

Abstract
We present statistical methods for analyzing replicated cDNA microarray expression data and report the results of a controlled experiment. The study was conducted to investigate inherent variability in gene expression data and the extent to which replication in an experiment produces more consistent and reliable findings. We introduce a statistical model to describe the probability that mRNA is contained in the target sample tissue, converted to probe, and ultimately detected on the slide. We also introduce a method to analyze the combined data from all replicates. Of the 288 genes considered in this controlled experiment, 32 would be expected to produce strong hybridization signals because of the known presence of repetitive sequences within them. Results based on individual replicates, however, show that there are 55, 36, and 58 highly expressed genes in replicates 1, 2, and 3, respectively. On the other hand, an analysis by using the combined data from all 3 replicates reveals that only 2 of the 288 genes are incorrectly classified as expressed. Our experiment shows that any single microarray output is subject to substantial variability. By pooling data from replicates, we can provide a more reliable analysis of gene expression data. Therefore, we conclude that designing experiments with replications will greatly reduce misclassification rates. We recommend that at least three replicates be used in designing experiments by using cDNA microarrays, particularly when gene expression data from single specimens are being analyzed.
Although the high-throughput technology now available enables genetic researchers to study expression for thousands of genes simultaneously, experiments by using microarrays may be costly and time consuming. The manufacturers of microarray equipment do not stress the need for replication of studies. Production of arrays can be slow and the supply limited. As a result, most current molecular genetic studies that use microarray technology are sometimes done without replication. However, statistical analyses in many settings have demonstrated that important insights into the nature of inherent variability are obtained by the replication of experiments.
In Section 1, we report the design of a controlled experiment involving replication of cDNA hybridizations. The study was conducted to investigate inherent variability in gene expression data and the extent to which replication in an experiment produces more consistent and reliable findings. In Sections 2.1 and 2.2, we introduce statistical models to describe the probability that an mRNA is contained in the target sample tissue, converted to probe, and ultimately detected on the slide as an observed expression. We use a mixed normal distribution to model the distribution of observed gene expressions. In Sections 2.3 and 2.4, we conduct a separate analysis for each replicate. In Sections 2.5 and 2.6, we introduce a model to provide a joint analysis based on the combined data collected from all replicates. In Section 2.7, we consider the reliability of the classification of gene expression as a function of the number of replicates.
Our results show that any single microarray output is subject to substantial variability. By pooling data from replicates, we can provide a more reliable classification of gene expression. Therefore, we conclude that designing experiments with replications will greatly reduce misclassification rates. We recommend that at least three replicates be used in designing experiments using cDNA microarrays. Although our results depend on specific instruments and techniques, the statistical models and methods that we propose in this article can be applied in general settings.
1. Materials and Methods
In this section, we provide a brief description of our experimental process. To check the consistency of microarray experiments, we conducted a study to investigate whether the unevenness of the surfaces of glass slides, the locations of cDNA spots on the slides, and other aspects of a microarray experiment may produce variation in measurements of transcriptions. To test these variables of cDNA microarrays generated in our facility, we printed triplicates of 288 cDNA sets (288 elements per set) at 3 locations on the same slide and performed hybridization experiments with probes from 1 source. By comparing the signals from these triplicates, we hoped to learn about the reproducibility of the array process and whether seemingly minor factors, such as the location of the spots in the array, can affect the outcome of analyses. Of the 288 genes considered in this experiment, 32 would be expected a priori to appear highly expressed because of structural features within the genes, namely Alu repeats that should crosshybridize to similar sequences widely distributed among expressed and nonexpressed portions of the genome.
1.1. Generation of Array-Ready cDNAs.
Frozen glycerol stocks of Escherichia coli containing individual cDNA clones in the IMAGE consortium distributed in 384-well plates were purchased from Genome Systems, St. Louis. Individual bacterial clones were selected and distributed into 96-well plates. Amplifications of DNA by PCR with primers specific to the vector sequences flanking the insert cDNA were performed in 96-well PCR plates in a Perkin–Elmer 9600 thermocycler in 50-μl reactions containing ×1 PCR buffer (Promega), 1.5 mM MgCl2, 0.2 mM dNTPs, 10 pmol of each primer, 5 units of Taq polymerase, and 0.5 μl of the bacterial culture. The annealing was at 55°C for 20 sec, and the extension was at 72°C for 90 sec for 30 cycles. Five microliters of the PCR reactions was analyzed in a 1% agarose gel to verify the success of PCR. The DNA in the remaining 45 μl was precipitated with addition of 45 μl of isopropanol and resuspended in 15 μl of ×3 SSC. Note that 32 of the 288 genes contained Alu repetitive sequences and therefore were expected to show a high level of signal because of crosshybridization of Alu containing messages.
1.2. Printing of the cDNAs on Glass Slides.
The array-ready cDNAs in 96-well plates were loaded into a 417 arrayer manufactured by Genetic Microsystems (Woburn, MA). Poly-l-lysine-coated slides from Sigma were used as the solid support for construction of the array. Triplicate arrays were placed on the slides at 6-mm intervals. After printing, the slides were hydrated over a steam bath and the DNA UV-crosslinked onto the slides. After blocking the slides with succinic anhydrate, the DNA was denatured by boiling for 2 min, and the slides were dehydrated with ethanol.
1.3. Preparation of Fluorescently Labeled Probes and Hybridization to Glass Slides.
Total RNA was isolated with Trizol reagents (Life Technologies, Grand Island, NY) from human tissue specimens obtained during surgical procedure. Fifty micrograms of total RNA was annealed to oligo(dT) and reverse transcribed in the presence of Cy3-labeled dUTP. The resulting cDNA was precipitated with ethanol, resuspended in 20 μl of hybridization solution [50% formamide/×5 SSC/0.5% SDS/1 μg of CoT1 DNA/10 μg of yeast tRNA/10 pmol of poly(dA)], heat denatured, applied to the slide, and sealed under a coverslip. The slide was placed in a humidified chamber at 42°C overnight. The washing was in ×0.5 SSC/0.2% SDS at room temperature twice for 5 min each, followed by three washes with ×0.2 SSC at room temperature for 2 min each. The slide was dried and scanned with a 418 array reader from Genetic Microsystems. The resulting image was quantified by using the software program scanalyze (1). The fluorescence of the Cy3 label is carried on Channel 1. Cy5 was not used in this experiment, and hence Channel 2 carried only background noise.
2. Statistical Model and Analytical Approach
For gene g in experimental replicate j, where g = 1, … , G, j = 1, … , J, let Xgj denote the median of the set of background-corrected single pixel values of Channel 1 to Channel 2 fluorescence for all pixels within the fluorescence spot. This measure is denoted by MRAT in Eisen et al. (2) and Eisen (1). We take the natural logarithm of MRAT as Ygj = ln(Xgj) and refer to Ygj as a log-ratio. In this experiment, three replications of expression measurements for 288 gene probes were obtained under the same experimental conditions from the same human tissue sample. Thus, G = 288 and J = 3.
2.1. The Probability of Observing Expressed Genes.
Consider any one replicate j among the three experimental replicates j = 1, 2, 3. Let ℰg represent the event that mRNA for gene g in the array is contained in the target sample tissue. In advance of observing the gene expression data, we attach a prior probability Pr{ℰg} = p to this event for each gene g that is under consideration. The fact that p is not indexed by g implies that, in advance of considering the experimental data, we are uniformly ignorant about whether any particular gene is contained in the sample tissue. We denote the complement of event ℰg by ℰ̄g.
For a gene to be detected on the slide, three hurdles must be cleared. First, the mRNA must be part of the sample from which the probe is prepared. Second, some of the mRNA in the sample must be converted to probe. Third, some of the probe must be detected by the cDNAs deposited on the slide. If any one of these hurdles is not cleared, the gene cannot be expressed in the microarray data.
The log-ratio Ygj for gene g in replicate j will have two distinct distributions, depending on whether gene g is contained in the sample tissue. First, if mRNA from gene g is not in the sample tissue (i.e., event ℰ̄g), its measured expression should reflect only experimental error. In this case, we assume that Ygj is normally distributed as N(μUj, σUj2), where subscript Uj refers to the anticipated outcome of being unexpressed. We denote the corresponding probability density function of conditional variable Ygj|ℰ̄g by fUj(y). Observe that the distribution parameter values may vary with the replicate j that is under consideration. On the other hand, if gene g is in the sample tissue (i.e., ℰg) and should therefore be detected on the slide, we assume that Ygj is distributed as N(μEj, σEj2), where subscript Ej refers to the anticipated outcome of being expressed. We denote the corresponding probability density function of the conditional variable Ygj|ℰg by fEj(y). Again, we note that the parameters may vary with the replicate j. By definition, we require μUj < μEj. For event ℰ̄g, Ygj is a measurement reflecting only background noise or inherent experimental error. For event ℰg, measurement Ygj reflects the actual expression of gene g in the sample tissue, obscured to some degree by the presence of background noise.
2.2. A Mixture Model for the Distribution of Observed Log Ratios.
Given the complementary events ℰg and ℰ̄g for any gene g, the observed log-ratio Ygj for replicate j will be distributed according to the following mixed normal probability density function.
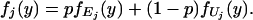 |
1 |
A simple manipulation of the two components of Eq. 1 gives posterior probabilities for whether gene g is expressed in the sample tissue based on a reading Ygj = y in replicate j. Specifically, if the microarray reading for the log-ratio of gene g is Ygj = y in replicate j, the posterior probability that the reading reflects expression of gene g in the sample tissue (and not simply background noise) is given by
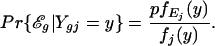 |
2 |
2.3. Separate Analysis for Each Replicate.
We now examine the problems of estimating the parameters p, μUj, σUj2, μEj, and σEj2 for model 1, interpreting the parameter estimates and using them to estimate the posterior probabilities in Eq. 2.
First we solved for the maximum likelihood estimates of the unknown parameters based on model 1.The estimates were calculated separately for the three replications to see how stable the results are from one replicate to another. The parameter estimates appear in Table 1.
Table 1.
Separate analysis for each experimental replicate
| Parameter | Replicate
|
||
|---|---|---|---|
| j = 1 | j = 2 | j = 3 | |
| p | 0.285 | 0.124 | 0.274 |
| μUj | 0.384 | 0.410 | 0.442 |
| μEj | 0.968 | 2.203 | 1.233 |
| σUj2 | 0.070 | 0.076 | 0.062 |
| σEj2 | 1.186 | 0.114 | 1.079 |
Parameter estimates of the mixed normal model (Eq. 1).
The estimates for replicate 2 in Table 1 are sharply different from those for the other two replicates. The estimate of mean parameter μEj is much larger than for replicates 1 and 3, and the estimates of variance parameter σEj2 and probability p are much smaller. It is unclear why replicate 2 is so different from the others, but it serves to remind us that replication does not ensure duplication of results, a fact that cannot be quantified when replication is not used. We also note in Table 1 that the estimate of p varies greatly from one replicate to another. Recall in our controlled experiment that only 32 of the 288 genes (fraction 0.111) should be classified as expressed. Thus, the estimates of p provided by replicates 1 and 3 are much too large.
We turn next to estimates of the posterior probabilities (Eq. 2). Table 2 summarizes a representative fragment of the results. We see generally that the posterior probability clearly indicates whether a gene is expressed in the sample tissue and that the results are quite uniform across the three replications. There are occasions, however, as illustrated by the results for gene no. 17, where the three replications do not give uniform results. Replicate 3 for this gene gives a very large posterior probability (1.0000) to the expression event ℰg|Yg3 = y, whereas the other two replicates give smaller probabilities (0.1404 and 0.0000).
Table 2.
Posterior probability of expression in sample tissue
| Gene g | Replicate 1
|
Replicate 2
|
Replicate 3
|
|||
|---|---|---|---|---|---|---|
| Yg1 = y | Pr{ℰg|Yg1 = y} | Yg2 = y | Pr{ℰg|Yg2 = y} | Yg3 = y | Pr{ℰg|Yg3 = y} | |
| 1 | 2.043 | 1.0000 | 1.6804 | 0.9993 | 2.6251 | 1.0000 |
| 2 | 0.6549 | 0.1356 | 0.5551 | 0.0000 | 0.6874 | 0.1134 |
| 3 | 0.4940 | 0.0877 | 0.3791 | 0.0000 | 0.5065 | 0.0682 |
| 17 | 0.6646 | 0.1404 | 0.2662 | 0.0000 | 1.7204 | 1.0000 |
| 18 | 2.4397 | 1.0000 | 2.3081 | 1.0000 | 2.2481 | 1.0000 |
| 19 | 2.2331 | 1.0000 | 2.0549 | 1.0000 | 2.5257 | 1.0000 |
Log ratios Ygj = y and estimates of posterior probabilities Pr{ℰg|Ygj = y} for a few illustrative genes g, for replicates j = 1, 2, 3.
2.4. Checking the Consistency of Results from the Three Replicates.
We next study the extent to which the three replications, analyzed separately, provide consistent classification with respect to gene expression. Using the posterior expression probabilities (such as those in Table 2) for each replicate j, we will classify a gene g as being expressed if Pr{ℰg|Ygj = y} is larger than 0.5 and as not being expressed otherwise. This classification is done independently for each replicate.
Table 3 contains the results of this classification process. Table 3 Left shows a three-way crossclassification, whereas, for ease of interpretation, Table 3 Right shows the three two-way crossclassifications corresponding to the three pairs of replicates. If the replicates were perfectly consistent, only two cells of Table 3 Left would have counts, namely, the cell counting unexpressed genes in all three replicates and the cell counting expressed genes in all three replicates. In fact, however, all of the cells in the table have counts, and four of these are sizeable. This is evidence that the replicates are not perfectly consistent. As one illustration of inconsistency, we note in Table 3 Left that 23 genes classified as expressed in replication 3 are classified as unexpressed in replications 1 and 2. As another illustration, we note in Table 3 Right that the numbers of genes classified as expressed in the three replicates are 55, 36, and 58, respectively. As 32 of the 288 genes should be classified as expressed, we are again reminded by these results that replicates 1 and 3 are providing a large number of false positives.
Table 3.
Comparing results of a separate analysis for each replicate
| Replicate 1 | Three-way crossclassification
|
Three two-way crossclassifications
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Replicate 3 | |||||||||||||||||
| U
|
E
|
||||||||||||||||
| Replicate 2 | Replicate 2 | Replicate 2 | Replicate 3 | Replicate 3 | |||||||||||||
| U | E | U | E | Total | Replicate 1 | U | E | Total | Replicate 1 | U | E | Total | Replicate 2 | U | E | Total | |
| U | 207 | 2 | 23 | 1 | 233 | U | 230 | 3 | 233 | U | 209 | 24 | 233 | U | 226 | 26 | 252 |
| E | 19 | 2 | 3 | 31 | 55 | E | 22 | 33 | 55 | E | 21 | 34 | 55 | E | 4 | 32 | 36 |
| Total | 226 | 4 | 26 | 32 | 288 | Total | 252 | 36 | 288 | Total | 230 | 58 | 288 | Total | 230 | 58 | 288 |
Crossclassification of the log-ratio for three replicates analyzed separately. A gene is classified as expressed if its posterior probability Pr{ℰg|Ygj = y} exceeds 0.5 and as unexpressed otherwise.
U, unexpressed; E, expressed.
To model the count data in Table 3, we again postulate a prior probability p that any given gene is expressed in the sample tissue. As discussed earlier, mRNA in the tissue must clear two further hurdles to appear “expressed” on the microarray slide. It must be converted to probe and hybridized to the cDNAs that are deposited on the slide. The stochastic behavior of this mechanical process can cause replications to differ. We let rj denote the conditional probability that a gene will be classified as “expressed” in replicate j, where j = 1, … , J, and assume that the corresponding conditional events are independent from one replicate to another. We also postulate that, by chance, a gene that is not expressed in the sample tissue may appear expressed on the slide because of background noise or other experimental artifacts. The conditional probability of such a spurious indication will be denoted by sj for the jth replicate, j = 1, … , J. Again, we assume that these conditional events are independent among the replicates. We can now estimate these probabilities from the count data in Table 3 using the method of maximum likelihood applied to the following likelihood function
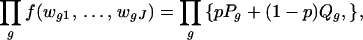 |
3 |
where
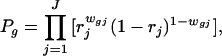 |
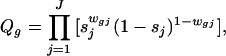 |
and wgj denotes the indicator variable for whether gene g is classified as expressed in replicate j = 1, … , J. For our experiment, the number of replicates is J = 3, and the maximum likelihood estimates based on the data in Table 3 are p̂ = 0.117, r̂1 = 1.000, r̂2 = 0.974, r̂3 = 0.946, ŝ1 = 0.084, ŝ2 = 0.013, and ŝ3 = 0.103. We note that r̂1 was numerically so close to the value 1 that it was set to 1 for the computation.
The probability estimates reveal several points of interest for microarray studies. First, the experimental design purposely selected 32 of 288 genes to be expressed, which is the exact fraction p = 0.111. Hence, the statistical analysis has reliably reproduced this fraction in the estimate of p. Second, the estimates of rj show that (i) it is not a certainty that an expressed gene will be classified as “expressed” on the slide, and (ii) the probability of doing so can vary from one experimental execution to another. Third, the estimates of sj show that “ghost” indications of genes (i.e., false positives) can occur with a frequency as large as 10% in a single experiment.
2.5. A Model for Analyzing the Combined Data from All Replicates.
We now seek to describe the microarray data from the three replications by a single model that will support a joint analysis. We use the following two-way linear model as a general statistical model for log-ratio data.
 |
4 |
Here E(Ygj) = μ + αg + βj + γgj is the mean log-ratio for gene g under experimental condition j. The component μ is the overall mean log-ratio for all genes and experimental conditions, αg is the main effect for gene g, βj is the main effect for experimental condition j, and γgj is an interaction term that reflects differential gene expression for gene g under experimental condition j. In this particular context, the experimental condition j refers to replicate j. The term ɛgj is a random error which, by definition, has a mean of zero. We assume that the error terms are independent, but we have no need in this study to make any assumption about their distributional form.
Following our earlier assumption for individual replicates, we define the main effect αg for gene g in model 4 as a random effect that follows one of two distinct distributions according to whether gene g is expressed in the sample tissue (event ℰg). The distribution of αg therefore follows a mixed normal model
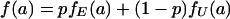 |
5 |
where we now use symbol a in place of y for the variable notation in model 1. We assume that the αg are independent effects for different genes g. We start our study by estimating the overall mean, the main effects for genes, and the main effects for experimental replicates in model 4, as follows.
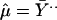 |
6a |
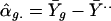 |
6b |
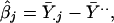 |
6c |
where Ȳg., Ȳ.j, and Ȳ.. denote average log-ratios for all j, all g, and all pairs (g, j), respectively.
It is conceivable that all of the effects in model 4 are random. The estimates in Eq. 6, however, are standard fixed-effect estimates. We choose these estimators because they are inherently free of any distributional assumption. In particular, the estimates α̂g provided by Eq. 6b are fixed-effect estimates that do not depend on the assumption of a normal mixture distribution. We now use these α̂g to estimate the parameters of the mixture distribution in Eq. 5 and subsequently use them again to examine the assumption of a normal mixture distribution. The parameter estimates of the mixed normal model 5 appear in Table 4, together with estimated standard errors. The standard errors are calculated from 100 bootstrap samples.
Table 4.
Analysis of the combined data from all three replicates
| Parameter | Estimate | Est. Std. Err. |
|---|---|---|
| p | 0.118 | 0.013 |
| μU | −0.204 | 0.009 |
| μE | 1.524 | 0.058 |
| σU2 | 0.044 | 0.003 |
| σE2 | 0.126 | 0.036 |
Parameter estimates of the mixed normal model (Eq. 5) derived from the estimated main effects for genes α̂g.
2.6. Analysis Results for the Combined Data.
As the main effects for genes are now estimated from three replications, the results are more sharply delineated than they are in Table 1, where the parameter estimates are calculated separately for each replicate. First, we see that the estimate of p̂ = 0.118 is very close to the known proportion of expressed genes in the sample tissue (32 of 288) and almost identical to the corresponding estimate derived from the count data in Table 3. Second, the estimates of the mean parameters μU and μE are well separated. Third, the variance estimates σU2 and σE2 are smaller than those obtained in separate analyses as listed in Table 1. In fact, they would be expected to be smaller by a factor of about 3. Fourth, the estimate of variance parameter σU2 is smaller than that of σE2. This difference is expected by the fact that, in the event of no expression (i.e., event ℰ̄g), variance parameter σU2 reflects the variability of the log-ratio of background noise on two channels. In the event of gene expression (i.e., event ℰg), variance parameter σE2 reflects two sources of variability: (i) the log-ratio of background noise on two channels, and (ii) the logarithm of gene expression itself.
The posterior probability that gene g is expressed, given the value of α̂g, can be calculated for each gene by using Eq. 2 with Yg replaced by α̂g. These posterior probabilities are all either close to 1 or close to zero. In fact, classifying the genes according to whether this probability is greater than 0.5, it is found that only 2 of the 288 genes are incorrectly classified as to whether they are expressed. Hence, based on the combined data, the classification gives only two false positives and no false negatives. Specifically, genes nos. 75 and 185 are classified as expressed when they were not included in the experimental set of genes. In contrast, recall from Table 3 that the individual replicates were far from perfect in their ability to classify genes.
Fig. 1 a and b show normal probability plots of the α̂g for the genes classified as expressed and unexpressed, respectively. According to the mixed normal model, these two plots should both be normal if the classification were perfect. The evidence seems quite supportive of the normality assumption in both plots. For the genes classified as expressed, there is some evidence of values being clustered. For those classified as unexpressed, there may be a little contamination of the normal distribution, leading to a few outlying observations relative to a pure normal distribution. Fig. 2 shows an overlay of a histogram of the α̂g and the mixed normal probability density function, as described in Eq. 5, based on the parameter estimates in Table 4. A comparison of the histogram and the density function shows that the mixed normal model is quite reasonable. Both the histogram and density function show that the expressed and unexpressed genes occupy well-separated locations on the scale. Note, however, the sharp difference in variability of the two component distributions.
Figure 1.

(a), Normal probability plot of main effect estimates for expressed genes. (b), Normal probability plot of main effect estimates for unexpressed. genes.
Figure 2.

Overlay of a histogram and mixed normal p.d.f. for gene expression main effect.
The interaction terms γgj in model 4 reflect differential gene expression among the experimental conditions and can be estimated as fixed effects, as follows.
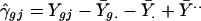 |
7 |
As the experimental conditions here represent replicates, the estimates in Eq. 7 should reflect simply the random noise contributed by the error terms ɛgj. We have discovered, however, that the replicates are not true duplicates and that some genes may be classified as expressed in one or two replicates but not in all three. The estimates of γ̂gj in these cases therefore indicate a differential expression of the genes. We do not need to study these estimates further because Table 3 describes the patterns of inconsistent expression among the three replicates. We note, however, that in microarray investigations of multiple tissues (or other varying experimental conditions), the estimates of differential expression in Eq. 7 are of central scientific interest in determining which genes are truly present in some tissues but not in others.
2.7. Reliability as a Function of the Number of Replicates.
How does the reliability of gene classification vary with the number of replicates? For this experiment, a partial answer is provided by Table 5, which shows the percentages of the 288 genes that are misclassified by this methodology for each possible combination of one, two, and three replicates in the experiment. The false-positive and false-negative components of the misclassification percentage are also shown in the table. First, we note that false positives dominate. This result could be anticipated from our earlier findings and suggests that false indications of expression may be prevalent in microarray studies. Second, the table shows how classification precision varies with the number of replicates. A single replicate, such as replicate 2, may happen to have a low misclassification percentage (1.4%) relative to other replicates but, unfortunately, this reliability cannot be anticipated in advance. For example, replicate 3 alone misclassified 9.0% of genes. As expected, Table 5 confirms that average reliability and the certainty of that reliability increase with the number of replicates. We might surmise that the maximum attainable precision has been achieved with three replicates in our experiment, because the error rate appears to be leveling out at 0.7%. We note that there is no assurance the error rate will go to zero with increasing replication unless all sources of experimental variability are replicated, which is not the case in this experiment.
Table 5.
Misclassification percentages for different combinations of replicates
| Classification Outcome | Combination of Replicates
|
||||||
|---|---|---|---|---|---|---|---|
| (1) | (2) | (3) | (1, 2) | (1, 3) | (2, 3) | (1, 2, 3) | |
| False positive, % | 8.3 | 1.4 | 9.0 | 1.0 | 2.1 | 0.7 | 0.7 |
| False negative, % | 0.3 | 0.0 | 0.0 | 0.3 | 0.3 | 0.0 | 0.0 |
| Misclassified, % | 8.7 | 1.4 | 9.0 | 1.4 | 2.4 | 0.7 | 0.7 |
The optimal number of replicates in a general microarray study will depend on many factors, including the type of array equipment, laboratory technique, and the condition and preparation of samples. If experimental resources and time permit, we see potential benefit from using a minimum of three replicates because three or more classification outcomes offer the possibility of triangulation of results. A comparison of classification outcomes for all possible combinations of replicates, as is done for pairs of replicates in Table 3 Right, for example, might show whether one or more replicates are rogues. A judgment might then be made whether such replicates should be discarded. Replicates might also be used with a majority voting rule to decide whether a gene is expressed. Such a rule is not beneficial in this experiment but might be useful in some applications.
Concluding Discussion
The findings of our simple experiment have three important implications for the generation, analysis, and interpretation of microarray data. First, we have shown that any single microarray output is subject to substantial variability even under the relatively controlled conditions of an experiment. By design, we have introduced only one potential source of variability, namely the location of spots on the slide. Variability from other sources, such as multiple preparations of probe, arrays on different slides, or arrays generated at different times, has not been admitted. Thus, our experiment is evaluating the minimum variability that is likely to be inherent in this system. Still the variation from this one source is considerable. A single output yields numerous misclassifications and, especially, numerous false positives. Replications of the experiment are not consistent and therefore produce different lists of expressed genes.
Second, in modeling the random variation in gene expression, we have found in any single replicate the probability may be as large as 5% that mRNA in the sample tissue either fails to be represented as probe or, if it is represented as probe, fails to be hybridized to the cDNAs that are deposited on the slide (false negatives). Also, the probability may be as large as 10% that ghost genes are expressed (false positives). When microarray data from several replications are combined, we have shown that, quite reasonably, a more accurate genetic picture is produced with a reduction of false positives and false negatives. Third, in the process of analyzing these experimental data, we introduced statistical methodology for microarray data. We have modeled gene expression measurements by using a mixture of normal distributions. From this mixture distribution, a posterior probability is calculated from the microarray reading that quantifies the likelihood that the gene is truly expressed in the tissue. This probability can be used to classify whether a gene transcript is present. A two-way linear statistical model is proposed for microarray data that can span a range of experimental conditions.
Although our results depend on specific instruments and techniques (e.g., RNA extraction method, probe synthesis and labeling, hybridization, array construction, use of glass slide as solid support, and use of only one channel Cy3), the statistical methods we propose can be extended to accommodate more general settings. For example, the methods can be used for experiments that use both channels Cy3 and Cy5. If the two-channel system is used in the standard way with mRNA from a test sample and a reference sample, differential gene expression becomes relevant (i.e., the interaction term of the two-way linear model). As there are then three states of expression (unexpressed, differentially expressed in favor of the test sample, and differentially expressed in favor of the reference sample), a three-component mixture model applies. The statistical methods also extend to data sets from experimental designs that involve additional sources of variability, such as variability introduced by multiple preparations of probes.
The main lesson to be learned from the study is that replication in microarray studies is not equivalent to duplication and hence is not a waste of scientific resources. Experimental replication is essential to reliable scientific discovery in genetic research. Understanding the sources of noise in the process, controlling it, and, if possible, eliminating it, are essential to drawing reliable inferences. By pooling data from replicates, we can provide a more reliable analysis of gene expression data.
Acknowledgments
We acknowledge with thanks the financial support provided for this research by National Institutes of Health grants HL40619–09 and EY12269–02 (M.-L.T.L.), and CA75354 (J.S.), by the National Foundation for Cancer Research (J.S.), by the Natural Sciences and Engineering Research Council of Canada (G.A.W.), and by the Social Sciences and Humanities Research Council of Canada (M.-L.T.L. and G.A.W.).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Eisen M. scanalyzeUser Manual. Stanford, CA: Stanford Univ.; 1999. , Ver. 2.32. [Google Scholar]
- 2.Eisen M B, Spellman P T, Brown P O, Bostein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]