

Abstract
Forward genetics, the phenotype-driven approach to investigating gene identity and function, has a long history in mouse genetics. Random mutations in the mouse transcend bias about gene function and provide avenues towards unique discoveries. The study of the peripheral nervous system is no exception; from historical strains such as the trembler mouse, which led to the identification of PMP22 as a human disease gene causing multiple forms of peripheral neuropathy, to the more recent identification of the claw paw and sprawling mutations, forward genetics has long been a tool for probing the physiology, pathogenesis, and genetics of the PNS. Even as spontaneous and mutagenized mice continue to enable the identification of novel genes, provide allelic series for detailed functional studies, and generate models useful for clinical research, new methods, such as the piggyBac transposon, are being developed to further harness the power of forward genetics.
Keywords: mouse genetics, forward genetics, mouse mutants, ENU mutagenesis, peripheral neuropathy
Introduction
The mouse has been an invaluable model system for studying human genetics and disease due to its many genetic and physiological similarities, genetic tractability, and short generation time. For the last two decades, much of the focus within the mouse genetics community has been on “reverse genetics”, or the genotype-to-phenotype approach, due to an ever-expanding repertoire of techniques that allow for more nuanced control of the mouse genome. In addition to knockouts and knockins that can target specific loci, a variety of methods have been developed that enable the spatial and temporal control of gene alteration and expression [1].
Despite the continuing success of reverse genetics, this decade has seen a resurgence of interest in the historical roots of mouse genetics: “forward genetics”, or the phenotype-to-genotype approach. This trend was largely complemented by the publication of the mouse genome [2]; the availability of the genomic sequence facilitates mapping and mutation identification, while it was hoped that a phenotype-driven approach might be faster and more effective than knockout technology in elucidating the functions of the thousands of genes predicted from the sequence but not yet studied [3, 4, 5, 6].
The study of the PNS, like many biological systems, has benefited from forward genetics in the mouse, historically as well as in the post-genome era. Long before the molecular underpinnings of peripheral nerve function and dysfunction could be effectively investigated, spontaneous mutants were serving as models of human disease, providing knowledge of physiology and pathogenesis and facilitating the development of treatments. Today, the genetic lesions responsible for the phenotypes of many historical strains have been identified, and new models continue to be discovered, leading to a better molecular understanding of the peripheral nerve.
Mutant Strain Origins: Spontaneity and Induction
The mouse community has been collecting spontaneous mouse mutants for over 100 years, obtaining these highly useful lines first from mouse fanciers, and later from scientific colonies [3]. The natural rate of mutation, however, is slow, prompting mouse geneticists to search for ways to speed up the process. Several agents of mutagenesis have been employed over the years, including X-rays and various chemicals. Random transgene insertions have also, unintentionally, contributed highly useful lines. More recently, as the completed mouse genome appeared on the horizon, geneticists began to think of mouse mutagenesis as a potential high-throughput method of addressing gene function throughout the genome. For these large-scale screens, N-ethyl-N-nitrosourea (ENU) has become the mutagen of choice among mouse geneticists
ENU’s popularity as a mutagen is due to its ease of use, its effectiveness, and the wide variety of mutations it can generate [5]. Because it does not require metabolization to be active, ENU can act readily in spermatogonial stem cells, and its alkylating activity is capable of producing point mutations that result in a wide variety of alleles [7]. The generation of hypomorphic, as well as dominant negative and other gain-of-function alleles is advantageous, as different alleles can produce unique phenotypes, providing insights into the multiple functions of a gene product, modeling human disease more closely, or circumventing an uninformative or embryonic lethal knockout phenotype. Also, unlike radiation mutagenesis, which can result in complicated multigenic chromosomal rearrangements, ENU results in single nucleotide polymorphisims that are unlikely to affect more than one gene, simplifying the correlation of phenotype to genotype.
The basic ENU mutagenesis procedure (Fig. 1a) involves treating male mice with ENU (usually three weekly doses of 100 mg/kg) and, after a period of sterility [8], mating them to wild type females of the same strain or, to facilitate mapping of recovered mutant alleles, to an outcross strain. The resulting G1 pups can be screened directly for dominant phenotypes. Alternatively, G1 animals can be crossed again to wild type to produce multiple G2 carriers that, when backcrossed to the G1 parent or intercrossed with littermates, produce G3 progeny that can be screened for recessive phenotypes [3, 4, 6].
Figure 1.
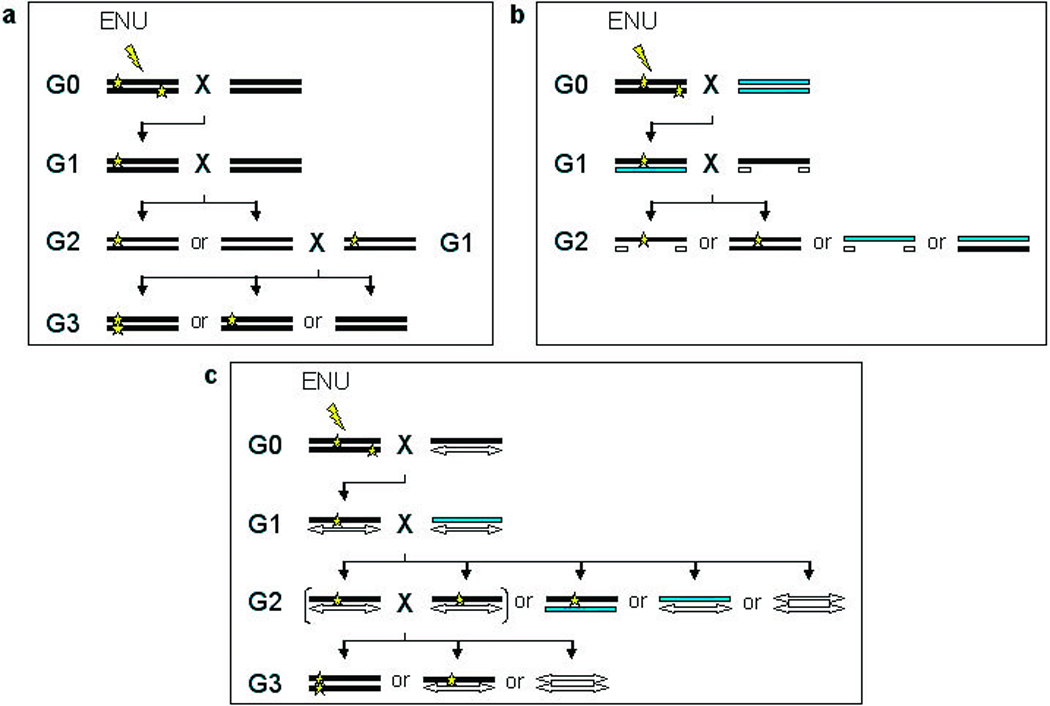
Methods for ENU mutagenesis. a, In a basic ENU mutagenesis screen, a male G0 mouse is treated with ENU and mated to a wild type female. The G1 progeny can be screened for dominant mutations. To screen for recessive mutations, the G1 carrier is mated to a wild type mouse to produce potential carriers in the G2 generation. The G2 mice can then be backcrossed to the original G1 parent to generate homozygotes in the G3 [3, 4, 6]. b, To more rapidly screen for recessive mutations within a particular region, a deletion chromosome may be used. The mutagenized G0 male is bred to a wild type female. The resulting G1 potential carriers can then be bred to mice carrying a deletion, and the G2 progeny can be screened for recessive mutations within the deleted region a generation earlier than the basic screen (a). The use of dominant markers, such as coat color, indicated by the grey and white chromosomes, can facilitate identification of potential mutants to be screened [3, 6, 9]. c, Additionally, chromosomes with a large inversion and a coat color marker, called balancer chromosomes, are useful for recovering and maintaining recessive mutations. The mutagenized G0 mouse is bred with a female carrying a balancer chromosome (white double-headed arrow). The resulting G1 progeny carrying the balancer are then bred to another mouse carrying the balancer chromosome and an alternative coat color marker (gray bar). The G2 progeny carrying both the balancer chromosome and the mutagenized chromosome can be identified by coat color and intercrossed, producing only carriers and potential mutants for screening, which are distinguishable by coat color. Progeny receiving two balancer chromosomes may not be viable [3, 4, 6, 10].
To reduce the number of potential mutants to be examined in a recessive screen, or to focus on a particular genomic region of interest, ENU screens have also taken advantage of chromosomal deletions and balancer chromosomes. In the case of a deletion screen (Fig. 1b), G1 progeny are mated to mice carrying a small deletion, allowing for recessive mutations to be identified a generation earlier, though only within the deleted region. Additionally, observable dominant markers, such as coat and eye color, can be used to identify progeny carrying the wild type and deletion chromosomes, eliminating uninformative mice from the screening process [3, 6, 9]. Balancer chromosomes (Fig. 1c), or chromosomes carrying an inversion that is recessive lethal, are equally useful in weeding out uninformative progeny, as only carrier and homozygous mutant mice survive and, again, dominant markers can be used to differentiate between the two. Nevertheless, because the genes within the inversion are still present, screening for recessive mutations requires the generation of G3 progeny, as described above. Like the deletion screen, balancer screens are also restricted to recovering mutations found within the targeted genomic region [3, 4, 6, 10].
Though time and labor intensive, generating a large pool of potential mutants is only the first step. The most important factor in the success of an ENU mutagenesis screen is the methodology used to identify interesting phenotypes. One method designed to address this concern, SHIRPA, was developed by the ENU Mutagenesis Programme [11] and later modified by researchers at RIKEN Genomic Sciences Center [12]. SHIRPA consists of a semi-quantitative three-tier approach that begins with generalized tests, then narrows in focus to assay a phenotypic category of interest, such as neurological mutants, as was the case in the Harwell screen. The primary screen includes several behavioral and visual examinations, where results are recorded on scales of 0–1 to 0–6 as detailed by the screen rubric [13]. Additional visual assessment of morphology was added by the modified protocol [12]. The secondary screen includes additional behavioral and functional tests, such as rota-rod to assess balance and hot plate to assess nociception, as well as biochemical examinations for metabolic defects. The tertiary screens are further specialized, though more directed tests can be used at the secondary level, depending on the goals of the screen. Neurological assays utilized in the Harwell study included MRI and electrophysiological tests, as well as behavioral assessments of anxiety, learning, and memory [11].
In addition to the ENU Mutagenesis Programme, several other mutagenesis centers, including those at Novartis’s Genomics Institute, the McLaughlin Research Institute, Oak Ridge, and the CMHD, have focused at least part of their screens specifically on finding neurological mutants [4, 5]. Three centers, the Neurogenomics Project at Northwestern University, the Neuroscience Mutagenesis Facility at Jackson Laboratory, and the Neuromutagenesis Project at the Tennessee Mouse Genome Consortium participated in an NIH initiative, organized through the website neuromice.org, intended both to develop neurological mutants and to make them available to the neuroscience community [3]. Phenotypic screens at these centers included those for movement/neuromuscular function, such as digital gait analysis [14], rota-rod, and grip strength, and for nociception, as well as assays for social and emotional behavior, hearing and vision, epilepsy, and drug and alcohol response [15]. Because of this emphasis on neurological phenotypes, many new ENU mutants relevant to the study of the PNS have been identified, paving the way to a more complete understanding of the PNS. Furthermore, many of the strains generated by ENU mutagenesis screens are as of yet unmapped, highlighting the potential for further important discoveries.
While mapping mutations, even in the postgenomic era, is not trivial, new tools and techniques are continuously being improved to increase mapping efficiency. In the 1990’s, simple sequence length polymorphisms (SSLP) markers, or microsatellites, regions of short repeats that vary in length among mouse strains, were developed, greatly increasing the number of markers available for linkage mapping [16]. SSLP markers are still commonly used, with great success, but to increase marker density and usefulness over a broader range of strains, effort has been made to develop large panels of single nucleotide polymorphism (SNP) markers [17, 18, 19]. SNP markers increase efficiency both in breeding, by reducing the number of mutants required to localize a mutation to a critical region, and in genotyping, by enabling the use of high-throughput methods such as MALDI-TOF mass spectrometry [20] and oligonucleotide arrays [19]. SNP panels are also valuable as mouse geneticists attempt to utilize the natural variation among mouse strains to find the genes behind quantitative traits [21]. This continued interest in dissecting the genetic factors underlying phenotypic traits indicates that forward genetics will continue to play an important role in gene discovery and functional elucidation.
PNS Mutants in the Mouse
A primary advantage of forward genetics is a lack of bias, which can lead to the identification of genes with nonobvious importance to a particular system and provide knowledge that might not have been gleaned otherwise. Additionally, the generation of allelic series can created new tools for the biochemical analysis of gene products and the investigation of isoform- and tissue-specific functions. Thirdly, mutants can aid in the identification of genes responsible for human disease and serve as models for the development of treatments. Forward genetics has benefited the study of the PNS in all of these areas; examples of these successes follow.
Gene Discovery and Functional Analysis
Several genes with functional importance in the PNS were first identified via mouse mutations, including dystonin (Dst) [22], Lpin1 [23], quaking (Qk) [24], Unc5c [25], and the channels Cacna1a [26, 27] and Scn8a [28]. After the mouse genome was sequenced, finding unknown genes was no longer a difficult task; rather, the sequence provided an abundance of newly annotated genes for which the functions are unknown or only suspected. One recent success was the identification of Lgi4 as the affected gene in the mutant strain claw paw (Fig. 2b), demonstrating Lgi4’s role in Schwann cell development [29]. Additionally, two mutant strains characterized by tetraparesis and early lethality have revealed a new link between mitochondrial function and peripheral nerve health. Both strains, paralysé (par) and Emv66, carry a mutation in the Afg3l2 gene, an m-AAA ATP-dependent mitochondrial metalloprotease that is required for proper axonal development [30]. It is expected that forward genetics will continue to identify new genes of interest in the study of the PNS.
Figure 2.
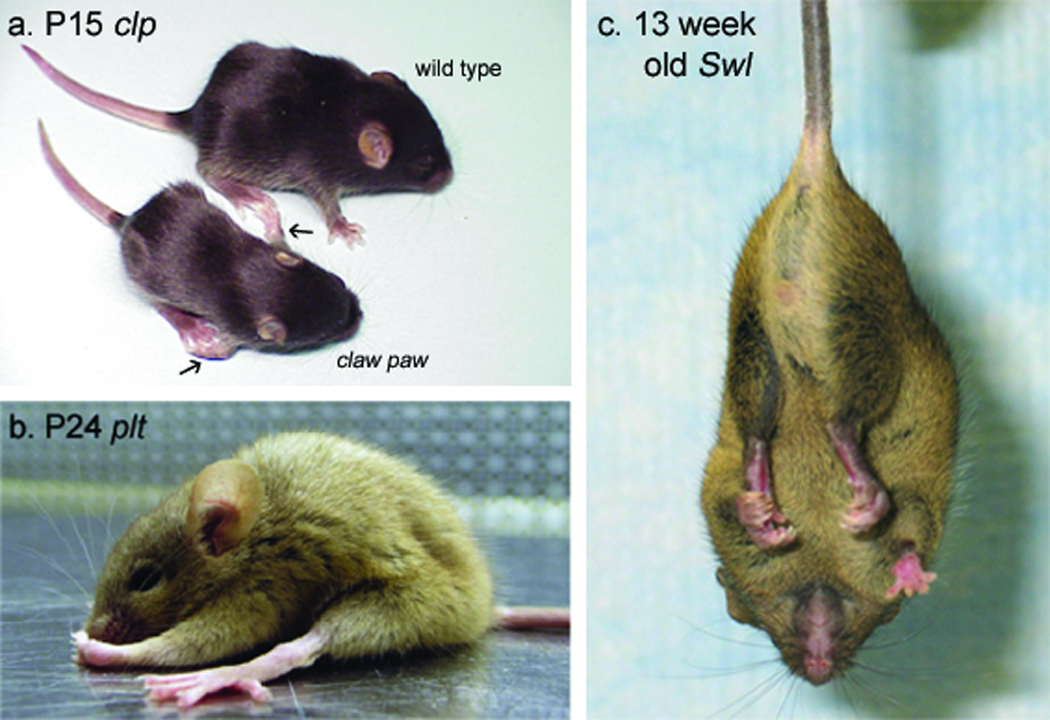
The causative mutations underlying several mouse models of peripheral nerve disorder have recently been identified. a, A P15 claw paw mutant and wild type littermate. The claw paw mutation, which results in abnormal forelimb posture (arrows) and hypomyelination in the PNS, was identified in Lgi4, a gene with no previous links to the PNS. The phenotype of the claw paw mouse suggests Lgi4 plays a role in myelination and peripheral nerve development. Reproduced with permission [23]. b, A P24 pale tremor mutant. Investigation of the pale tremor mouse, which is characterized by weakness, an abnormal gait, and loss of large-diameter axons in the PNS, led to the identification of a mutation in Fig4, a PtdIns(3,5)P25-phosphatase, as well as mutations in four unrelated human patients with a peripheral neuropathy that is now designated CMT4J. Reproduced with permission [31]. c, A 13 week-old sprawling mutant. The sprawling mouse’s sensory neuropathy is characterized by an abnormal posture, retraction of the hind limbs when suspended by the tail (shown here), and reduced numbers of muscle spindles. The sprawling genetic defect was found in the cytoplasmic dynein heavy chain 1 gene (Dync1h1), demonstrating a new function for a gene previously linked to late-onset motor neuropathy. Reproduced with permission [29].
In addition to locating novel genes, forward genetics is particularly useful in pinpointing previously studied genes with unexpected functional or pathological relevance to the PNS. In some cases, a specifically neurological phenotype is surprising because the affected gene is widely expressed. This is true for Gars, which encodes the ubiquitously required glycyl tRNA synthetase, and which has been shown to cause peripheral neuropathy in both the mouse and human patients [31, 32]. Similarly, the product of Usp14 is a deubiquitinating enzyme that is expressed in all tissues, but the phenotype of the Usp14 mutant, ataxia, is largely due to loss of neuronal functions [33], such as the regulation of acetylcholine (Ach) release at the neuromuscular junction (NMJ) [34]. Tbce, which encodes a tubulin cofactor necessary for proper microtubule assembly, results specifically in motoneuron degeneration and apoptosis when mutated [35]. Mutations in Dync1h1, the dynein heavy chain 1 gene, affect retrograde transport and lead to sensory and/or motor neuron disease, depending on the allele [36, 37]. Finally, the dysfunction of two endosome-associated genes, Fig4 and Vps54, causes neuropathy in mouse models. The pale tremor mouse (Fig. 2a) exhibits motor and sensory neurodegeneration, and carries a mutation in the phosphatase Fig 4. The substrate of Fig4, phosphatidylinositol-2,5-bisphosphate (PtdIns(3,5)P2), is important to endosomal membrane trafficking [38]. Vps54 is a member of the Golgi Associated Retrograde Protein (GARP) complex, which functions in endosomal trafficking [39]; the Vps54 mutation in the wobbler (wr) strain results in motoneuron disease [40].
Explanations for why ubiquitously expressed genes can result in specifically neuronal phenotypes tend to invoke the unique physiology of neurons and Schwann cells; for the former, the relatively long distances axons extend can pose challenges for intracellular trafficking [41], while the latter are more susceptible to deficiencies in the production of proteins and lipids, as they must generate large quantities of both to produce and maintain the myelin sheath [42]. Alternatively, in the case of gain of function mutations, ectopic activity or interactions may be to blame, as is proposed for Gars. While mice that are heterozygous for a gene-trapped Gars allele show halved transcript expression and significantly reduced aminoacylation activity, the dominant neuropathy phenotype seen in the spontaneous P278KY allele is absent, suggesting that the missense mutation phenotype results from a pathological mechanism other than loss of function [31], which could potentially have tissue-specific effects.
Forward genetics has also provided information on the tissue-specific functions of less broadly expressed genes. One example of this is the dystonin (Dst) gene. Current understanding of Dst comes from the convergence of two lines of research: the dystonia musculorum (dt) sensory neuropathy mouse model and the human autoimmune disorder bullous pemphigoid, which affects epithelial cells [22]. The tissue-specific functions of Dst appear to be regulated by the expression of tissue-specific isoforms. While the epithelial form, Bpag1 (bullous pemphigoid antigen 1), is known to be necessary for linking keratin intermediate filaments to hemidesmosomes in keratinocytes, the role of the neuronal isoforms is yet to be determined and does not appear to involve the binding of intermediate neurofilaments [43].
Another example is Lpin1, the recently identified mammalian phosphatidate phosphatase. Its spontaneous null, the fatty liver dystrophy (fld) mouse, has primarily been used to study Lpin1’s role in adipose and liver cells, but Lpin1 is also expressed in Schwann cells and fld mice exhibit striking myelin defects. In addition to its enzymatic activity, Lpin1 has been shown to act as a transcriptional coactivator in hepatic cells; whether it possesses a similar role in Schwann cells has yet to be determined [44].
Within the nervous system, the shiverer (shi) mouse, which lacks myelin basic protein (MBP), has highlighted differences in myelin structure between the central nervous system (CNS) and PNS. The loss of MBP in the CNS, where it makes up a higher percentage of myelin proteins, results in myelin that fails to compact. PNS myelin in the shi mouse, however, appears more structurally normal, though closer examination reveals abnormal paranodal loop-axolemma interactions, a slight reduction in axon diameter, and an increased number of Schmidt-Lanterman incisures (SLI) [45]. Interestingly, MBP appears to inversely impact the level of SLI components Cx32 and MAG postranscriptionally in a dose-dependant manner, though the mechanism is not known, nor has the relationship been investigated in the CNS, where MBP expression is greater and SLI number is lower [46].
Finally, random mutation has generated a truly novel model system useful for functional studies in the PNS. The Wallerian degeneration slow (WldS) mouse arose from a tandem triplication event. The altered locus produces a chimeric protein that is a combination of the N-terminal of ubiquitination factor E4B (Ube4b) and the whole product of nicotinamide nucleotide adenylyltransferase 1 (Nmnat1). The WldS mouse is characterized by delayed Wallerian degeneration of distal axons after injury. The biological activity of the ectopic protein is not fully understood. Nicotinamide adenine dinucleotide (NAD) synthesis, the enzymatic activity of Nmnat1, is retained by WldS, but this activity does not explain the full activity of the chimeric protein. Overexpression of wild type Nmnat1 in neurons confers only mild protection against axonal degeneration, a WldS allele with a targeted mutation abolishing NAD synthesis retains mild protection, and overexpression of the Ube4b N-terminal fragment also confers mild protection. Therefore, the novel function exhibited by the WldS protein appears to be intrinsic to the whole chimera [47]. While the molecular mechanism of WldS protection remains enigmatic, crossing the WldS mutant with other models of neuropathy, such as progressive motor neuronopathy [48], gracile axonal dystrophy [49], and the myelin protein zero (Mpz) knockout [50], has helped probe the role of axon degeneration in these disorders.
Allelic Series
One benefit that distinguishes random mutations from knockouts is the wide variety of alleles that may be generated beyond a null. Partial loss of function alleles can increase viability, allowing for the analysis of later-onset phenotypes, impact expression and function in a tissue- or isoform-specific manner, separate out multiple functions of a protein, aid biochemical studies, and model human disease. A series of such alleles, each with its own unique properties or phenotype, can be a valuable tool for understanding the subtleties of gene function. Multiple mutations have been discovered in a majority of the genes listed in Table 1.
Table 1.
| Gene | Allele Distribution3 (Nontargeted) | Notable Alleles | Associated Human Peripheral Neuropathies | Molecular Function | Affected Regions of PNS | References |
|---|---|---|---|---|---|---|
| ATPase family gene 3-like 2 (Afg3l2) | 1 spontaneous 1 proviral insertion | paralysé (par) | motoneuron disease | m-AAA mitochondrial ATP- dependent metalloprotease | neuronal mitochondria | 30 |
| ecotropic murine leukemia virus insertion 66 (Emv66) | ||||||
| calcium channel, voltage-dependent, P/Q type, alpha 1A subunit (Cacna 1a) | 4 spontaneous 3 ENU mutagenesis | tottering (tg) | calcium channel, initiates acetylcholine release at the NMJ | NMJ | 26, 27, 58–64 | |
| leaner (la) | ||||||
| rolling Nagoya (RN) | Lambert-Eaton myasthenic syndrome (LEWIS) | |||||
| dystonin (Dst) | 14 spontaneous 3 ENU mutagenesis 1 other mutagenesis 1 transgene insertion | dystonia musculorum (dt) | unknown, possible structural function | sensory axons | 22, 43, 52, 53 | |
| dtJ | ||||||
| dtAlb | ||||||
| dtTg4 | ||||||
| dt24J | ||||||
| dt27J | ||||||
| dtFrkordt29J | ||||||
| dynein cytoplasmic 1 heavy chain 1 (Dync1h1) | 2 ENU mutagenesis 1 X-ray mutagenesis | cramping 1 (cral) | retrograde axonal transport | 36,37 | ||
| legs at odd angles (loa) | muscle spindles, sensory axons, DRG cell bodies | |||||
| sprawling (swl) | human sensory neuropathy (HSN) | |||||
| FIG4 homolog (Fig4) | 1 spontaneous | pale tremor (plt) | Charcot-Marie-Tooth (CMT4J) | Ptdlns(3,5)P25-phosphatase | sensory and motor cell bodies, lage-diameter axons | 38 |
| galactosyl-ceramidase (Gale) | 3 spontaneous | twitcher (twi) | globoid cell leukodystrophy (Krabbe’s disease) | glycoside hydrolase, several glycosphingolipid substrates | Schwann cells | 77–82 |
| glycyl-tRNA synthetase (Gars) | 1 spontaneous 1 gene-trapped | Nmf249 | Charcot-Marie-Tooth (CMT2D) | only aminoacyl tRNA synthetase for glycine | motor and sensory axons | 31,32,92 |
| laminin, alpha 2 (Lama2) | 4 spontaneous 1 ENU mutagenesis | dystrophia muscularis (dy) | congenital muscular dystrophy (MDC1A) | structural component of basal lamina | Schwann cells and Schwann cell basal lamina | 57, 83, 85, 87–89 |
| dy2J | ||||||
| dvPas | ||||||
| Large | 2 spontaneous 1 transgene insertion | myodystrophy (myd) originally “froggy” (fg) | congenital muscular dystrophy (MDC1D) | glycosyltransferase for α-dystroglycan | Schwann cells, NMJ | 67, 84, 86, 90, 91 |
| enervated (enr) | ||||||
| leucine-rich repeat LGI family, member 4 (Lgi4) | 1 spontaneous | claw paw (dp) | signaling molecule necessary for myelination | Schwann cells and possibly neurons | 29 | |
| Lipin1 (Lpin1) | 2 spontaneous | fatty liver dystrophy (fld) | phosphatidate phosphatase | myelin in the peripheral nerves | 23,44 | |
| myelin basic protein (MBP) | 4 spontaneous 1 spontaneous revertant | shiverer (Shi) | structural myelin protein | Schwann cells | 45,46 | |
| myelin deficient (mld) | ||||||
| peripheral myelin protein (Pmp22) | 3 spontaneous 3 ENU mutagenesis | trembler (Tr) | Dejerine-Sottas syndrome (DSS) | membrane protein necessary for myelination, possibly involving interaction with the basal lamina | Schwann cells | 74–76 |
| trembler-J (Tr-J) | Charcot-Marie Tooth (CMT1A) | |||||
| Tr-Ncnp | ||||||
| Tr-m1H | Dejerine-Sottas syndrome (DSS) | |||||
| Tr-m2H | ||||||
| Trm-3H | ||||||
| quaking (qk) | 5 ENU mutagenesis 2 spontaneous | quaking quaking viable (qkv) | RNA binding protein involved in multiple pathways, including glial cell fate determination | Schwann cells | 24, 54–56 | |
| qkkt1 | ||||||
| qkk2 | ||||||
| qkkt3/4 | ||||||
| qklethal-1 | ||||||
| qke5 | ||||||
| sodium channel, voltage-gated, type VIII, alpha (Scn8a) | 5 spontaneous 4 ENU mutagenesis 1 gene-trapped 1 transgene insertion | motor endplate disease (med) | sodium channel, primary channel at the node of Ranvier | motor axons and NMJ | 28, 65, 66, 96–98 | |
| medJ | ||||||
| jolting (jo) | ||||||
| med-TgA4Bs | ||||||
| degenerating muscle (dmu) | ||||||
| tubulin-specific chaperone e (Tbce) | 1 spontaneous | progressive motor neuronopathy (pmn) | motoneuron disease | tubulin assembly | motor axons | 35, 48, 71 |
| ubiquitin carboxy-terminal hydrolase L1 (Uchl1) | 1 spontaneous | gracile axonal dystrophy (gad) | ubiquitin hydrolase and/or ligase, ubiquitin monomer stabilization | motor and sensory axons, DRGs | 49, 72, 73 | |
| ubiquitin specific peptidase 14 (Usp14) | 1 spontaneous | ataxia Jackson (axJ) | deubiquitinating enzyme, regulates neurotransmitter release | NMJ | 33,34 | |
| unc-5 homolog C (Unc5c) | 1 spontaneous 2 transgene insertion | rostral cerebellar malformation (rem) | netrin-1 receptor, axon guidance | trochlear and phrenic nerves | 25,95 | |
| rcmTg(Ucp)1.23Kz | ||||||
| vacuolar protein sorting 54 (Vsp54) | 1 spontaneous 1 gene-trapped | wobbler (wr) | motoneuron disease | member of Golgi Associated Retrograde Protein (GARP) complex, endosomal trafficking, anterograde and retrograde axonal transport | motoneurons | 39,40 |
| leptin (Lep) | 2 spontaneous | obese (Ob) | type II diabetic neuropathy | leptin, control of blood gluscose levels | large motor and sensory fibers, small sensory fibers | 69 |
| leptin receptor (Lepr) | 12 spontaneous 1 ENU mutagenesis 1 transgene insertion | diabetes (db) | type II diabetic neuropathy | leptin receptor, control of blood glucose levels | large motor and sensory fibers | 68,70 |
| wallerian degeneration (WId) | 1 spontaneous | Wallerian degeneration slow (Wlds) | chimeric protein, delays Wallerian degeneration | axons | 47–50 | |
Allele numbers from Mouse Genome Data base FMGD
Dst is notable for its susceptibility to mutation (14 spontaneous mutations, 4 induced, and 1 random transgene insertion) [51], which is likely due, in part at least, to the large size of the locus [52]. As described above, Dst has seemingly different functions in epithelial cells and neurons. These functional differences arise from the expression of multiple tissue-specific isoforms, as illustrated by the phenotypes of the various dt alleles. Comparison of two alleles, dtJ and dtAlb, both of which display the neuropathy phenotype, demonstrated protein expression in the epithelium of the former, but not the latter, consistent with the respective lack and presence of hemidesmosome abnormalities in the two strains [53]. Further analysis of mRNA expression levels among various dt alleles has shown that transcription is reduced for all Dst regions in dtAlb, dtTg4, and dt24J mice, but in dt27J mice, reductions were observed for regions found only in neural and muscle isoforms. It seems likely that the neuronal isoforms of Dst have a regulatory region unique from the epithelial isoforms; unfortunately, the identity of the genetic lesion has only been determined in two null alleles, dtAlb and dtTg4. Identifying the lesions in dt, dtJ, and dt27J should prove useful in understanding the regulatory mechanisms of Dst [52].
Another gene exhibiting alternate regulation of isoforms is quaking (Qk), an RNA binding protein expressed in multiple cell types, though restricted to Schwann cells within the PNS, and with targets that include the Mbp transcript [54]. For the quaking mouse, the existence of an allelic series has been crucial to identifying the affected gene, as well as investigating its function. The original quakingviable (qkV) allele, which causes myelin defects in both the CNS and PNS, resulted from a large spontaneous deletion that affects three loci: parkin (Park2), Park2-coregulated gene (Pacrg), and Qk. The multigenic lesion complicated genotype-phenotype analysis until the generation of additional alleles by ENU mutagenesis confirmed that alterations to the Qk locus are responsible for the neurological phenotype of quakingviable [55]. All but one of the ENU-generated mutants are recessive embryonic lethal; like the original qkV mutants, qke5 mice survives to adulthood, though with a reduced lifespan not seen in qkV. The variation in phenotype is tied to the isoform affected. In embryonic lethal mutants, the embryonically-required QKI-5 isoform is deficient in function or expression. In qkV and qke5, QKI-6 and QKI-7 are expressed in astrocytes, but not in Schwann cells or oligodendrocytes. QKI-5 expression is normal in qkV, but reduced in qke5, perhaps explaining its more severe phenotype [55, 56]. Therefore, the Qk allelic series provides a model to study both temporal and tissue-specific regulation of isoform expression.
Lama2 is also expressed in multiple cell types, specifically muscle and Schwann cells. The series of Lama2 alleles includes those characterized by reduced expression (dystrophic, dy), truncation (dy2J), reduced expression and truncation (the partial knockout dyW), and complete loss of expression (dyPas and the complete knockout dy3K). Comparison of effect on muscle tissue among the alleles has indicated that a reduced level of expression can be more detrimental than truncation. This result has not been corroborated in Schwann cells, however, because all of the known dy alleles lack expression in the peripheral nerve, suggesting that Lama2, like Dst, possesses nerve-specific regulatory sequences that are yet to be identified [57].
Allelic series can also provide a system in which to probe the function of a gene product and the repercussions of dysfunction on physiology. Functional comparison of three alleles of Cacna1a, the gene which encodes the α1A pore-forming subunit of the Cav2.1 high voltage-activated calcium channel, demonstrated three different sets of electrophysiological properties affecting Ach release at the NMJ [58, 59, 60]. This variation in channel function is accompanied by variation at the clinical phenotype level: while the tottering mouse displays only a mild phenotype with no CNS pathology [61], the leaner mouse has cerebellar defects and a shortened lifespan [62, 63], and the rolling Nagoya mouse uniquely lacks seizures and demonstrates muscle weakness [64].
Similarly, an allelic series promises to provide insight into the functioning of the sodium channel encoded by Scn8a, the primary channel found at mature nodes of Ranvier. While two null mutations, motor endplate disease (med) and TgNa4Bs, result in motor axon degeneration and muscle atrophy, the minimal expression of Scn8a by the medJ allele avoids paralysis, demonstrating weakness and dystonia instead [65]. Interestingly, the degenerating muscle (dmu) mutant, also showing reduced expression, is characterized muscle degeneration without motor fiber loss [66], while the jolting (jo) missense mutant’s phenotype is limited to the cerebellum [65]. As the natures of each of these molecular defects becomes clearer, they will be useful for investigating the specific role of the channel in the affected tissue types and why they are variably affected among the models.
Finally, while forward genetics can remove the bias towards genotype, it can foster bias in the analysis of phenotype; the investigator, focused on a particular phenotype of interest, may initially miss additional pathological clues. In cases such as this, multiple alleles, studied independently, can result in a more complete understanding of a gene’s function. For example, mice carrying the enervated (enr) allele of Large, which arose from the random insertion of a transgene, were demonstrated to have NMJ abnormalities, leading to observations of similar pathology in the older mutant strain, myd, previously shown to have defects in Schwann cells and muscle [67]. More recently, the localization of the sprawling (Swl) mutation (Fig. 2c) to the cytoplasmic dynein heavy chain 1 gene (Dync1h1 prompted a second look at two previously described ENU-induced mutations in Dync1h1, legs at odd angles (Loa) and cramping 1 (Cra1). Loa and Cra1 had been shown to cause late-onset motor neuron degeneration [37]. In contrast, Swl has a long history of investigation as a model of early-onset sensory neuropathy, characterized by a reduction in muscle spindles and a loss of DRG neurons. The mapping of Swl to Dync1h1 led to a fresh examination of the Loa phenotype and similar early-onset sensory deficits were discovered. Nevertheless, the three alleles do not share all phenotypic characteristics; the Swl mutation does not result in progressive motor neuron loss, nor can it delay the progression of the amyotrophic lateral sclerosis-like phenotype of the human superoxide dismutase transgenic model SODG93A, as has been shown for Loa and Cra1 [36]. Therefore, comparison of Swl with Loa and Cra1 may also yield information on the tissue-specific importance of Dync1h1 retrograde transport.
Models of Human Disease
Mutant strains are highly valuable because they can be used to model various aspects of physiological dysfunction. Nevertheless, strains developed as models of human disease based upon phenotypic similarity do not always mirror the disease at the molecular level. For example, the Cacna1a mutant rolling Nagoya has been proposed as a model of Lambert-Eaton myasthenic syndrome (LEMS). While the Cav2.1 channel is at the heart of this disorder in humans, the phenotype arises from autoimmune antibodies targeting the channel, not a molecular defect in the channel itself [60]. Similarly, though type 2 diabetes is a human disease with a complex genetic and environmental etiology, two monogenic mutants, diabetes (db) and obese (ob), with mutations in the leptin receptor and leptin, respectively, are employed as models. Both models have been used to investigate diabetic peripheral neuropathy [68, 69], a highly important line of research, given that 45–50% percent of the growing population with type 2 diabetes will experience neuropathic complications [70].
Nor do strains carrying lesions in known human disease genes always mimic the human disease, as is the case for the mouse mutant progressive motor neuronopathy (pmn). Pmn mice are homozygous for a mutation in Tbce and are characterized by distal-to-proximal mononeuron degeneration. Meanwhile, mutations in the human TBCE gene result in Kenny-Caffey and Sanjad-Sakati syndromes, which present with hypoparathyroidism, retardation, and a variety of deformities [71]. Additionally, the mutant of the ubiquitin hydrolase Uchl1, gracile axonal dystrophy (gad), is characterized by neuron degeneration that initiates at the ends of the peripheral and central axons of DRG neurons, before progressing to motor neurons and the upper tracts of the CNS. Human UCHL1, in contrast, may be associated with Parkinson disease [72], though this association is controversial [73]. Reasons for these discrepancies could be due to differences at the allelic or organismal level.
Nevertheless, a subset of the strains listed in Table 1 does share a known molecular defect with a specific human disorder affecting the PNS. Among these strains, the allelic trembler (Tr) and trembler-J (Tr-J), dystrophia muscularis (dystrophic, dy) and its allele dy2J, myodystrophy (myd), and twitcher (twi) mice, have been providing clues to human disorders for decades.
The Tr and Tr-J mice carry autosomal dominant mutations in peripheral myelin protein (PMP22). PMP22 is one of the most studied genes associated with human peripheral neuropathy, since abnormalities in PMP22 can lead to three different diseases: Charcot-Marie-Tooth type 1A (CMT1A), Dejerine-Sottas syndrome (DSS), and hereditary neuropathy with liability to pressure palsies (HNPP). In fact, it was the discovery of the PMP22 mutation in the original trembler mouse that first led to the discovery of PMP22 defects in human patients [74]. Both Tr and Tr-J, as well as the more recently discovered ENU-induced alleles Tr-m1H, Tr-m2H, and Tr-m3H are missense mutations. Tr-Ncnp is an in-frame deletion of exon 4. All six of these alleles model the most severe of the three phenotypes, DSS, though there is a gradient of severity among them [75]. As indicated by transgenic models of PMP22 overexpression and absence, HNPP results from haploinsufficiency, while CMT1A results from a moderate 2-fold increase in expression; greater than 2-fold overexpression results in DSS. This genotype-phenotype correlation, along with the observation of PMP22-containing protein aggregates in Tr-J mice, supports a mechanism of perturbed intracellular trafficking [76].
Interestingly, from a clinical standpoint, the Tr and Tr-J mutations are seen in DSS and CMT1A patients, respectively. While, Tr-J is the less severely affected of the two mutant strains, it is still classified as DSS rather than CMT1A, suggesting genetic background or other mouse/human differences might play a role in phenotype expressivity. The residues altered in Tr-m1H and Tr-m3H are also altered in human patients, though the substituted residue is not the same. In particular, mutations at the Tr-m3H position have been found in several patients, suggesting its importance [75]. Such an extended series of alleles that mirrors both human genotype and phenotype promises to be useful in both understanding the molecular pathogenesis more fully and providing a model in which to develop treatments.
Another mouse mutant with clinical relevance is twitcher (twi), which has proven to be a highly faithful model of human globoid cell leukodystrophy (GLD, Krabbe disease). In addition to sharing histological features, such as loss of myelin in both the CNS and PNS and abnormal cell morphology, the twi mouse demonstrates the biochemical features of human Krabbe disease: loss of galactosylceramidase activity resulting in the accumulation of the substrate galactosylsphingosine (psychosine), believed to be toxic [77]. Unsurprisingly, the genetic defect in the twi mouse was identified as a nonsense mutation in the galactosylceramidase (Galc) gene [78], the same gene affected in human patients [79].
The molecular fidelity of the twi mutant has allowed it to serve as a model for both investigating the pathogenesis of GLD and developing treatments. Grafts of mutant nerve into wild type mice and vice versa revealed the twicher PNS phenotype to be intrinsic to the Schwann cells and not the axon. The eventual disappearance of the leukodystraphy phenotype from the grafted mutant Schwann cells suggested enzyme replacement was a feasible clinical treatment [80]. Indeed, bone marrow transplantation (BMT) was able to reduce psychosine levels, partially alleviate symptoms, and increase myelination and survival in mice [81]. Transplantation of healthy umbilical-cord blood into affected human babies has since been shown to be a successful treatment [82].
In the case of the dy, dy2J, and myd strains, their initial phenotypic descriptions were recognized as being highly similar to human muscular dystrophy. Muscular dystrophy, as suggested by the name of the disorder and its models, was originally thought to be a primary myopathy [83, 84]. Nevertheless, detailed phenotypic investigation that took advantage of the dy and myd models demonstrated that peripheral nerve myelination defects were also present, due to abnormalities in the basal lamina laid down by Schwann cells [85, 86].
The mutation in the dy and dy2J strains were linked to the Lama2 gene, encoding a laminin chain, formerly called merosin, which was known to be a ligand of α-dystroglycan and an integral component of the basal lamina of striated muscle and peripheral nerves [87]. Not long after, mutations in the human LAMA2 gene were identified in muscular dystrophy patients [88]. Muscular dystrophy caused by LAMA2 deficiency (MDC1A) is currently the most common form of the disorder [89].
Several years after the dy mutation was mapped, the myd mouse was shown to carry a defect in the Large gene [90], a glycosyltransferase that is believed to be responsible for glycosylating α-dystroglycan. This posttranslational modification is potentially important to α-dystroglycan’s affinity for its ligands, including Lama2 [86, 89]. Such a relationship, if correct, would nicely explain the similar phenotypes of the dy and myd mice. So far, only one human patient with congenital muscular dystrophy (MDC1D) has been reported to carry a mutation in LARGE [89]. Meanwhile, another LARGE mutation was found in a patient diagnosed with the more severe Walker-Warburg Syndrome (WWS), suggesting phenotypic variation due to allele differences or genetic background that has not yet been observed in mouse models [91].
More recently, two new genetically faithful models of human disease have been discovered with mutations in Gars and Fig4. The dominant mutation in Gars, though not identical to any alleles seen in human patients, models CMT2D, one of the two human diseases associated with human GARS. Analysis of the mouse allele has the potential to complement recent biochemical and structural analysis of the CMT2D-associated human alleles [92]. Meanwhile, the mutation in Fig4 strain pale tremor (Fig. 2b) is particularly exciting because Fig4 was not previously associated with PNS pathology. In addition to demonstrating essential Fig4 function in the PNS, the mapping of the pale tremor lesion led to the discovery of four independent mutations in human patients with CMT, now designated CMT4J [38].
Modifier Loci
Several factors, including the availability of the mouse genome and the development of genomic analysis techniques, have fostered a more global perspective on gene function. To paraphrase John Donne, no gene is an island; the genetic environment of any given allele can affect the translation of genotype to phenotype. It is not uncommon, in the course of mapping a gene, to discover the phenotype of progeny on the outcrossed background demonstrates a broader gradient of phenotype expressivity. When an allele is moved to a new background, the phenotype can become more severe, making it easier to observe or causing early lethality, it can gain new features and affect new tissue types, or in some unlucky cases, it can even disappear, depending on the strain. The existence of such genetic modifiers, though they complicate phenotype analysis, can be a boon to forward geneticists, offering an even greater yield of knowledge about gene regulation, interaction, or function [93, 94].
Some of the variation among the commonly used laboratory strains is likely due to complex, multigenic differences [94]. Because more statistical approaches are required to dissect the potentially small effects of multiple genes in these cases of genetic variation, investigators involved with mapping Mendelian traits may not be inclined to follow up on them. Nevertheless, there are several instances of modifiers demonstrating clear Mendelian inheritance, including two that were observed in the course of investigating two PNS-associated mutations, rostral cerebellar malformation (rcm) and medJ. The rcm mutation in netrin-1 receptor Unc5c, which results in axon projection abnormalities, was originally maintained on a C57Bl/6J × SJL/J hybrid background and mutants lived a normal lifespan. When the allele was transferred to a congenic C57Bl/6J background, however, axon misprojections became so severe that pups could no longer survive until weaning, suggesting a suppressor of the rcm phenotype was present in the SJL/J strain. While the modifier was mapped to chromosome 17, it has not been identified yet [95].
Meanwhile, the modifier discovered in conjunction with the Scn8a allele medJ, has been identified. The medJ allele is characterized by a small deletion that removes a splice donor site, resulting in incorrect splicing in 90% of transcripts on the initially investigated C3H background. When the allele was moved to the C57Bl/6J background, the percentage of incorrectly spliced transcripts rose to 95% and the mutation resulted in early lethality, as seen in the Scn8a null mouse [96]. Genetic mapping of the modifier identified a predicted gene, given the name sodium channel modifier 1 (Scnm1), which may be involved in the splicing of transcripts with minor class U12 (AT-AC) introns. The C57Bl/6J allele of Scnm1 is truncated, potentially interfering with its ability to interact with the spliceosome. It therefore appears that impaired non-consensus splicing ability in C57Bl/6J sensitizes the strain to splicing-related mutations [97, 98].
As discussed above, a good deal of study has been devoted to allelic series; indeed, allelic variation has been shown to influence disease phenotype. But genetic variation is also highly contributory to clinical variation in human patients. Investigating the natural variation present among mouse strains has the potential of identifying susceptibility factors that play a role in patients, perhaps leading to more individually tailored courses of treatment. Additionally, the identification of modifiers can be considered a secondary forward genetics method of gene discovery. While the susceptibility alleles of modifier genes may be present in wild type strains, the corresponding phenotype is subclinical, relying on additional genetic modification for detection. By being attentive to phenotypic variability in mutant strains, geneticists have the opportunity to discover genes and gene functions that, due to reasons such as redundancy [99] or a role that is only essential under physiological or environmental stress, would not be detected under normal, healthy conditions. Similarly, the use of known modifier alleles in future mutagenesis screens can enhance the phenotype of new mutations, resulting in the discovery of alleles that might otherwise go undetected in a standard screen.
Future Directions
It is clear that forward genetics has a long, successful history of advancing the study of the peripheral nervous system. Before genome manipulation was an option, spontaneous and mutagenesis-derived mouse mutants provided an avenue for elucidating gene functions and modeling human disease. More recently, large ENU mutagenesis screens have provided mutations such as the Dync1h1 alleles loa and cra1 [37], which have led to a new PNS phenotype-genotype association, and new alleles for previously known PNS genes, most notably Pmp22 [75] and Qk [55], which have furthered the analysis of these genes.
Additional methods for forward genetics are also being utilized. Gene trap mutagenesis is a useful technique. Though gene trapping cannot generate a wide variety of alleles like ENU mutagenesis, it retains the lack of genetic bias that forward genetics is prized for, while permitting fast localization of mutations via 5’ or 3’ rapid amplification of cDNA ends (RACE). Additionally, constructs have been generated to allow for conditional mutations [100]. In the same vein, transposable elements such as Sleeping Beauty (SB) [101] and piggyBac (PB) [102] can be used for random mutagenesis and gene trapping, with the same benefit of rapid localization. The ability of the PB construct to excise itself also offers a method of confirming genotype-phenotype associations. Additionally, the PB transposon has been used successfully to insert loxP sites randomly throughout the mouse genome, allowing the generation of large duplications and translocations, with the goal of investigating the regulatory functions of conserved noncoding elements (CNEs) [102].
As knowledge about the genome has grown, geneticists have gained greater respect for the complexity of gene regulation and interaction that exists. Moving forward in our understanding of the genome, gene by gene and as a whole, will require a broad range of techniques; there is no single catch-all method that will provide every answer. Though it has been a long time since forward genetics was the only game in town, it’s still a vital player, able to provide answers to the questions geneticist have yet to think of.
Acknowledgements
We thank John Bermingham Jr. and Miriam Meisler for the mouse images and Maria Traka for critically reading the manuscript.
References
- 1.Gao X, Kemper A, Popko B. Advanced transgenic and gene-targeting approaches. Neurochem Res. 1999;24:1181–1188. doi: 10.1023/a:1020772706279. [DOI] [PubMed] [Google Scholar]
- 2.Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 3.Balling R. ENU Mutagenesis: Analyzing Gene Function in Mice. Annu. Rev. Genomics. Hum. Genet. 2001;2:463–492. doi: 10.1146/annurev.genom.2.1.463. [DOI] [PubMed] [Google Scholar]
- 4.Clark AT, Goldowitz D, Takahashi JS, et al. Implementing large-scale ENU mutagenesis screens in North America. Genetica. 2004;122:51–64. doi: 10.1007/s10709-004-1436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cordes SP. N-Ethyl-N-Nitrosourea Mutagenesis: Boarding the Mouse Mutant Express. Microbiol. Mol. Biol. Rev. 2005;69:426–439. doi: 10.1128/MMBR.69.3.426-439.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown SD, Balling R. Systematic approaches to mouse mutagenesis. Curr. Opin. Genet. Dev. 2001;11:268–273. doi: 10.1016/s0959-437x(00)00189-1. [DOI] [PubMed] [Google Scholar]
- 7.Noveroske JK, Weber JS, Justice MJ. The mutagenic action of N-ethyl-N-nitrosurea in the mouse. Mamm. Genome. 2000;11:478–483. doi: 10.1007/s003350010093. [DOI] [PubMed] [Google Scholar]
- 8.Justice MJ, Carpenter DA, Favor J, et al. Effects of ENU dosage on mouse strains. Mamm. Genome. 2000;11:484–488. doi: 10.1007/s003350010094. [DOI] [PubMed] [Google Scholar]
- 9.Rinchik EM, Carpenter DA. N-ethyl-N-nitrosourea mutagenesis of a 6- to 11-cM subregion of the Fah-Hbb interval of mouse chromosome 7: Completed testing of 4557 gametes and deletion mapping and complementation analysis of 31 mutations. Genetics. 1999;152:373–383. doi: 10.1093/genetics/152.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hentges KE, Nakamura H, Furuta Y, et al. Novel lethal mouse mutants produced in balancer chromosome screens. Gene. Expr. Patterns. 2006;6:653–665. doi: 10.1016/j.modgep.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Rogers DC, Fisher EMC, Brown SDM, et al. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm. Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- 12.Masuya H, Inoue M, Wada Y, et al. Implementation of the modified-SHIRPA protocol for screening of dominant phenotypes in a large-scale ENU mutagenesis program. Mamm. Genome. 2005;16:829–837. doi: 10.1007/s00335-005-2430-8. [DOI] [PubMed] [Google Scholar]
- 13. http://www.mgu.har.mrc.ac.uk/facilities/mutagenesis/mutabase/shirpa_1.html.
- 14.Wooley CM, Sher RB, Kale A, et al. Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve. 2005;32:43–50. doi: 10.1002/mus.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. http://www.neuromice.org/browseAssays.do.
- 16.Love JM, Knight AM, McAleer MA, et al. Towards construction of a high resolution map of the mouse genome using PCR-analysed microsatellites. Nucleic Acids Res. 1990;18:4123–4130. doi: 10.1093/nar/18.14.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pletcher MT, McClurg P, Batalov S, et al. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2004;2:e393. doi: 10.1371/journal.pbio.0020393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frazer KA, Eskin E, Kang HM, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 19.Szatkiewicz JP, Beane GL, Ding Y, et al. An imputed genotype resource for the laboratory mouse. Mamm. Genome. 2008;19:199–208. doi: 10.1007/s00335-008-9098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moran JL, Bolton AD, Tran PV, et al. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome Res. 2006;16:436–440. doi: 10.1101/gr.4563306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stylianou IM, Affourtit JP, Shockley KR, et al. Applying Gene Expression, Proteomics and SNP Analysis for Complex Trait Gene Identification. Genetics. 2008;178:1795–1805. doi: 10.1534/genetics.107.081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown A, Bernier G, Mathieu M, et al. The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nature. 1995;10:301–306. doi: 10.1038/ng0795-301. [DOI] [PubMed] [Google Scholar]
- 23.Péterfy M, Phan J, Xu P, et al. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat. Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- 24.Ebersole TA, Chen Q, Justice MJ, et al. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat. Genet. 1996;12:260–265. doi: 10.1038/ng0396-260. [DOI] [PubMed] [Google Scholar]
- 25.Acerman SL, Kozak LP, Przborski SA, et al. The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature. 1997;386:838–842. doi: 10.1038/386838a0. [DOI] [PubMed] [Google Scholar]
- 26.Fletcher CF, Lutz CM, O'Sullivan TN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 27.Doyle J, Ren X, Lennon G, et al. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mamm. Genome. 1997;8:113–120. doi: 10.1007/s003359900369. [DOI] [PubMed] [Google Scholar]
- 28.Burgess DL, Kohrman DC, Galt J, et al. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant 'motor endplate disease'. Nat. Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 29.Bermingham JR, Jr, Shearin H, Pennington J, et al. The claw paw mutation reveals a role for Lgi4 in peripheral nerve development. Nat. Neurosci. 2006;9:76–84. doi: 10.1038/nn1598. [DOI] [PubMed] [Google Scholar]
- 30.Maltecca F, Aghaie A, Schroeder DG, et al. The mitochondrial protease AFG3L2 is essential for axonal development. J. Neurosci. 2008;28:2827–2836. doi: 10.1523/JNEUROSCI.4677-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seburn KL, Nangle LA, Cox GA, et al. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 32.Antonellis A, Lee-Lin SQ, Wasterlain A, et al. Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J. Neurosci. 2006;26:10397–10406. doi: 10.1523/JNEUROSCI.1671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crimmins S, Jin Y, Wheeler C, et al. Transgenic rescue of ataxia mice with neuronal-specific expression of ubiquitin-specific protease 14. J. Neurosci. 2006;26:11423–11431. doi: 10.1523/JNEUROSCI.3600-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson SM, Bhattacharyya B, Rachel RA, et al. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 2002;32:420–425. doi: 10.1038/ng1006. [DOI] [PubMed] [Google Scholar]
- 35.Bommel H, Xie G, Rossoll W, et al. Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J. Cell. Biol. 2002;159:563–569. doi: 10.1083/jcb.200208001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen XJ, Levedakou EN, Millen KJ, et al. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J. Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafezparast M, Klocke R, Ruhrberg C, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- 38.Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quenneville NR, Chao TY, McCaffery JM, et al. Domains within the GARP subunit Vps54 confer separate functions in complex assembly and early endosome recognition. Mol. Biol. Cell. 2006;17:1859–1870. doi: 10.1091/mbc.E05-11-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt-John T, Drepper C, Mussmann A, et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 2005;37:1213–1215. doi: 10.1038/ng1661. [DOI] [PubMed] [Google Scholar]
- 41.Züchner S, Vance JM. Mechanisms of Disease: a molecular genetic update on hereditary axonal neuropathies. Nat. Clin. Pract. Neurol. 2006;2:45–53. doi: 10.1038/ncpneuro0071. [DOI] [PubMed] [Google Scholar]
- 42.Berger P, Niemann A, Suter U. Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease) Glia. 2006;54:243–257. doi: 10.1002/glia.20386. [DOI] [PubMed] [Google Scholar]
- 43.Young KG, Kothary R. Dystonin/Bpag1--a link to what? Cell. Motil. Cytoskeleton. 2007;64:897–905. doi: 10.1002/cm.20235. [DOI] [PubMed] [Google Scholar]
- 44.Reue K, Zhang P. The lipin protein family: Dual roles in lipid biosynthesis and gene expression. FEBS. Lett. 2008;582:90–96. doi: 10.1016/j.febslet.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gould RM, Byrd AL, Barbarese E. The number of Schmidt-Lanterman incisures is more than doubled in shiverer PNS myelin sheaths. J. Neurocytol. 1995;24:85–98. doi: 10.1007/BF01181552. [DOI] [PubMed] [Google Scholar]
- 46.Smith-Slatas C, Barbarese E. Myelin basic protein gene dosage effects in the PNS. Mol. Cell. Neurosci. 2000;15:343–354. doi: 10.1006/mcne.1999.0829. [DOI] [PubMed] [Google Scholar]
- 47.Jia H, Yan T, Feng Y, et al. Identification of a critical site in Wld(s): essential for Nmnat enzyme activity and axon-protective function. Neurosci. Lett. 2007;413:46–51. doi: 10.1016/j.neulet.2006.11.067. [DOI] [PubMed] [Google Scholar]
- 48.Ferri A, Sanes JR, Coleman MP, et al. Inhibiting Axon Degeneration and Synapse Loss Attenuates Apoptosis and Disease Progression in a Mouse Model of Motoneuron Disease. Curr. Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- 49.Mi W, Beirowski B, Gillingwater TH, et al. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2005;128:405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- 50.Samsam M, Mi W, Wessig C, et al. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J. Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eppig JT, Bult CJ, Kadin JA, et al. The Mouse Genome Database (MGD): from genes to mice—a community resource for mouse biology . Nucleic. Acids. Res. 2005;33:D471–D475. doi: 10.1093/nar/gki113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pool M, Lariviere CB, Bernier G, et al. Genetic alterations at the Bpag1 locus in dt mice and their impact on transcript expression. Mamm. Genome. 2005;16:909–917. doi: 10.1007/s00335-005-0073-4. [DOI] [PubMed] [Google Scholar]
- 53.Guo L, Degenstein L, Dowling J, et al. Gene targeting of BPAG1: abnormalities in mechaical strength and cell migration in stratified epithelia and neurological degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- 54.Chénard CA, Richard S. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 2008;86:233–242. doi: 10.1002/jnr.21485. [DOI] [PubMed] [Google Scholar]
- 55.Noveroske JK, Hardy R, Dapper JD, et al. A new ENU-induced allele of mouse quaking causes severe CNS dysmyelination. Mamm. Genome. 2005;16:672–682. doi: 10.1007/s00335-005-0035-x. [DOI] [PubMed] [Google Scholar]
- 56.Hardy RJ, Loushin CL, Friedrich VL, et al. Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J. Neurosci. 1996;16:7941–7949. doi: 10.1523/JNEUROSCI.16-24-07941.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo LT, Zhang XU, Kuang W, et al. Laminin α2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul. Disord. 2003;13:207–215. doi: 10.1016/s0960-8966(02)00266-3. [DOI] [PubMed] [Google Scholar]
- 58.Plomp JJ, Vergouwe MN, Van den Maagdenberg AM, et al. Abnormal transmitter release at neuromuscular junctions of mice carrying the tottering alpha(1A) Ca(2+) channel mutation. Brain. 2000;123:463–471. doi: 10.1093/brain/123.3.463. [DOI] [PubMed] [Google Scholar]
- 59.Kaja S, van de Ven RC, Broos LA, et al. Characterization of acetylcholine release and the compensatory contribution of non-Ca(v)2.1 channels at motor nerve terminals of leaner Ca(v)2.1-mutant mice. Neuroscience. 2007;144:1278–1287. doi: 10.1016/j.neuroscience.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Kaja S, van de Ven RC, van Dijk JG, Verschuuren JJ, et al. Severely impaired neuromuscular synaptic transmission causes muscle weakness in the Cacna1a-mutant mouse rolling Nagoya. Eur. J. Neurosci. 2007;25:2009–2020. doi: 10.1111/j.1460-9568.2007.05438.x. [DOI] [PubMed] [Google Scholar]
- 61.Green MC, Sidman RL. Tottering--a neuromusclar mutation in the mouse. And its linkage with oligosyndacylism. J. Hered. 1962;53:233–237. doi: 10.1093/oxfordjournals.jhered.a107180. [DOI] [PubMed] [Google Scholar]
- 62.Yoon CH. Disturbances in developmental pathways leading to a neurological disorder of genetic origin, “leaner,” in mice. Dev. Biol. 1969;20:158–181. doi: 10.1016/0012-1606(69)90011-6. [DOI] [PubMed] [Google Scholar]
- 63.Seyfried TN, Itoh T, Glaser GH, et al. Cerebellar gangliosides and phospholipids in mutant mice with ataxia and elilepsy: the tottering/leaner syndrome. Brain. Res. 1981;216:429–436. doi: 10.1016/0006-8993(81)90145-1. [DOI] [PubMed] [Google Scholar]
- 64.Oda S. [The observation of rolling mouse Nagoya (rol), a new neurological mutant, and its maintenance (author's transl)] Jikken Dobutsu. 1973;22:281–288. doi: 10.1538/expanim1957.22.4_281. [DOI] [PubMed] [Google Scholar]
- 65.Meisler MH, Plummer NW, Burgess DL, et al. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica. 2004;122:37–45. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- 66.De Repentigny Y, Côté PD, Pool M, et al. Pathological and genetic analysis of the degenerating muscle (dmu) mouse: a new allele of Scn8a. Hum. Mol. Genet. 2001;10:1819–1827. doi: 10.1093/hmg/10.17.1819. [DOI] [PubMed] [Google Scholar]
- 67.Levedakou EN, Chen XJ, Soliven B, et al. Disruption of the mouse Large gene in the enr and myd mutants results in nerve, muscle, and neuromuscular junction defects. Mol. Cell. Neurosci. 2005;28:757–769. doi: 10.1016/j.mcn.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 68.Wright DE, Johnson MS, Arnett MG, et al. Selective changes in nocifensive behavior despite normal cutaneous axon innervation in leptin recoptor--null mutant (db/db) mice. J. Peripher. Nerv. Syst. 2007;12:250–261. doi: 10.1111/j.1529-8027.2007.00144.x. [DOI] [PubMed] [Google Scholar]
- 69.Drel VR, Mashtalir N, Ilnytska O, et al. The leptin-deficient (ob/ob) mouse: a new animal model of peripheral neuropathy of type 2 diabetes and obesity. Diabetes. 2006;55:3335–3343. doi: 10.2337/db06-0885. [DOI] [PubMed] [Google Scholar]
- 70.Yagihashi S, Yamagishi S, Wada R. Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes. Res. Clin. Pract. 2007;77:S184–S189. doi: 10.1016/j.diabres.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 71.Parvari R, Hershkovitz E, Grossman N, et al. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat. Genet. 2002;32:448–452. doi: 10.1038/ng1012. [DOI] [PubMed] [Google Scholar]
- 72.Saigoh K, Wang YL, Suh JG, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 73.Hutter CM, Samii A, Factor SA, et al. Lack of evidence for an association between UCHL1 S18Y and Parkinson's disease. Eur. J. Neurol. 2008;15:134–139. doi: 10.1111/j.1468-1331.2007.02012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patel PI, Roa BB, Welcher AA, et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 75.Oliver PL, Davies KE. Analysis of human neurological disorders using mutagenesis in the mouse. Clin. Sci. (Lond) 2005;108:385–397. doi: 10.1042/CS20050041. [DOI] [PubMed] [Google Scholar]
- 76.Meyer ZuHörste G, Nave KA. Animal models of inherited neuropathies. Curr. Opin. Neurol. 2006;19:464–473. doi: 10.1097/01.wco.0000245369.44199.27. [DOI] [PubMed] [Google Scholar]
- 77.Suzuki K, Taniike M. Murine model of genetic demyelinating disease: the twitcher mouse. Microsc. Res. Tech. 1995;32:204–214. doi: 10.1002/jemt.1070320304. [DOI] [PubMed] [Google Scholar]
- 78.Sakai N, Inui K, Tatsumi N, et al. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe's disease. J. Neurochem. 1996;66:1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- 79.Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum. Mutat. 1997;10:268–279. doi: 10.1002/(SICI)1098-1004(1997)10:4<268::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 80.Scaravilli F, Jacobs JM. Enzyme replacement in grafted nerve of twitcher mouse. Nature. 1983;305:713–715. doi: 10.1038/305713a0. [DOI] [PubMed] [Google Scholar]
- 81.Luzi P, Rafi MA, Zaka M, et al. Biochemical and pathological evaluation of long-lived mice with globoid cell leukodystrophy after bone marrow transplantation. Mol. Genet. Metab. 2005;86:150–159. doi: 10.1016/j.ymgme.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 82.Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N. Engl. J. Med. 2005;352:2124–2126. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- 83.Michelson AM, Russell ES, Harman PJ. Dystrophia muscularis: a hereditary primary myopathy in the house mouse. Proc. Natl. Acad. Sci. U. S. A. 1955;41:1079–1084. doi: 10.1073/pnas.41.12.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lane PW, Beamer TC, Myers DD. Myodystrophy, a new myopathy on chromosome 8 of the mouse. J. Hered. 1976;67:135–138. doi: 10.1093/oxfordjournals.jhered.a108687. [DOI] [PubMed] [Google Scholar]
- 85.Court FA, Wrabetz L, Feltri ML. Basal lamina: Schwann cells wrap to the rhythm of space-time. Curr. Opin. Neurobiol. 2006;16:501–507. doi: 10.1016/j.conb.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 86.Levedakou EN, Popko B. Rewiring enervated: thinking LARGEr than myodystrophy. J. Neurosci. Res. 2006;84:237–243. doi: 10.1002/jnr.20896. [DOI] [PubMed] [Google Scholar]
- 87.Sunada Y, Bernier SM, Kozak CA, et al. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J. Biol. Chem. 1994;269:13729–13732. [PubMed] [Google Scholar]
- 88.Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin alpha2−chain gene (LAMA2) cause merosin−deficient congenital muscular dystrophy. Nat. Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- 89.Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr. Dev. Pathol. 2006;9:427–443. doi: 10.2350/06-07-0127.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grewal PK, Holzfeind PJ, Bittner RE, et al. Mutant glycosyltransferase and altered glycosylation of alpha-dystroglycan in the myodystrophy mouse. Nat. Genet. 2001;28:151–154. doi: 10.1038/88865. [DOI] [PubMed] [Google Scholar]
- 91.Godfrey C, Clement E, Mein R, et al. Refining genotype−phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 92.Nangle LA, Zhang W, Xie W, et al. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl. Acad. Sci. U. S. A. 2007;104:11239–11244. doi: 10.1073/pnas.0705055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nadeau JH. Modifier genes in mice and humans. Nat. Rev. Genet. 2001;2:165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- 94.Yoshiki A, Moriwaki K. Mouse phenome research: implications of genetic background. ILAR J. 2006;47:94–102. doi: 10.1093/ilar.47.2.94. [DOI] [PubMed] [Google Scholar]
- 95.Burgess RW, Jucius TJ, Ackerman SL. Motor axon guidance of the mammalian trochlear and phrenic nerves: dependence on the netrin receptor Unc5c and modifier loci. J. Neurosci. 2006;26:5756–5766. doi: 10.1523/JNEUROSCI.0736-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buchner DA, Trudeau M, George AL, Jr, et al. High-resolution mapping of the sodium channel modifier Scnm1 on mouse chromosome 3 and identification of a 1.3-kb recombination hot spot. Genomics. 2003;82:452–459. doi: 10.1016/s0888-7543(03)00152-6. [DOI] [PubMed] [Google Scholar]
- 97.Buchner DA, Trudeau M, Meisler MH. SCNM1, a putative RNA splicing factor that modifies disease severity in mice. Science. 2003;301:967–969. doi: 10.1126/science.1086187. [DOI] [PubMed] [Google Scholar]
- 98.Howell VM, Jones JM, Bergren SK, et al. Evidence for a direct role of the disease modifier SCNM1 in splicing. Hum. Mol. Genet. 2007;16:2506–2516. doi: 10.1093/hmg/ddm206. [DOI] [PubMed] [Google Scholar]
- 99.Barbaric I, Miller G, Dear TN. Appearances can be deceiving: phenotypes of knockout mice. Brief. Funct. Genomic. Proteomic. 2007;6:91–103. doi: 10.1093/bfgp/elm008. [DOI] [PubMed] [Google Scholar]
- 100.Abuin A, Hansen GM, Zambrowicz B. Gene Trap Mutagenesis. Handb. Exp. Pharmacol. 2007;178:129–147. doi: 10.1007/978-3-540-35109-2_6. [DOI] [PubMed] [Google Scholar]
- 101.Takeda J, Keng VW, Horie K. Germline mutagenesis mediated by Sleeping Beauty transposon system in mice. Genome Biol. 2007;8(Suppl 1):S14.1–S14.7. doi: 10.1186/gb-2007-8-s1-s14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu S, Ying G, Wu Q, et al. Toward simpler and faster genome-wide mutagenesis in mice. Nat. Genet. 2007;39:922–930. doi: 10.1038/ng2060. [DOI] [PubMed] [Google Scholar]