 1). As shown in Fig. 1A, preincubation of RAW264.7 macrophages with apoptotic THP-1 cells suppressed PMA-induced superoxide generation. Secondary necrotic THP-1 cells also significantly attenuated ROS formation. This observation indicates that plasma membrane integrity during cell demise is not a prerequisite for attenuating superoxide generation. Interestingly, cells dying in response to oxLDL did not interfere with the oxidative burst in macrophages. Although the percentage of apoptotic cells after oxLDL treatment was reduced compared with staurosporine-treated cells, we noticed that apoptotic cells generated in response to staurosporine suppressed the oxidative burst even when added at a 1:4 ratio with viable cells (Supplementary Fig. II). This excludes the possibility that simply reducing the amount of apoptotic cells after oxLDL treatment is responsible for their inability to attenuate the oxidative burst.
Fig. 1.

oxLDL interferes with the ability of apoptotic cells to block ROS formation. A: RAW264.7 macrophages were incubated with apoptotic THP-1 cells generated in the absence (AC THP) or presence (oxLDL-AC THP) of 75 µg/ml oxLDL as well as with secondary necrotic cells (SN THP) or cells exposed to oxLDL alone (oxLDL THP) for 1 h prior to stimulation with 1 µM PMA. B: Jurkat cells were treated with staurosporine in the absence (AC Jurkat) or presence (oxLDL-AC Jurkat) of 25 µg/ml oxLDL or with oxLDL alone (oxLDL Jurkat). RAW264.7 macrophages were incubated with different AC alone or in combination with 100 µg/ml oxLDL for 1 h prior to stimulating ROS production with 1 µM PMA. C: Human primary macrophages were incubated with apoptotic THP-1 cells, generated in the absence (AC) or presence (oxLDL-AC) of 75 µg/ml oxLDL for 1 h prior to stimulating ROS formation with 1 µM PMA. Superoxide generation was analyzed by staining with 2 µM HE and flow cytometry. *, P < 0.05 vs. PMA; #, P < 0.05 vs. AC (n = 5).
The failure of oxLDL-treated cells to attenuate phagocyte ROS generation suggests that oxLDL may inhibit pathways in apoptotic cells, which reduce the oxidative burst upon their phagocytosis. To examine whether oxLDL affects the ability of apoptotic cells to lower PMA-induced ROS formation, we exposed cells to staurosporine in the presence or absence of oxLDL. Cotreatment with oxLDL shifted cell death toward necrosis while retaining caspase activity (Table 1). In contrast to preincubations with apoptotic THP-1 cells, cells treated with the combination of staurosporine plus oxLDL failed to inhibit the oxidative burst (Fig. 1A). However, cells cotreated with staurosporine and unmodified LDL still suppressed ROS generation (Supplementary Fig. II). Thus, oxLDL apparently suppressed changes in apoptotic cells that interfered with superoxide generation in phagocytes. The observations made with apoptotic/necrotic THP-1 cells could be reproduced with Jurkat T-cells. As shown in Fig. 1B, apoptotic Jurkat cells suppressed superoxide generation in RAW264.7 macrophages. Treatment of Jurkat cells with oxLDL alone or together with staurosporine reversed their oxidative burst-suppressing properties. Interestingly, adding oxLDL at concentrations that prevent apoptotic cell phagocytosis (12, 13) directly during coincubations of RAW264.7 macrophages with apoptotic cells left the inhibition of ROS formation intact. We then extended observations made in mouse macrophages to human primary macrophages (Fig. 1C). Preincubating human macrophages with apoptotic THP-1 cells decreased PMA-stimulated superoxide generation to levels observed in untreated cells. In contrast, when human macrophages were preincubated with cells cotreated with staurosporine and oxLDL, ROS formation was no longer suppressed, confirming results obtained in the murine system.
oxLDL-treated cells are phagocytized normally and activate PPARγ
Phagocytosis of apoptotic cells is important in shaping macrophage responses toward dying cells. Although oxLDL inhibited phagocytosis of apoptotic material when added simultaneously with apoptotic cells, the ability of macrophages to ingest apoptotic cells generated by oxLDL treatment remained intact (18). To examine whether the effect of oxLDL on ROS formation reflects differences in phagocytosis, we generated apoptotic cells in the presence or absence of oxLDL and studied their uptake by human primary macrophages. As shown in Fig. 2, we observed no significant changes in the uptake of apoptotic Jurkat cells exposed to oxLDL during cell death induction. Similarly, there was no difference in phagocytosis of apoptotic cells obtained in the presence or absence of oxLDL by RAW264.7 macrophages (Supplementary Fig. III). Thus, the failure of oxLDL-treated apoptotic cells to attenuate ROS formation is not caused by impaired uptake by phagocytes.
Fig. 2.

Phagocytosis of apoptotic cells. Jurkat cells were prelabeled with 0.5 µM CMF-DA prior to the treatment with 0.5 µg/ml staurosporine in the absence (AC) or presence (oxLDL-AC) of 25 µg/ml oxLDL. Resulting apoptotic cells as well as untreated (viable) cells were incubated for 1 h with human primary macrophages labeled with 5 µM CellTracker Orange. Phagocytosis was analyzed by microscopy. Representative images of macrophages engulfing AC (A) or oxLDL-AC (B) are shown. C: Quantitation of phagocytosis using the phagocytic index as described in the Methods section. *, P < 0.05 versus viable cells (n = 8).
Previous observations suggested an important role of PPARγ in suppressing the oxidative burst by apoptotic cells (3). Therefore, we examined whether the presence of oxLDL during apoptosis modulates the ability of apoptotic material to activate PPARγ by using a PPAR luciferase reporter assay. Figure 3 shows that apoptotic Jurkat cells caused robust PPAR reporter activation in RAW264.7 macrophages. There were no differences in the reporter response when apoptotic cells were generated in the presence of oxLDL. Similar results were obtained with apoptotic THP-1 cells. These observations exclude that differences in PPARγ activation may account for defective inhibition of ROS production by oxLDL-exposed apoptotic cells.
Fig. 3.
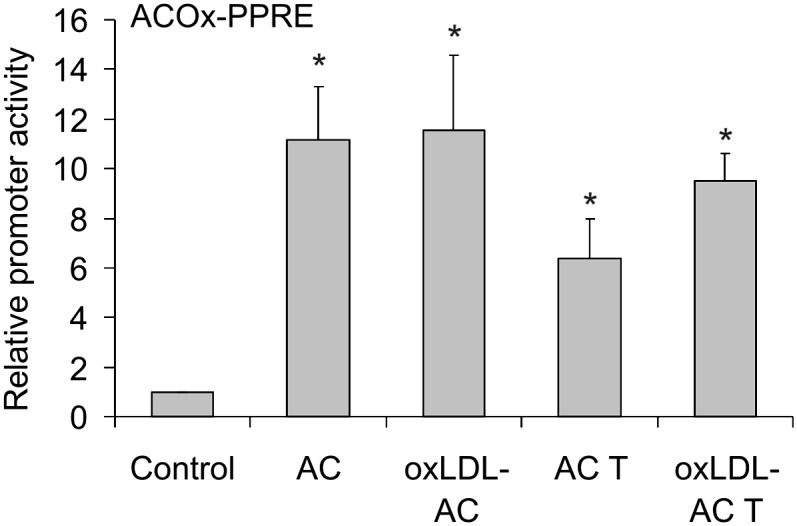
Activation of PPARγ by apoptotic cells. RAW264.7 macrophages were transfected with the plasmids encoding ACOx-PPRE luciferase and Renilla luciferase and subsequently stimulated for 15 h with apoptotic Jurkat cells (AC) or THP-1 cells (AC T) generated in the absence or in the presence of 25 µg/ml oxLDL (oxLDL-AC/oxLDL-AC T). Renilla-normalized reporter activity is presented. *, P < 0.05 versus control (n = 4).
Cells exposed to oxLDL do not suppress PMA-induced ERK activation
Mitogen-activated protein kinases ERK and p38 modulate the phagocyte oxidative burst (21, 22). Moreover, apoptotic cells may affect activation of these kinases (23, 24). Thus, we tested whether apoptotic and oxLDL-treated cells differentially regulate ERK and p38 phosphorylation in RAW264.7 cells after PMA stimulation using phospho-specific antibodies. As seen in Fig. 4A, PMA caused robust stimulation of ERK phosphorylation. Preincubation with apoptotic Jurkat cells reduced PMA-induced ERK phosphorylation. This reduction was significantly lower when macrophages were preincubated with oxLDL-treated cells (Fig. 4B shows quantification of ERK phosphorylation). p38 was not phosphorylated after PMA treatment.
Fig. 4.

Effects of apoptotic and oxLDL-treated cells on PMA-induced ERK and p38 phosphorylation. RAW264.7 cells were incubated with apoptotic or oxLDL-treated Jurkat cells for 1 h followed by 5 min stimulation with 100 nM PMA. A: Phosphorylation of ERK and p38 was evaluated using Western blotting. B: Quantification of ERK phosphorylation using the Odyssey imaging system. *, P < 0.05 versus PMA; #, P < 0.05 versus AC (n = 3).
Inhibition of ROS formation by apoptotic cells is independent of phosphatidylserine exposure
Considering the importance of phosphatidylserine (PS) exposure on apoptotic cells and the relevance of PS oxidation for the interaction with phagocytes (16), we questioned whether externalization of PS might affect ROS formation. Experimentally, we treated THP-1 cells with staurosporine in the presence or absence of oligomycin. Oligomycin lowers PS exposure, while leaving caspase-3/7 activation intact (25). As expected, staurosporine-induced PS externalization was reduced in the presence of oligomycin from 83.3 ± 1.2% to 26.1 ± 7.4%, while caspase-3 activity remained comparable to values observed in staurosporine-exposed cells (20,470 ± 2,800 units in staurosporine-treated cells vs. 21,800 ± 250 units in staurosporine plus oligomycin-treated THP-1). Exposing staurosporine versus staurosporine plus oligomycin-treated apoptotic cells to RAW264.7 macrophages revealed a more potent inhibition of the oxidative burst by oligomycin-treated cells (Fig. 5). Therefore, inhibition of ROS formation in macrophages does not require PS exposure on the surface of apoptotic cells. Moreover, oxidative events at the surface of phagocytes and/or apoptotic cells may interfere with apoptotic cell recognition by phagocytes (17). Toward this possibility, treatment of apoptotic cells with 1 mM of the oxidative, i.e., peroxynitrite-generating, compound SIN-1 did not affect their ability to block the phagocyte oxidative burst (Supplementary Fig. IV). This suggests that oxLDL is unlikely to act via a simple oxidative process in reversing the ability of apoptotic cells to block the oxidative burst in macrophages.
Fig. 5.

PS exposure does not interfere with the oxidative burst. THP-1 cells were treated for 3 h with 0.5 µg/ml staurosporine (STS) with or without 10 µg/ml oligomycin. RAW264.7 cells were incubated with apoptotic THP-1 cells (AC), generated in the absence or presence of oligomycin, for 1 h prior to stimulation of the oxidative burst. *, P < 0.001 versus PMA; #, P < 0.05 versus AC (n = 4).
Phagocytosis of oxLDL-treated cells does not suppress phagocyte cytokine production
To obtain more information how apoptotic cells versus cells dying in response to oxLDL might affect the balance between pro- and antiinflammatory signals, we analyzed LPS-induced proinflammatory cytokine expression in RAW264.7 and human primary macrophages. Apoptotic cells attenuated mRNA expression of MCP-1, TNFα, and IL-6 in response to LPS in RAW264.7 cells (Fig. 6A). This inhibition was absent (TNFα and IL-6) or only slightly visible (MCP-1) when apoptotic cells were generated in the presence of oxLDL. In human macrophages, expression of MCP-1 mRNA by LPS was suppressed by apoptotic cells generated in the absence, but not in the presence, of oxLDL (Fig. 6B, AC vs. oxLDL-AC). When macrophages were exposed to Jurkat cells treated with oxLDL (oxLDL Jurkat), MCP-1 mRNA induction was enhanced compared with cells treated with LPS alone. In addition, the exposure of macrophages to oxLDL Jurkat cells did not affect TNFα mRNA induction by LPS, while exposure to apoptotic cells significantly suppressed it. Thus, cells dying in response to or in the presence of oxLDL did not share the ability of apoptotic cells to lower inflammatory cytokine production after LPS treatment. This suggests that oxLDL-induced cell death may contribute to a proinflammatory microenvironment in the atherosclerotic plaque.
Fig. 6.

oxLDL treatment of apoptotic cells reverses their ability to suppress macrophage proinflammatory cytokine gene expression. A: RAW264.7 cells were incubated with apoptotic Jurkat cells, generated in the absence (AC) or presence (oxLDL-AC) of 25 µg/ml oxLDL for 90 min prior to stimulation with 1 µg/ml LPS for 3 h. B: Human primary macrophages were incubated with apoptotic Jurkat cells, generated in the absence (AC) or presence (oxLDL-AC) of 25 µg/ml oxLDL or with cells treated with oxLDL alone (oxLDL-Jurkat) for 90 min prior to stimulation with 1 µg/ml LPS for 3 h. Cytokine mRNA expression was analyzed by quantitative PCR. *, P < 0.05 versus LPS; #, P < 0.05 versus AC (n = 6).
DISCUSSION
Advanced atherosclerosis is characterized by a defective apoptotic cell clearance (9, 11). It was suggested that oxLDL attenuates phagocytosis of apoptotic material, thus provoking accumulation of cells progressing toward secondary necrosis and necrotic core formation in the advanced plaques. It is also established that oxLDL induces death of vascular cells (26). What remains unknown is how oxLDL affects properties of dying cells regarding their phagocytic recognition and their ability to balance pro- vs. antiinflammatory signals in phagocytes. We show that cells dying in the presence of oxLDL are defective in suppressing NADPH oxidase activation and concomitant ROS formation. Blocking the phagocyte oxidative burst appears to be an essential component of the antiinflammatory program elicited by apoptotic cells (3, 4). Thus, the failure of oxLDL-treated cells to inhibit NADPH oxidase activity may support a continuing inflammatory process in the lesion during advanced atherosclerosis.
oxLDL-induced cell death reveals characteristics of both apoptosis and necrosis, depending on the cell type and the concentration of oxLDL. In mouse peritoneal macrophages, oxLDL initiates an apoptotic program (18, 27, 28), while human macrophages die by necrosis (29). Moreover, pro-survival effects of oxLDL were also observed, with the notion that the degree of LDL oxidation appears important in balancing pro- versus antiapoptotic responses (19, 28). Under the conditions used here, oxLDL induced a mixed type of cell death based on PS exposure, plasma membrane permeabilization, or caspase-3 activation. Increased plasma membrane permeability in oxLDL-treated cells indicated a switch to necrosis. There are indications that both necrotic and apoptotic cells are phagocytized and irrespective of the mode of cell death may elicit antiinflammatory responses (30, 31). However, other studies point to fundamental differences between apoptotic and necrotic cells in polarizing macrophages (24, 32), including their ability to inhibit the oxidative burst (3). Some of the reported discrepancies are likely to result from differences in generating necrotic cells, e.g., necrosis induced by ATP depletion (30) as opposed to freeze-thawing or heating (3, 24, 32). Based on our observations with secondary necrotic cells, it appears unlikely that membrane permeabilization observed in cells treated in the presence of oxLDL would per se determine burst inhibition. Although necrotic cells may release a number of soluble proinflammatory factors such as proteases or HMGB1 (33, 34), our procedure to generate apoptotic cells includes several washing steps, which would eliminate soluble factors. As oxLDL-treated cells also retained caspase activity, the differences in affecting ROS generation elicited by cells dying in the presence or absence of oxLDL appear to be unrelated to caspase activity. We also noticed that oxLDL had to be continuously present during the time course of cell death induction to reverse burst inhibition. A 10 min exposure of staurosporine-treated cells to oxLDL followed by its washout did not interfere with inhibition of macrophage ROS production by apoptotic cells (data not shown).
The uptake of apoptotic cells may affect macrophage polarization. During atherosclerosis, oxLDL was reported to prevent phagocytosis of apoptotic cells by macrophages (13, 15, 17), probably by competing for common phagocyte recognition receptors (15, 16). In contrast, we observed that the presence of oxLDL during apoptosis does not interfere with the uptake of apoptotic material by macrophages, corroborating a previous report demonstrating normal uptake of oxLDL-treated cells by mouse peritoneal macrophages (18). Our data showing that oxLDL, coincubated with apoptotic cells, does not prevent inhibition of the oxidative burst concurs with previous results indicating that the ability to impair NADPH oxidase is unrelated to the uptake of apoptotic cells (3).
Phosphorylation events regulate NADPH oxidase assembly and activity (35). In this regard, MAP kinase ERK was reported to phosphorylate and activate the cytosolic NADPH oxidase components p47phox and p67phox (21, 22). As we observed inhibition of PMA-induced ERK phosphorylation by apoptotic cells, but not by oxLDL-treated cells, these differences may explain altered ROS formation. Further studies are needed to elucidate the mechanism whereby apoptotic cells attenuate ERK and thus to explain the divergent effects of apoptotic versus oxLDL-treated cells.
PS exposure on the surface of apoptotic cells was reported to suppress phagocyte inflammatory responses, including ROS generation (4). In addition, PS oxidation affects the interaction of apoptotic cells with phagocyte recognition receptors (16), and oxLDL may influence the degree of PS oxidation on the surface of apoptotic cells. Using oligomycin-treated apoptotic cells showing reduced PS exposure, we noticed that the ability of apoptotic cells to attenuate ROS formation was unrelated to PS externalization during apoptosis. Thus, although PS enrichment in the outer leaflet of the plasma membrane may provide an inhibitory signal for ROS production (4), the degree of PS exposure may not explain discrepancies in NADPH oxidase inhibition by apoptotic versus oxLDL-treated cells.
Analysis of proinflammatory cytokine expression showed a reduced ability of oxLDL-treated cells to block induction of proinflammatory cytokines in both murine and human macrophages. These results strengthen the conclusion on a proinflammatory character of oxLDL-induced cell death. Surprisingly, the diminished inhibitory effect of oxLDL-exposed cells on macrophage cytokine expression is not paralleled by changes in PPARγ reporter activation. Previously, we showed an important role of PPARγ in macrophage polarization in response to apoptotic cells regarding both oxidative burst and cytokine production (3, 36). These discrepancies may reflect different mechanisms governing PPARγ activation and suppression of gene expression or suggest PPARγ-independent pathways accounting for a proinflammatory character of oxLDL-treated cells.
Although excluding a number of possibilities to explain why apoptotic cells generated in the presence of oxLDL failed to block ROS formation compared with classically induced apoptotic cells, our observations might be of medical importance. Considering that oxLDL is a major cell death inducer in atherosclerosis, our findings support the notion that macrophages in the atherosclerotic plaque remain activated and do not polarize toward an antiinflammatory phenotype. It will be challenging to determine the molecular mechanisms of how oxLDL induces a proinflammatory character of cell death in order to identify potential targets for preventing chronic inflammatory responses during atherosclerosis progression.
Supplementary Material
Acknowledgments
The authors thank Christine von Hayn and Franz-Josef Streb for technical assistance.
Footnotes
Abbreviations:
- CMF-DA
- 5-chloromethylfluorescein diacetate
- ERK
- extracellular signal-regulated kinase
- HE
- hydroethidine
- IL
- interleukin
- MCP
- mouse monocyte chemotactic protein
- oxLDL
- oxidized LDL
- PMA
- phorbol 12-myristate 13-acetate
- PPAR
- peroxisome proliferator-activated receptor
- PPRE
- PPAR response element
- PS
- phosphatidylserine
- ROS
- reactive oxygen species
- TNF
- tumor necrosis factor
This study was supported by Deutsche Forschungsgemeinschaft (BR999, ECCPS), the European Union (PROLIGEN), and LOEWE (LiFF).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of four figures.
REFERENCES
- 1.Savill J., Dransfield I., Gregory C., Haslett C. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2: 965–975. [DOI] [PubMed] [Google Scholar]
- 2.Erwig L. P., Henson P. M. 2007. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 171: 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johann A. M., von Knethen A., Lindemann D., Brune B. 2006. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-gamma and attenuates the oxidative burst. Cell Death Differ. 13: 1533–1540. [DOI] [PubMed] [Google Scholar]
- 4.Serinkan B. F., Gambelli F., Potapovich A. I., Babu H., Di Giuseppe M., Ortiz L. A., Fabisiak J. P., Kagan V. E. 2005. Apoptotic cells quench reactive oxygen and nitrogen species and modulate TNF-alpha/TGF-beta1 balance in activated macrophages: involvement of phosphatidylserine-dependent and -independent pathways. Cell Death Differ. 12: 1141–1144. [DOI] [PubMed] [Google Scholar]
- 5.Johann A. M., Barra V., Kuhn A. M., Weigert A., von Knethen A., Brune B. 2007. Apoptotic cells induce arginase II in macrophages, thereby attenuating NO production. FASEB J. 21: 2704–2712. [DOI] [PubMed] [Google Scholar]
- 6.Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101: 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voll R. E., Herrmann M., Roth E. A., Stach C., Kalden J. R., Girkontaite I. 1997. Immunosuppressive effects of apoptotic cells. Nature. 390: 350–351. [DOI] [PubMed] [Google Scholar]
- 8.Weigert A., Tzieply N., von Knethen A., Johann A. M., Schmidt H., Geisslinger G., Brune B. 2007. Tumor cell apoptosis polarizes macrophages: role of sphingosine-1-phosphate. Mol. Biol. Cell. 18: 3810–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabas I. 2005. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 25: 2255–2264. [DOI] [PubMed] [Google Scholar]
- 10.Shah P. K. 2007. Molecular mechanisms of plaque instability. Curr. Opin. Lipidol. 18: 492–499. [DOI] [PubMed] [Google Scholar]
- 11.Schrijvers D. M., De Meyer G. R., Herman A. G., Martinet W. 2007. Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc. Res. 73: 470–480. [DOI] [PubMed] [Google Scholar]
- 12.Sambrano G. R., Steinberg D. 1995. Recognition of oxidatively damaged and apoptotic cells by an oxidized low density lipoprotein receptor on mouse peritoneal macrophages: role of membrane phosphatidylserine. Proc. Natl. Acad. Sci. USA. 92: 1396–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang M. K., Bergmark C., Laurila A., Horkko S., Han K. H., Friedman P., Dennis E. A., Witztum J. L. 1999. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. USA. 96: 6353–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan M., Pelengaris S., Cooper M., Smith C., Evan G., Betteridge J. 2003. Oxidised lipoproteins may promote inflammation through the selective delay of engulfment but not binding of apoptotic cells by macrophages. Atherosclerosis. 171: 21–29. [DOI] [PubMed] [Google Scholar]
- 15.Miller Y. I., Viriyakosol S., Binder C. J., Feramisco J. R., Kirkland T. N., Witztum J. L. 2003. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J. Biol. Chem. 278: 1561–1568. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg M. E., Sun M., Zhang R., Febbraio M., Silverstein R., Hazen S. L. 2006. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 203: 2613–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrijvers D. M., De Meyer G. R., Kockx M. M., Herman A. G., Martinet W. 2005. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25: 1256–1261. [DOI] [PubMed] [Google Scholar]
- 18.Li Y., Gerbod-Giannone M. C., Seitz H., Cui D., Thorp E., Tall A. R., Matsushima G. K., Tabas I. 2006. Cholesterol-induced apoptotic macrophages elicit an inflammatory response in phagocytes, which is partially attenuated by the Mer receptor. J. Biol. Chem. 281: 6707–6717. [DOI] [PubMed] [Google Scholar]
- 19.Namgaladze D., Kollas A., Brune B. 2008. Oxidized LDL attenuates apoptosis in monocytic cells by activating ERK signaling. J. Lipid Res. 49: 58–65. [DOI] [PubMed] [Google Scholar]
- 20.Schrijvers D. M., Martinet W., De Meyer G. R., Andries L., Herman A. G., Kockx M. M. 2004. Flow cytometric evaluation of a model for phagocytosis of cells undergoing apoptosis. J. Immunol. Methods. 287: 101–108. [DOI] [PubMed] [Google Scholar]
- 21.Dang P. M., Morel F., Gougerot-Pocidalo M. A., El Benna J. 2003. Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochemistry. 42: 4520–4526. [DOI] [PubMed] [Google Scholar]
- 22.Dewas C., Fay M., Gougerot-Pocidalo M. A., El-Benna J. 2000. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J. Immunol. 165: 5238–5244. [DOI] [PubMed] [Google Scholar]
- 23.Reddy S. M., Hsiao K. H., Abernethy V. E., Fan H., Longacre A., Lieberthal W., Rauch J., Koh J. S., Levine J. S. 2002. Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J. Immunol. 169: 702–713. [DOI] [PubMed] [Google Scholar]
- 24.Patel V. A., Longacre A., Hsiao K., Fan H., Meng F., Mitchell J. E., Rauch J., Ucker D. S., Levine J. S. 2006. Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: Implications for the delayed clearance model of autoimmunity. J. Biol. Chem. 281: 4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhuang J., Ren Y., Snowden R. T., Zhu H., Gogvadze V., Savill J. S., Cohen G. M. 1998. Dissociation of phagocyte recognition of cells undergoing apoptosis from other features of the apoptotic program. J. Biol. Chem. 273: 15628–15632. [DOI] [PubMed] [Google Scholar]
- 26.Salvayre R., Auge N., Benoist H., Negre-Salvayre A. 2002. Oxidized low-density lipoprotein-induced apoptosis. Biochim. Biophys. Acta. 1585: 213–221. [DOI] [PubMed] [Google Scholar]
- 27.Nhan T. Q., Liles W. C., Chait A., Fallon J. T., Schwartz S. M. 2003. The p17 cleaved form of caspase-3 is present within viable macrophages in vitro and in atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 23: 1276–1282. [DOI] [PubMed] [Google Scholar]
- 28.Boullier A., Li Y., Quehenberger O., Palinski W., Tabas I., Witztum J. L., Miller Y. I. 2006. Minimally oxidized LDL offsets the apoptotic effects of extensively oxidized LDL and free cholesterol in macrophages. Arterioscler. Thromb. Vasc. Biol. 26: 1169–1176. [DOI] [PubMed] [Google Scholar]
- 29.Asmis R., Begley J. G. 2003. Oxidized LDL promotes peroxide-mediated mitochondrial dysfunction and cell death in human macrophages: a caspase-3-independent pathway. Circ. Res. 92: e20–e29. [DOI] [PubMed] [Google Scholar]
- 30.Hirt U. A., Leist M. 2003. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ. 10: 1156–1164. [DOI] [PubMed] [Google Scholar]
- 31.Brouckaert G., Kalai M., Krysko D. V., Saelens X., Vercammen D., Ndlovu M., Haegeman G., D'Herde K., Vandenabeele P. 2004. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell. 15: 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cvetanovic M., Ucker D. S. 2004. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 172: 880–889. [DOI] [PubMed] [Google Scholar]
- 33.Fadok V. A., Bratton D. L., Guthrie L., Henson P. M. 2001. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J. Immunol. 166: 6847–6854. [DOI] [PubMed] [Google Scholar]
- 34.Scaffidi P., Misteli T., Bianchi M. E. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418: 191–195. [DOI] [PubMed] [Google Scholar]
- 35.Sheppard F. R., Kelher M. R., Moore E. E., McLaughlin N. J., Banerjee A., Silliman C. C. 2005. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 78: 1025–1042. [DOI] [PubMed] [Google Scholar]
- 36.Jennewein C., Kuhn A. M., Schmidt M. V., Meilladec-Jullig V., von Knethen A., Gonzalez F. J., Brune B. 2008. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 181: 5646–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.