

Abstract
Selective 20S proteasomal inhibition and apoptosis induction were observed when several lines of cancer cells were treated with a series of copper complexes described as [Cu(LI)Cl] (1), [Cu(LI)OAc] (2), and [Cu(HLI)(LI)]OAc (3), where HLI is the ligand 2,4-diiodo-6-((pyridine-2-ylmethylamino)methyl)phenol. These complexes were synthesized, characterized by means of ESI spectrometry, infrared, UV-visible and EPR spectroscopies, and X-ray diffraction when possible. After full characterization species 1-3 were evaluated for their ability to function as proteasome inhibitors and apoptosis inducers in C4-2B and PC-3 human prostate cancer cells and MCF-10A normal cells. With distinct stoichiometries and protonation states, this series suggests the assignment of species [CuLI]+ as the minimal pharmacophore needed for proteasomal chymotryspin-like activity inhibition and permits some initial inference of mechanistic information.
Three well characterized discrete copper complexes with asymmetric phenol-substituted ligands are able to inhibit the proteolytic activity of the 20s proteasome. Evidence for a minimal pharmacophore suggests a potential basis for new cancer therapies with tunable and cost-effective metallodrugs.
Keywords: proteasome inhibition, anticancer therapy, copper(II), metallodrugs, bioinorganic
1. Introduction
The investigation of metal-based complexes in anticancer drug discovery has been precipitated by the clinical effectiveness [1] and understanding of the mechanisms[2] of platinum-containing drugs. Nonetheless, toxicity and acquired drug resistance have been hampering the widespread use of such metallodrugs.[3] These findings have fostered efforts toward the rational development of metal-containing agents that present modes of action distinct from those of cisplatin and its derivatives.[4]
A modern target in cancer treatment, the 26S Proteasome is a large multicatalytic protease complex composed of two terminal 19S regulatory caps and a 20S proteolytic core.[5] This complex is responsible for the degradation of many endogenous proteins including misfolded or damaged proteins to ensure normal cellular function.[6] The ubiquitin-proteasome pathway plays an essential role in multiple cellular processes, including cell cycle progression, apoptosis, differentiation and senescence. It has been shown that human cancer cells are more sensitive to proteasome inhibition than normal cells, indicating that proteasome inhibitors could be used as novel anticancer drugs.[7] Therefore, inhibition of the 20S core particle in the 26S proteasome by substrate modifications such as alkylations, and ring formations has been demonstrated by a number of organic species such as lactacystin,[8] macrocyclic esters,[9] epoxyketones,[10] and several peptide derivatives,[11] including recently FDA-approved boronates.[12,13] The premise that stoichiometric mixtures of copper ions with organic chelators can form a new class of proteasome inhibitors has been investigated by the Dou group in recent years.[14,15] Similarly, discrete gallium complexes with the asymmetric NpyridineN'amineOphenolate 2,4-di-X-6-((pyridine-2-ylmethylamino)methyl)phenol ligands HLX (where X = H, tert-butyl, bromo, or iodo) were synthesized by the Verani group. The bromo or iodo substituted species rendered effective against cisplatin-resistant neuroblastoma,[16] as well as for a number of other tumors.[17] The link between the in vitro and in vivo activity of these gallium complexes and proteasome inhibition has been established recently in a joint article.[18] In the course of this work, we observed that in situ complexation of HLBr or HLI with bivalent transition metal salts leads to the generation of species that display superior performance in cell death assays when compared to the equivalent gallium species.
Because detailed knowledge of the nature of the pharmacophore is pivotal to an understanding of the underlying mechanisms for metal-based proteasome inhibition, we present in this article a thorough study that encompasses the synthesis, spectrometric and spectroscopic characterization, and pharmacological evaluation of a series of copper complexes with the ligand HLI. The systems investigated in this article are [Cu(LI)Cl] (1), [Cu(LI)OAc] (2), and [Cu(HLI)(LI)]OAc (3), as displayed in Scheme 1. These copper complexes were found to be able to induce proteasomal inhibition and apoptosis in cultured human prostate cancer and leukemia cells Based on these results, we conclude with suggestions for some initial mechanistic insights on how these complexes may act. The findings described in this paper might have an impact in the development of a novel route to cancer therapy.
Scheme 1.

Copper(II) complexes.
2. Results and Discussion
2.1. Ligand Design and in-situ Copper Complexation
The iodo-substituted ligand 2,4-diiodo-6-((pyridine-2-ylmethylamino)methyl)phenol, HLI was synthesized by treatment of 2-hydroxy-3,5-diiodobenzaldehyde with 2-aminomethylpyridine followed by reduction with sodium borohydride.[16] It can be considered as an evolution from its terbutylated analogues inspired by biomimetic efforts to model redox-active enzymes such as galactose-oxidase.[19] The complexes were designed considering that a metal ion coordinated to the ligand could bind to the 20S core of the proteasome, possibly via the terminal threonine residue Thr1 or another available coordination site. Initial exploratory studies on human C4-2B prostate cancer cells, comprised of cell death induced by a stoichiometric mixture of HLI and copper(II) chloride in DMSO and toward proteasomal activity in whole-cell extracts. These assays showed that the resulting HLI:CuCl2:DMSO mixture was fourfold more potent than the recently reported gallium species.
2.2. Syntheses, Spectrometry, and Spectroscopic Characterization of 1-3
Spectrometric evaluation of the stoichiometric HLI:CuCl2:DMSO mixture using ESI in the positive mode led to the identification of monomeric and dimeric fragments that may act as pharmacophores to the inhibition of the proteasome complex. These fragments fit well with the expected distribution anticipated in systems containing copper and iodine isotopes. The relative ESI-MS profile for the monomeric [CuLI]+ with an m/z = 527.9 is shown in Figure 1.
Figure 1.

Experimental (bars) and simulated (continuum) ESIMS m/z data for monomeric [CuLI]+.
A minor peak at m/z = 994 is also detectable in this mixture suggesting a 2:1 ligand-to-copper complex, where either two fully deprotonated ligands are coordinated to the metal ion as in{[Cu(LI)2]+H+}+ or one of the ligands remains protonated as in [Cu(HLI)(LI)]+, respecting a 5-coordination preference imposed by the Jahn-Teller effect expected by a 3d9 species such as the copper(II) ion. Based on similar systems,[20] the latter proposition is favored.
With the intent of isolating and testing these species as anticancer agents, reactions with 1:1, and 2:1 ligand-to-metal ratios were performed. Treatment of 1 equiv. of the ligand with 1 equiv. of CuCl2·2H2O in DMSO yielded a green solution that was precipitated with ethanol in 30% yield as a crystalline material.
The isolated product was characterized as [Cu(LI)Cl] (1). It is noteworthy that 1 can also be obtained using methanol or ethanol as solvents, and the choice of DMSO was intended to match the experimental conditions of the initially used stoichiometric mixture. The chloride anions from the copper salt seem able to deprotonate the ligand with subsequent formation of hydrochloric acid. Hence, copper chloride was replaced by copper acetate, primarily in order to increase the yield of this reaction, as well as a cautionary measure to avoid HCl build up.[21] The ligand HLI (1 equiv.) was treated with Cu(OAc)2·2H2O (1 equiv.) in presence of triethylamine as a base to support ligand deprotonation yielding the species [Cu(LI)OAc] (2) in 90% yield. The ESI(pos) MS spectrum shows the characteristic m/z = 527.8 associated with the fragment [Cu(LI)]+, whereas the acetate species was detected by infrared spectroscopy at 1586 and 1402 cm-1 as a monodentate ligand. Elemental analysis showed excellent agreement with the proposed formula.
The EPR spectra taken at 77 K for 1 and 2 reinforce this notion with values of g‖ ≈ 2.26, g⊥ ≈ 2.06, A∥ ≈ 174 G and A⊥ ≈ 19 G, thus typical of 3d9 copper(II) ions in nearly square planar environments.[19d,f] Furthermore, the UV-visible spectra of species 1 and 2 in methanol show the presence of a phenolate-to-copper charge transfer band at around 450 nm (ε ≈ 1250 L·mol-1·cm-1).[19]
When a reaction of 2:1 stoichiometry between HLI and the copper acetate salt was carried out in methanol, a green precipitate described as [Cu(HLI)(LI)]OAc (3) was obtained. The compound was recrystallized in dichloromethane yielding a microcrystalline material. Although crystals suitable for X-ray diffraction were not obtained, infrared spectroscopy reveals the presence of acetate counterions, as displayed by prominent peaks at 1564 and 1439 cm-1. Additional peaks at 3448 are attributed to the presence of OH stretches belonging to a protonated HLI ligand. ESI mass spectra in the positive mode shows m/z = 527.9 characteristic of the [Cu(LI)]+ fragment. In good agreement with the elemental analysis, these data confirm the presence of two ligands coordinated to copper, one of which remaining protonated. This coordination mode for copper has been observed in our laboratories with the analogous ligand HLtBu, where tertiary butyl groups occupy the 2- and 4- positions of the phenol ring.[20,22] Species 3 shows EPR parameters g⊥, A∥, and A⊥ similar to those of 1 and 2 but with g∥ = 2.30, thus, evidencing bonding along the z-axis typical of five-coordinate copper(II) ions.[19d]
2.3. Molecular Structure of1
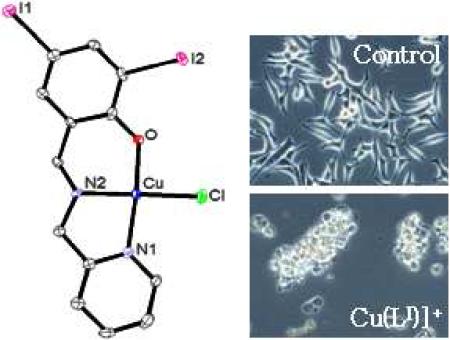
Needle-like crystals of [Cu(LI)Cl] (1) were isolated and solved diffractometrically by means of X-ray diffraction. The crystal structure of 1 is shown in Figure 2 and confirms its mononuclear nature. One copper(II) ion is coordinated to a deprotonated ligand with distances of 1.93 Ǻ to the oxygen of the phenolate group, 1.99 Ǻ to the amine nitrogen, and 2.02 Ǻ for the pyridine nitrogen, resembling similar systems.[19,22] The coordination sphere is completed with the anionic chloro ligand occupying the fourth position 2.24 Ǻ away from the metal. The copper center adopts a distorted square-planar environment. One can, therefore, conclude that contrary to the tetradentate phenol-containing ligands described by Zurita,[19f,23] tridentate ligands such as HLI do not foster the formation of stable dimers in the solid state.
Figure 2.

ORTEP diagram at 50% probability level for [Cu(LI)Cl] (1) with selected bond lengths (Å) and angles (°). Cu(1)-O(1) = 1.929(3), Cu(1)-N(2) = 1.990(4), Cu(1)-N(1) = 2.018(4), Cu(1)-Cl(1) = 2.2488(14), N(2)-Cu(1)-Cl(1) = 162.63(14), O(1)-Cu(1)-N(1) = 66.40(17).
2.4. Antiproliferative Effect of 1-3 in Tumor Cells
Our results thus far have allowed us to gain detailed understanding on the coordination chemistry of copper complexes as candidate drugs or prodrugs for cancer therapy. Human leukemia Jurkat T cells were treated with complexes 1-3 at different concentrations for 18 h, followed by trypan blue assay to assess cell death. This is a reliable assay that is reproducible and frequently used in our lab. All species tested demonstrated a dose-dependent increase in cell-killing activity with nearly 100% cell death at 15 µM, compared to a control treated with DMSO. The values given in Table 1 and Figure 2 reflect the concentration that each compound induces 50 % cell death.
Table 1.
IC50 values for cell death induction by copper compounds. (a) Human leukemia Jurkat T-cells were treated with copper compounds 1-3 for 18 h, followed by measurement of cell death in a trypan blue exclusion assay. Standard deviations are indicated.
| Compound | Cell death induction IC50 μ mol/L |
|---|---|
| 1 | 3.82 ± 0.01 |
| 2 | 4.46 ± 0.01 |
| 3 | 3.98 ± 0.01 |
| HLI | no activity |
| CuCl2 | no activity |
The 1:1 copper species 1 and 2 demonstrated IC50 values of 3.82 and 4.46 μmol/L. Interestingly, complex 3 exerted a similar degree of potency (IC50 = 3.98 μmol/L). Because the cation [CuII(LI)]+ of 1 is isostructural with that of 2, this observation suggests the 1:1 ligand-to-metal ratio as the possible pharmacophore and potential therapeutic agent. Furthermore, treatment of the cells with up to 50 μM of the ligand HLI or the copper salt failed to induce death ratios greater than 10%. Due to the small standard deviation values between 1 and 2, ascertaining the role played by the nature of the anionic ligand (chloride vs. acetate) remains unclear and further analysis will be necessary to provide a more conclusive result.
2.5. In-vitro Inhibition of Proteasomal Chymotryspin-like Activity by1-3
In order to test the role of complexes 1-3 in targeting the cellular proteasome, we compared the independent activity of the salt CuCl2·2H2O, the ligand HLI, and that of a 1:1 mixture of copper chloride and the ligand HLI towards C4-2B prostate cancer cell extract. The results indicate that both the copper chloride salt and the copper chloride:ligand mixture inhibit the chymotrypsin-like activity of the 26S proteasome with IC50 values of ~3.5 μM (Figure 4a). In contrast, the ligand HLI alone had a negligible effect, even at concentrations as high as 25 μM. This seems to be consistent with prior work in our laboratories in which the copper ion [14,15] is the driving force underlying proteasome inhibition. This suggests that the ligand is necessary as a carrier to preclude undesired nonspecific interactions of copper in the cell extract.[23] In order to underscore the role of the ligand, we tested complexes 1-3 under similar conditions. All species inhibited the proteasomal chymotrypsin-like activity in a concentration-dependent manner as shown in Figure 4b.
Figure 4.

In vitro proteasome-inhibitory activity of 1-3 in C4-2B cell extracts: (a) Proteasomal chymotrypsin-like activity (%CT) of the ligand HLI, the salt CuCl2, and an 1:1 HLI:CuCl2 mixture in DMSO at 25 mmol/L stock solution. (b) Concentration dependent inhibition by 1-3 at 0.1, 1, 5, 10, and 25 μM.
2.6. Inhibition of Chymotrypsin-like Activity and Induction of Apoptosis by 1-3 in Multiple Prostate Cancer Cell Lines
In the previous section we have discussed the inhibition of proteasomal chymotryspin-like activity under cell-free conditions. When intact prostate cancer cells were treated with HLI or the Cu+2 ion at the 15 μmol/L concentration, less than 20% proteasome inhibition was evident. In order to confirm the ability of the copper complexes to inhibit the proteasomal activity in cancer cells, two distinct prostate cell lines were tested, namely androgen receptor-positive C4-2B and androgen receptor-negative PC-3.
The androgen receptor-positive C4-2B prostate cells: The cells were treated with compound 1 at 5, 10 and 15 μM concentrations for 18 h. The cells were harvested and proteins extracted, followed by measurement of proteasomal inhibition and apoptosis. Cells were also treated with the vehicle DMSO, the ligand HLI, and the salt CuCl2·2H2O at the same concentrations to serve as negative controls. Compound 1 inhibits the proteasomal chymotrypsin-like activity in a dose dependent manner by 35% at 5 μM, 62% at 10 μM and 85% at 15 μM (Figure 5). Consistent with proteasome inhibition by 1, levels of ubiquitinated proteins were also increased in a dose-dependent fashion in the treated C4-2B cells (Figure 6). In a sharp contrast, cells treated either with the solvent, the ligand, or the copper salt failed to inhibit significantly the proteasome activity (Figures. 5-6 and data not shown). This fact provides strong evidence that the combination of copper(II) ion and ligand is a necessary requirement in order to cross the cellular membrane and reach the proteasome.
Figure 5.

Dose-dependent effects of 1-3 in C4-2B cells. The results were also compared to three negative controls (DMSO, HLI, and CuCl2·2H2O) for 18 h followed by measuring inhibition of proteasomal chymotrypsin-like activity.
Figure 6.

Dose-dependent effects of 1-3 in C4-2B cells. Accumulation of ubiquitinated proteins (Ub-Prs), and cleavage of PARP.
It has been shown that inhibition of the proteasomal chymotrypsin-like activity is associated with apoptosis of tumor cells.[24] To investigate whether the proteasomal inhibition by 1 is associated with apoptotic cell death, cleavage of the DNA repair protein, poly-(ADP-ribose) polymerase (PARP) and cellular morphological changes were measured in the same experiment. Cells showed a significant decrease in levels of PARP protein (Figure 5) when treated with 1 at 10 and 15 μM. Consistently, aberrant morphological changes, namely the rounded up shape and characteristic apoptotic blebbing shown in Figure 6, were also observed in 1 in a concentration-dependent manner. Again, cells treated with DMSO, the ligand HLI, or the salt CuCl2 individually were unable to perturb the full length PARP fragment and no visible aberrant morphological changes were observed up to 15 μM tested (Figures 6-7 and data not shown).
Figure 7.

Dose-dependent effects of 1-3 in C4-2B cells. Micrographs of apoptotic morphologic changes in HLI, CuCl2, 1, 2, and 3. (low definition PNG file)
Similar results were also obtained from the C4-2B cells treated with compounds 2 and 3: these two compounds inhibited about 80% of proteasomal activity at concentrations of 10 and 15 μM (Figure 4), associated with increased levels of ubiquitinated proteins, decreased levels of PARP protein and the appearance of characteristic apoptotic cell morphology (Figures 6-7).
These results show that the inhibition of proteasomal chymotrypsin-like activity in C4-2B cells is associated with the induction of apoptosis. Due to similar activity, it is also possible that in the 2:1 species 3, the protonated ligand be labile and easily interchangeable suggesting that the active pharmacophore in both 2 and 3 is the cupric cation [Cu(LI)]+
The androgen receptor-negative PC-3 prostate cells: Upon demonstrating the ability of copper compounds to inhibit the proteasomal chymotrypsin-like activity in androgen receptor-positive C4-2B cells, we then tested the effect of compounds 2 and 3 on androgen receptor-negative PC-3 prostate cancer cells. PC-3 cells were treated respectively with different concentrations of 2 and 3, copper salt, and HLI for 18 h, followed by measurement of the proteasome activity, accumulated ubiquitinated proteins and apoptosis induction. We found that 3 could inhibit the chymotrypsin-like activity by ~30% and ~85% at 5 and 10 μmol/L, respectively, whereas 2 could inhibit the chymotrypsin-like activity by ~88% at 10 μmol/L (Figure S1a). Additionally, cells treated with either CuCl2 or HLI showed no effect toward proteasome activity compared to the DMSO control.
Consistent with the inhibition of proteasomal chymotrypsin-like activity, significantly increased levels of ubiquitinated proteins were detected in the PC-3 cells treated with 2 and 3 but not metal salt or HLI (Figure S1b). In the same experiment, treatment with 2 and 3 resulted in significant cellular detachment and cleavage of PARP, associated with apoptosis induction (Figure S1c). These results show that as in C4-2B prostate cancer cells, copper compounds 2 and 3 could target and inhibit the proteasome in PC-3 cells. Our results remain consistent in that HLI or the metal salt alone had little effect on proteasome activity and overall cellular integrity.
2.7. Kinetic Effect of 1-3 on Proteasome Inhibition and Apoptosis Induction
The previous results show that complexes 1-3 demonstrated potent proteasome-inhibitory and apoptosis-inducing activities in multiple prostate cancer cell lines. In order to study the kinetic effect of copper complex-induced proteasome inhibition, C4-2B prostate cancer cells were treated with 15 μM of 2 for 2-18 h. The proteasomal chymotrypsin-like activity was inhibited by 45%, 60%, 70% and 80% after 2, 4, 8, and 18 h, respectively, as shown in the graph on Figure 8a. The proteasomal chymotrypsin-like activity inhibition was associated with accumulated levels of ubiquitinated proteins (Figure 7b). Additionally, higher levels of the proteasomal target p27 protein are visible at later time points (data not shown). Consistently, apoptosis-specific PARP cleavage was visible after 8 h treatment and full length PARP was completely cleaved into its respective p65 fragment after 18 h (Figure 8b). This observation is also supported by abnormal morphological changes during later time points, as well as the appearance of rounded up and detached cells indicative of apoptosis, as shown by selected micrographs in Figure 8c. These results clearly show that apoptosis induction occurs following inhibition of proteasomal activity. Therefore, proteasome inhibition appears to be a requirement for apoptosis induction.
Figure 8.

Kinetic effect of proteasome inhibition and apoptosis induction by 2 in C4-2B cells. (a) measurement of proteasomal chymotrypsin-like activity over time, (b) accumulated ubiquitinated proteins (UB-Prs) and PARP cleavage with Actin as a loading control, and (c) apoptotic cellular morphological changes.
2.8. Nontoxic Effect of 1-3 in Immortalized Human Breast Cells
The ability to distinguish normal from malignant cells is imperative for developing successful anticancer drugs. To determine whether inhibition of proteasome activity achieved by copper compounds is selective toward malignant cells and not normal cells, we used normal-immortalized human breast cell line, MCF-10A to test this effect. MCF-10A cells were treated with different concentrations of 2 and 3 up to 10 μmol/L for 18 h, followed by measurement of proteasomal chymotrypsin-like activity and apoptosis. We found that when these nontransformed cells were treated with 2 and 3, no proteasome inhibition was detected (Figure S2a). Other treatments also had no or little effect on MCF-10A cells. To determine whether the inability of copper compounds to inhibit the proteasome activity is associated with the lack of apoptosis induction in these normal immortalized breast cells, apoptosis-associated morphological changes were then assessed in the same experiment. These normal, immortalized MCF-10A cells showed only little, if any, such cell death-related detachment after treatment with 2 and 3 up to 18 h (Figure S2b). Our data suggests that these copper compounds could inhibit the proteasome activity and induce apoptosis selectively in human cancer cells but not in normal immortalized breast cells.
3. Conclusions
Following the encouraging results observed for similar gallium(III) complexes,[18] this article presented the results of proteasome inhibition by copper(II) complexes of the iodosubstituted ligand HLI as a potential anticancer therapeutic route. Three compounds with distinctive stoichiometries and nature of the anionic monodentate ligand were compared. On one hand, it has been demonstrated that the non-metallated ligand is incapable of inhibiting the 20S proteasome activity. On the other hand, inhibition does occur by the copper(II) chloride salt, but only in cell-free conditions. Therefore, one could conclude that the primary function of the copper complexes is to serve as a carrier to cross the cell membrane. Once inside the cell, this complex is likely to cause proteasome inhibition by coordination of the metal center to available amino acids capable of forming Cu-N, Cu-S, or Cu-O bonds. If this hypothesis is correct, the stoichiometry of the complex is of paramount importance. Consistently, the 1:1 metal-to-ligand complexes 1 and 2 are comparable or slightly more effective than the 1:2 species 3. Nonetheless, due to the small SD values in terms of cytotoxicty, it seems premature to make such a comparison. Copper(II), being a labile species, can gain in stability by bonding to deprotonated and negatively charged phenolates and the nature of the anionic monodentate ligand per se may or may not be relevant. Species 1 and 2 have shown comparable proteasome-inhibitory activity in vitro. Therefore, it can be suggested that the pharmacophore to act as an active species can be described as [CuLI]+ or, more likely, some equivalent solvated species such as [CuLI(H2O)]+ or [CuLI(H2O)2]+, where one or more water molecules replace the chloro or acetato ligands. Granted that more detailed studies are necessary to provide final evidence, this pharmacophore would present an open coordination that facilitates interaction with available amino acids with high affinity for copper. It is viable that the active form of the copper complex binds to the amino-terminal threonine residue of the chymotryptic active center in the 20S proteasome, This hypothesis is based on known mechanisms for lactacystin, esters, epoxyketones, peptide derivatives, and boronates.8-13 Recent data25 indicating that the JAMM domain of the 19S caps in the 26S proteasome is another target for copper complexes suggests that further studies are necessary to assess this possibility and to ascertain the precise locus of coordination. It is also possible that 3 is a pro-drug and in order to become active, the loss of a ligand must occur. This process would convert this compound into a similar [CuLI(H2O)n]+ species, as described in Scheme 2.
Scheme 2.

Suggested conversion of 3 into a [CuLI(H2O)n]+ species.
The depth of the therapeutic potential for metal-based proteasome inhibitors is yet to be determined. It is evident, however, that this can become a viable novel route to anticancer therapy. Ongoing work in our laboratories focus on (i) the metabolic stability and the pharmacokinetic behavior of these compounds, (ii) assessment of the role of anionic ligands, and (iii) on the inclusion of other bivalent metal centers such as cobalt, nickel, and zinc, and trivalent ions such as iron and ruthenium to assess the roles of ligand dissociation, the 1:1 and 1:2 metal-to-ligand stoichiometry, redox activity, and charge. We are also developing biomimetic models of the 1:1 gallium and copper species towards complexation with threonine and other amino acids and short peptides in order to determine the precise coordination loci within the proteasome.
4. Experimental Section
4.1. Chemicals
All reagents were obtained from commercial sources. CuCl2·2H2O was purchased from Sigma-Aldrich (St. Louis, MO). Solvents were purified by means of an I.T. solvent purification system. RPMI 1640, penicillin and streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from Aleken Biologicals (Nash, TX). Fluorogenic peptide substrate Suc-LLVY-AMC (for the proteasomal chymotrypsin-like activity) were from Calbiochem (San Diego, CA). Mouse monoclonal antibody against human poly(AP-ribose) polymerase (PARP), p27, ubiquitin and secondary antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
4.2. Methods
ESI spectra were measured on a Micromass QuattroLC triple quadrupole mass spectrometer with an electrospray/APCI source and Walters Alliance 2695 LC, autosampler and photodiode array UV detector. Experimental assignments were simulated based on peak location and isotopic distributions. Infrared spectra were measured from 4000 to 400 cm-1 as KBr pellets on a Tensor 27 FTIR-spectrophotometer. UV-visible spectroscopy from 1.0 × 10-4 methanol or methanol:DMSO solutions were performed using a Cary 50 spectrometer in the range 250 to 1000 nm. The samples were mortar-ground and heat-dried under vacuum overnight to eliminate solvent molecules. First derivative X-Band EPR spectra of 1.0x10-3 M methanol solutions were performed with a Bruker ESP 300 spectrometer using liquid helium as the coolant. Elemental analyses were performed by Midwest Microlab, Indianapolis, Indiana. Cellular morphology analyses and imaging with phase contrast were performed on a Zeiss Axiovert 25 microscope. X-ray Structural Determination for [Cu(LI)(Cl)] (1). Diffraction data were measured at 100 K on a Bruker X8 APEX-II kappa geometry diffractometer with Mo radiation and a graphite monochromator. Frames were collected as a series of sweeps with the detector at 40 mm and 0.3 degrees between each frame, recorded for 10 or 20 s. APEX-II [26] and SHELX-97 [27] software were used in the collection and refinement (Table 2). Crystals of 1 [C13H11N2O1Cl1I2Cu1]were blue-green needles; the diffraction sample was 0.4 × 0.02 × 0.01 mm3. 50251 total reflections were recorded, yielding 7828 independent hkl data. Hydrogen atoms were placed in calculated positions. The asymmetric unit consists of two independent neutral complexes.
Table 2.
Crystal data and structure refinements for [Cu(LI)C1] (1).
| Formula | C13H11N2O1Cl1I2Cu1 |
|---|---|
| FW | 564.03 |
| Space group | P21/c |
| a (Å) | 6.8589(2) |
| b (Å) | 16.9913(5) |
| c (Å) | 27.0658(8) |
| β (deg) | 95.822(2) |
| V (Å3) | 3138.0(2) |
| Z | 8 |
| Temp (K) | 100(2) |
| λ (Å) | 0.71073 |
| Density, calcd (g cm-3) | 2.388 |
| μ (mm-1) | 5.493 |
| R(F) (%)a | 4.12 |
| Rw(F) (%)a | 6.51 |
R(F) = Σ ‖Fo|-|Fc‖/Σ|Fo|; Rw(F) = [Σw(Fo2 - Fc2)2/ Σw(Fo2)2]1/2 for I>2σ(I).
4.3. Syntheses
The ligand 2,4-diiodo- 6-((pyridine-2- ylmethylamino) methyl)phenol was synthesized according to a previously published procedure.[16]
[Cu(LI)Cl] (1)
(0.50 g, 1.1 mmol) of HLI was dissolved in 15 mL of DMSO. After 5 minutes (0.18 g, 1.2 mmol) of CuCl2 2H2O was dissolved in 15 mL of DMSO and the resulting solution was added drop wise and stirred at room temperature for 45 min. The green solution was added to 15 mL of cold ethanol to afford dark green needle-like crystals after 48 hours. Yield = 0.15 g (27%). Elemental analysis calc. (%) for C13H11ClCuI2N2O: C 27.68; H 1.97; N 4.97. Found: C, 27.72; H, 1.88; N, 4.86. IR data (KBr, cm-1): 3073 ν(N-H); ESI+ MS data (MeOH): m/z = 527.9 for [CuL]+, 605.9 [CuL-DMSO]+.
[Cu(LI)OAc] (2)
(0.50 g, 1.1 mmol) of HLI was dissolved in 15 mL of DMSO and treated with 1.1 equivalents of triethylamine. While stirring for 5 minutes at room temperature, (0.27 g, 1.2 mmol) of Cu(OAc)2 2H2O was dissolved in 15 mL of DMSO and the ligand solution was added drop wise and allowed to react for 45 min. The complex was isolated using suction filtration and washed with cold methanol and ether to afford a green precipitate. Yield = 0.505 g (86%). Elemental analysis calc. (%) for C15H14CuI2N2O3: C 30.66; H 2.40; N 4.77. Found: C, 30.82; H, 2.45; N, 4.70. IR data (KBr, cm-1): 3073 ν(N-H); 1586 νasym(OAc¨); 1402 νvsym(OAc¨). ESI+ MS data (MeOH): m/z = 527.8 for [CuL]+ 606.0 [CuL-DMSO]+.
[Cu(HLI)(LI)]OAc (3)
A 15 mL methanol solution of HLI (1.05 g, 2.1 mmol) was added drop wise to a 15 mL methanol solution of Cu(OAc)2 2H2O (0.27 g, 1.2 mmol) at 45 °C. After 45 minutes a green precipitate was obtained, isolated by frit filtration, and washed with cold methanol and ether. The solid was recrystallized in dichloromethane. Yield = 0.900 g (79%). Elemental analysis calc (%) for 3•CH2Cl2 C29H28Cl2CuI4N4O4: C 30.59; H 2.48; N 4.92. Found: C, 30.82; H, 2.42; N, 4.76. IR data (KBr, cm-1) 3448 ν(OH); 3076 ν(N-H); 1564 νasym(OAc¨);1439 νsym(OAc¨). ESI+ MS data (MeOH): m/z = 527.9 for [CuL]+, 994 (minor) for [Cu(HL)(L)]+.
4.4. Trypan Blue Assay
The trypan blue dye exclusion assay was performed by mixing 100 μl of cell suspension with 50 μl of 0.4% trypan glue dye before injecting into a hemocytometer and counting. The number of cells that absorbed the dye and those that excluded the dye were counted, from which the percentage of nonviable cell number to total cell number was calculated.
4.5. Cell Cultures and Whole-cell Extract Preparation
Human prostate cancer cells, C4-2B and PC-3 were grown in RPMI 1640 medium supplemented with 10% FBS and maintained at 37°C and 5% CO2. MCF-10A cells (normal, derived from benign human breast tissue) were obtained and cultured as previously described. A whole cell extract was prepared.[28]
4.6. Analysis of the Proteasomal Activity in Whole-cell Extract
C4-2B, PC-3 and MCF-10A whole-cell extract (8 μg) was incubated with 10 μmol/L chymotrypsin-like-substrate (Suc-LLVY-AMC) in 100 μL assay buffer [50 mmol/L Tris-HCl (pH 7.5)] in the presence of different copper compounds, ligand, and inorganic copper salt at various concentrations or solvent DMSO as control. After a 2 h incubation at 37° C, production of hydrolyzed AMC groups was measured using a Wallac Victor3™ multilabel counter with an excitation filter of 365 nm and an emission filter of 460 nm.[28]
4.7. Western Blot Analysis
Cell extracts were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Western blot analysis was performed using specific antibodies to p27, ubiquitin, or PARP (Santa Cruz Biotechnology Inc, Santa Cruz, CA) followed by visualization using the HyGLO reagent (Denville Scientific, Metuchin, NJ)
Supplementary Material
Figure 3.

IC50 values for cell death induction by 1 - 3. (a) Human leukemia Jurkat Tcells were treated with copper compounds 1-3 for 18 h, followed by measurement of cell death in a trypan blue exclusion assay. HLI, CuCl2, and DMSO are used as controls. Standard deviations are shown as error bars.
Acknowledgements
S.S.H. and M.F. contributed equally to this publication. C.N.V. gratefully acknowledges the Wayne State University and partial support from the National Science Foundation (CHE-0718470). Q.P.D. acknowledges the Karmanos Cancer Institute of Wayne State University, the Department of Defense Breast Cancer Research Program (W81XWH-04-1-0688 and DAMD17-03-1-0175) and the National Cancer Institute (1R01CA120009). M.F. acknowledges a training grant, “Ruth L. Kirschstein National Service Research Award” (T32-CA009531).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kelland L. Nature Rev. Cancer. 2007;7:573. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- [2].(a) Pavelka M, Lucas MFA, Russo N. Chem. Eur. J. 2007;13:10108. doi: 10.1002/chem.200700887. [DOI] [PubMed] [Google Scholar]; (b) Todd RC, Lovejoy KS, Lippard SJ. J. Am. Chem. Soc. 2007;129:6370. doi: 10.1021/ja071143p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Beljanski V, Villanueva JM, Doetsch PW, Natile G, Marzilli LG. J. Am. Chem. Soc. 2005;127:15833. doi: 10.1021/ja053089n. [DOI] [PubMed] [Google Scholar]; (d) Fuertes MA, Alonso C, Pérez JM. Chem. Rev. 2003;103:645. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]; (e) Deubel DV. J. Am. Chem. Soc. 2002;124:5834. doi: 10.1021/ja012221q. [DOI] [PubMed] [Google Scholar]; (f) Jamieson ER, Lippard SJ. Chem. Rev. 1999;99:2467. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- [3].(a) Zhang CX, Lippard SJ. Curr. Opin. Chem. Biol. 2003;7:481. doi: 10.1016/s1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]; (b) Kelland LR, Farrell N, editors. Platinum-based Drugs in Cancer Therapy. Humana Press; Totowa: 2000. [Google Scholar]
- [4].(a) Jakupec MA, Galanski M, Arion VB, Hartinger CG, Keppler BK. Dalton Trans. 2008:183. doi: 10.1039/b712656p. [DOI] [PubMed] [Google Scholar]; (b) Fricker SP. Dalton Trans. 2007;43:4903. doi: 10.1039/b705551j. [DOI] [PubMed] [Google Scholar]; (c) Kalinowski DS, Sharpe PC, Bernhardt PV, Richardson DR. J. Med. Chem. 2007;50:6212. doi: 10.1021/jm070839q. [DOI] [PubMed] [Google Scholar]; (d) Tardito S, Bussolati O, Maffini M, Tegoni M, Giannetto M, Dall'Asta V, Franchi-Gazzola R, Lanfranchi M, Pellinghelli MA, Mucchino C, Mori G, Marchio L. J. Med. Chem. 2007;50:1916. doi: 10.1021/jm061174f. [DOI] [PubMed] [Google Scholar]; (e) Schatzschneider U, Metzler-Nolte N. Angew. Chem., Int. Ed. 2006;45:1504. doi: 10.1002/anie.200504604. [DOI] [PubMed] [Google Scholar]
- [5].(a) Goldberg AL. Biochem. Soc. Trans. 2007;35:12. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]; (b) Goldberg AL. Science. 1995;268:522. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]; (c) Borissenko L, Groll M. Chem. Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- [6].Dou QP, Li B. Drug Resist. Updates. 1999;2:215. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- [7].Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. J. Clin. Oncology. 2005;23:630. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- [8].Masse CE, Morgan AJ, Adams J, Panek JS. Eur. J. Org. Chem. 2000;14:2513. [Google Scholar]
- [9].(a) Basse N, Piguel S, Papapostolou D, Ferrier-Berthelot A, Richy N, Pagano M, Sarthou P, Sobczak-Thepot J, Reboud-Ravaux M, Vidal J. J. Med. Chem. 2007;50:2842. doi: 10.1021/jm0701324. [DOI] [PubMed] [Google Scholar]; (b) Groll M, Goetz M, Kaiser M, Weyher E, Moroder L. Chem. Biol. 2006;13:607. doi: 10.1016/j.chembiol.2006.04.005. [DOI] [PubMed] [Google Scholar]; (c) Koguchi Y, Kohno J, Nishio M, Takahashi K, Okuda T, Ohnuki T, Komatsubara S. J. Antibiot. 2000;53:105. doi: 10.7164/antibiotics.53.105. [DOI] [PubMed] [Google Scholar]
- [10].(a) Groll M, Kim KB, Kairies N, Huber R, Crews CM. J. Am. Chem. Soc. 2000;122:1237. [Google Scholar]; (b) Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Chem. Biol. 1999;6:811. doi: 10.1016/s1074-5521(99)80128-8. [DOI] [PubMed] [Google Scholar]
- [11].(a) Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott P. Clin. Chem. 2000;46:673. [PubMed] [Google Scholar]; (b) Loidi G, Groll M, Musiol HJ, Huber R, Moroder L. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5418. doi: 10.1073/pnas.96.10.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lum RT, Nelson MG, Joly A, Horsma AG, Lee G, Meyer SM, Wick MM, Schow SR. Bioorg. Med. Chem. Lett. 1998;8:209. doi: 10.1016/s0960-894x(98)00015-8. [DOI] [PubMed] [Google Scholar]; (d) Lynas JF, Harriott P, Healy A, Mckarvey MA, Walker B. Bioorg. Med. Chem. Lett. 1998;8:373. doi: 10.1016/s0960-894x(98)00030-4. [DOI] [PubMed] [Google Scholar]; (e) Vinitsky A, Cardozo C, Sepp-Lorenzio L, Machaud C, Orlowski M. J. Biol. Chem. 1994;269:29860. [PubMed] [Google Scholar]
- [12].(a) Labutti J, Parsons I, Huang R, Miwa G, Gan L-S, Daniels JS. Chem. Res. Toxicol. 2006;19:539. doi: 10.1021/tx050313d. [DOI] [PubMed] [Google Scholar]; (b) Vivier M, Jarrousse A-S, Bouchon B, Galmier M-J, Auzeloux P, Sauzieres J, Madelmont J-C. J. Med. Chem. 2005;48:6731. doi: 10.1021/jm050181l. [DOI] [PubMed] [Google Scholar]; (c) Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, Schlossman R, Podar K, Munshi NC, Mitsiades N, Anderson KC. Blood. 2003;101:1530. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]; (d) Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Cancer Res. 1999;59:2615. [PubMed] [Google Scholar]
- [13].Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure. 2006;14:451. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- [14].(a) Daniel KG, Gupta P, Harbach RH, Guida C, Dou QP. Biochem. Pharmacol. 2004;67:1139. doi: 10.1016/j.bcp.2003.10.031. [DOI] [PubMed] [Google Scholar]; (b) Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Breast Cancer Res. 2005;7:R897. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen D, Dou QP. Expert Opin. Ther. Targets. 2008;12:739. doi: 10.1517/14728222.12.6.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].(a) Shakya R, Peng F, Liu J, Heeg MJ, Verani CN. Inorg. Chem. 2006;45:6263. doi: 10.1021/ic060106g. [DOI] [PubMed] [Google Scholar]; (b) Shakya R, Imbert C, Hratchian HP, Lanznaster M, Heeg MJ, McGarvey BR, Allard MM, Schlegel HB, Verani CN. Dalton Trans. 2006:2517–2525. doi: 10.1039/b514190g. [DOI] [PubMed] [Google Scholar]
- [17].Frezza M, Verani CN, Chen D, Dou QP. Lett. Drug Design & Discov. 2007;4:311. [Google Scholar]
- [18].Chen D, Frezza M, Shakya R, Cui CQ, Milacic V, Verani CN, Dou QP. Cancer Res. 2007;67:9258. doi: 10.1158/0008-5472.CAN-07-1813. [DOI] [PubMed] [Google Scholar]
- [19].(a) Michel F, Thomas F, Hamman S, Philouze C, Saint-Aman E, Pierre J-L. Eur. J. Inorg. Chem. 2006;I-18:3684. [Google Scholar]; (b) Shimazaki Y, Huth S, Odani A, Yamauchi O. Angew. Chem. 2000;112:1732. doi: 10.1002/(sici)1521-3773(20000502)39:9<1666::aid-anie1666>3.0.co;2-o. Angew. Chem., Int. Ed. 2000, 39, 1666. [DOI] [PubMed] [Google Scholar]; (c) Itoh S, Taki M, Takayama S, Nagatomo S, Kitagawa T, Sakurada N, Arakawa R, Fukuzumi S. Angew. Chem. Int. Ed. 1999;38:2774. doi: 10.1002/(sici)1521-3773(19990917)38:18<2774::aid-anie2774>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (d) Neves A, Verani CN, de Brito MA, Vencato I, Mangrich A, Oliva G, Souza DDHF, Batista AA. Inorg. Chim. Acta. 1999;290:207. [Google Scholar]; (e) Vaidyanathan M, Viswanathan R, Palaniandavar M, Balasubramanian T, Prabhaharan P, Muthiah TP. Inorg. Chem. 1998;37:6418. doi: 10.1021/ic971567s. [DOI] [PubMed] [Google Scholar]; (f) Zurita D, Gautier-Luneau I, Ménage S, Pierre J-L, Saint-Aman E. J. Biol. Inorg. Chem. 1997;2:46. [Google Scholar]
- [20].Hindo SS. Synthesis, Structure, Electrochemistry, Spectroscopy, and Reactivity of Phenolate-Based Copper(II) Archetypes and Modules for Magnetic Soft Materials. M.Sc. Dissertation. Wayne State University; Detroit, MI: 2005. [Google Scholar]
- [21].Kim CJ, Lee SJ, Seo MH, Cho NY, Sohn UD, Lee MY, Shin YK, Sim SS. Arch. Pharm. Res. 2002;25:675. doi: 10.1007/BF02976943. [DOI] [PubMed] [Google Scholar]; (b) Olaleye SB, Farombi EO. Phytother. Res. 2006;20:14. doi: 10.1002/ptr.1793. [DOI] [PubMed] [Google Scholar]
- [22].Lesh FD, Hindo SS, Heeg MJ, Allard MM, Jain P, Peng B, Hryhorczuk L, Verani CN. Eur. J. Inorg. Chem. 2009:345. [Google Scholar]
- [23].Brewer GJ. Exp. Biol. Med. 2007;232:323. [PubMed] [Google Scholar]
- [24].Lopes UG, Erhardt P, Yao R, Cooper GM. J. Biol. Chem. 1997;272:12893. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- [25].Cvek B, Milacic V, Taraba J, Dou QP. J. Med. Chem. 2008;51:6256–6258. doi: 10.1021/jm8007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].SMART and APEX II collection and processing programs are distributed by the manufacturer. Bruker AXS Inc.; Madison WI, USA: [Google Scholar]
- [27].Sheldrick G. SHELX-97. University of Gottingen; Germany: 1997. [Google Scholar]
- [28].Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Breast Cancer Res. 2005;7:R897. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.