

Abstract

A catalytic enantioselective propargylation in the presence of 10 mol % Cr-catalyst prepared from Cr(III) bromide and (R)-sulfonamide E furnishes homopropargyl alcohol 8 in 78% yield with 90% ee. Coupled with the workup based on Amano-lipase, this method provides a practical synthesis of optically pure 8 in multi-gram scale. With maintenance of its optical purity, 8 has been converted to 5b, the C14-C19 building block of halichondrins and E7389, in 2 steps.
In the first generation synthesis of halichondrin B, we synthesized the C14–C26 building block from 2-deoxy-L-arabinose diethylthioacetal 4,5-acetonide in 17 steps in approximately 20% overall yield (Scheme 1). 1, 2 This synthesis used three C-C bond-forming reactions highlighted in Scheme 1. Although lengthy, it served well for not only the first generation total synthesis of the halichondrins, but also the discovery and development of E7389.1, 3 For the past several years, we have been exploring a new synthetic route, resulting in a concise and stereoselective synthesis of the same building block. The new synthesis relied on three catalytic asymmetric Cr-mediated couplings in a consecutive manner (Scheme 1).4 With one chromato-graphic separation, the new synthesis provides us with stereochemically homogeneous C14–C26 building block in 6 steps from (t-Bu)(Ph)2SiO(CH2)3CHO.
Scheme 1.

Structure of halichondrin B and C-C bond-forming sites in the synthesis of C14-C26 building block. Panel I: the first generation synthesis using three C-C bond-forming reactions indicated.1 Panel II: the new synthesis using three catalytic asymmetric Cr-mediated coupling reactions indicated.5
We were pleased with the new synthesis in terms of the overall yield and stereoselectivity, except for the catalyst-loading for the C19–C20 coupling (Scheme 2). This bond was formed via a catalytic asymmetric Ni/Cr-mediated coupling of 1a with 2 in the presence of the Cr-catalyst derived from (R)-sulfonamide A, with a useful level of stereoselectivity (dr = 22:1). However, this process required 20 mol % of the catalyst to maintain an acceptable level of coupling-rate. We speculated that the poor catalyst-loading might be attributed to that one of the O=S bonds of the sulfate group present in the nucleophile 1a could coordinate to the Cr-metal, cf. B in Scheme 2, thereby slowing down or shutting down the coupling. This notion encouraged us to study the Ni/Cr-mediated coupling with the nucleophile bearing no O=S or O=C bond, such as the chloride 1b. Experimentally, it was found that the efficiency of Cr-mediated coupling, i.e., coupling rate and catalyst loading, was noticeably improved in the 1b-series (cat. loading: ≤10 mol %) over the 1a-series (cat. loading: ≥20 mol %), while the same level of the asymmetric induction was maintained.5
Scheme 2.

New synthesis of C14-C23 building block 3. Note that the SN2-cyclization of the C20-OH to the C17 carbon takes place in situ.
Originally, we planned to synthesize 1b from the vinyl iodide alcohol 6, readily available via a catalytic enantioselective Co/Cr-mediated 2-haloallylation (Scheme 3).6 However, we soon realized that the SN2 OH → Cl substitution is accompanied with partial racemization under various conditions tested (Scheme 3). No literature precedent related to the observed partial-racemization was found.7 However, because the corresponding homoallyl alcohols are known to maintain the optical purity in the SN2 OH → Cl substitution,7 we speculated that the observed partial racemization might be due to formation of the (partial) carbocation C stabilized via participation of the lone pair electron of iodine. For this reason, we modified the original plan to perform the SN2 OH → Cl substitution on homopropargyl alcohol 8 (Scheme 4).
Scheme 3.

Attempted SN2 OH → Cl substitution. Representative reactions tested include: (1) CCl4, PPh3, CH2Cl2, rt; 93% ee → 50% ee, (2) Cl3CCONH2, PPh3, CH2Cl2, rt; 93% ee → 70% ee, and (3) a. MsCl, Et3N, CH2Cl2, rt and b. LiCl, DMF, rt; 93% ee → 75% ee.
Scheme 4.

New synthesis of C14-C19 building block. Reagents and conditions: (a) 1. catalytic enantioselective Cr-mediated propargylation in the presence of the Cr-catalyst (10 mol %) prepared from CrBr3 and (R)-sulfonamide E. 2. Ac2O/pyr/rt. 3. Amano lipase PS-800 (10% wt), phosphate buffer:acetone (1:9), 50 °C; the overall yield of optically pure 8 from 5: 73%. (b) Cl3CCONH2, PPh3, CH2Cl2, rt (94%). (c) B-iodo-9-BBN, CH2Cl2, 0 °C (85%).15
Among a number of methods reported for synthesis of chiral homopropargyl alcohols,8,9 we were interested in a catalytic enantioselective Cr-mediated propargylation of an aldehyde with a propargyl halide, primarily because of the directness of preparation. The work by Inoue and Nakada is most relevant to the proposed catalytic enantioselective Cr-mediated propargylation; with use of carbazole-based ligands, they demonstrated that the catalytic enantioselective Cr-mediated propargylation is a valuable synthetic method. 10 We were encouraged with their propargylation of hexanal because of its structural similarity to the aldehyde 5. However, we wished to improve its overall efficiency (55% yield; 58% ee). In particular, considering a possible mechanistic similarity between Cr-mediated propargylation and allylation/2-haloallylation, we were anxious in testing the sulfonamide ligand developed for catalytic enantioselective Cr-mediated 2-haloallylation and allylation in this laboratory.6b
In the presence of the Cr-catalyst prepared from CrBr3 and the (R)-sulfonamide D (Scheme 4),6b catalytic enantioselective propargylation of 5 with propargyl bromide (7) proceeded smoothly to furnish 8 in 83% yield with 87% ee. The optical purity of 8 was estimated from a 1H NMR analysis of its Mosher ester and, based on the previous examples,6 the absolute configuration was predicted as indicated, and then confirmed by a chemical correlation with the authentic sample prepared via a different route.11
As shown for the allylation/2-haloallylation, this reaction was easy to scale up. Unlike the allylation/2-haloallylation, however, the asymmetric induction of this process varied between 80% and 87%. Despite extensive efforts, we were unable to establish an experimental protocol to reproducibly perform the coupling at the level of 87% ee. Under this circumstance, we shifted our focus to search for a new sulfonamide ligand.
Using the ligand-optimization strategy recently developed,12 we searched for an alternative ligand within the pool of chiral sulfonamides, whose performance is better than, or comparable with, that of (R)-sulfonamide D.13 Among them, (R)-sulfonamide E was found to be the best ligand in terms of the asymmetric induction, although the coupling yield was modest (65%). In order to improve the coupling efficiency, we then attempted various modifications on the coupling conditions used for allylation/2-haloallylation and eventually found that addition of LiCl significantly improves the coupling yield. The mechanistic explanation for the LiCl effect is not clear at this time. However, we should note that addition of LiCl slightly slows down the coupling rate.
With this modification, it is now possible to prepare 8 in approximately 80% yield with 90% ee in a multi-gram scale with excellent reproducibility (Scheme 4). 14 It is worthwhile adding that sulfonamide E can be recovered in at least 50% yield (80% before recrystallization) by simple operation.
To reveal the scope and limitation, we studied the catalytic asymmetric propargylation on representative aliphatic and aromatic aldehydes in the presence of the Cr-catalysts (10 mol %) derived from two sulfonamides D and E (Table 1). Not surprisingly, it was found that the Cr-catalyst derived from E performs well for aliphatic aldehydes with no α-substituent (see entries 1–3). Interestingly, unlike the 2-haloallylations,6b the Cr-catalysts derived from both E and D recognize the chirality of the β-methyl group present in (S)- and (R)-citronellals (entries 4a,b and 5a,b). For α-substituted aliphatic aldehydes, the Cr-catalyst derived from D gives a better result than the catalyst from E (entries 6 and 7). Overall, the Cr-catalysts derived from E and D match well with aliphatic aldehydes. However, their performance on benzaldehyde and trans-cinnamaldehyde is slightly lower than, or comparable with, that by Nakada’s catalyst (entries 8 and 9).10
Table 1.
Catalytic asymmetric Cr-mediated propargylations.a
 | ||||
|---|---|---|---|---|
| entry | R1 | sulfonamide | yield (%)b | ee or de (%)c |
| 1a | -CH2CH2CH2OTBDPS | E | 78 | 90 |
| 1b | D | 85 | 83 | |
| 2a | -CH2CH2CH2CH2Me | E | 82 | 81 |
| 2b | D | 81 | 67 | |
| 3a | -CH2CH2C6H5 | E | 91 | 89 |
| 3b | D | 94 | 72 | |
| 4a | CH2CH(Me)CH2CH2CH=C(Me)2-S | E | 70 | 93 |
| 4b | D | 81 | 93 | |
| 5a | -CH2CH(Me)CH2CH2CH=C(Me)2-R | E | 73 | 80 |
| 5b | D | 86 | 84 | |
| 6a | -(CH2)6-c | E | 67 | 78 |
| 6b | D | 88 | 90 | |
| 7a | -C(Me)3 | E | 55 | 92 |
| 7b | D | 75 | 73 | |
| 8a | -C6H5 | E | 80 | 46 |
| 8b | D | 92 | 73 | |
| 9a | -CH=CHC6H5-E | E | 84 | 51 |
| 9b | D | 89 | 70 | |
All reactions were performed with CrBr3 (10 mol %), D or E (11 mol %) at 0 °C. Based on the previous examples,6 the absolute configuration of the major product was predicted as indicated and confirmed from the 1H NMR analysis of Mosher esters.
Yields based on chromatographically isolated products.
Ee and de were estimated by 1H-NMR analysis of its Mosher ester. For details, see Supporting Information.
In order to use the reported catalytic asymmetric reaction for the preparative purpose, it is important to have a practical means to enrich the optical purity of 8. To achieve this goal, we took two different approaches. First, we attempted to convert 8 into a crystalline derivative suitable for fractional recrystallization, but with no significant success. 16 Second, we tested use of Amano lipase to discriminate 8 from its enantiomer at either esterification or saponification. Eventually, it was found that the acetate derived from only (S)-8 is hydrolyzed by Amano lipase PS-800, to furnish optically pure (S)-8, readily separable from the remaining acetate via filtration through a silica gel plug. Amano lipase PS-800 was found to be equally effective for the crude acetate directly prepared from the crude product obtained in the catalytic enantioselective Cr-mediated propargylation. Thus, the step of lipase-based optical purity enrichment was coupled with the workup of the catalytic enantioselective Cr-mediated propargylation, to furnish optically pure (S)-8 in 73% overall yield from 5 (5-gram scale).17 Overall, the catalytic enantioselective Cr-mediated propargylation provides us with a practical and scalable synthesis of (S)-8.14
With optically pure homopropargyl alcohol (S)-8 in hand, we studied the SN2 OH → Cl substitution, which was uneventfully achieved on treatment with trichloro-acetamide and triphenylphosphine.18 Upon treatment with 9-iodo-9-borabicyco[3.3.1]nonane (B-iodo-9-BBN),15 the terminal acetylene in 9 was cleanly converted to the corresponding vinyl iodide 1b. As mentioned earlier, our concern was the optical purity of 1b obtained from (S)-8. Among several methods studied, HPLC analyses on a chiral column of the 4-acetylphenyl urethane of 1b gave the most reliable result. 19 With this method, we demonstrated no detectable loss of the optical purity in the process of 8 → 9 → 1b.
In summary, we have reported a catalytic enantio-selective Cr-mediated propargylation in the presence of 10 mol % of Cr-catalyst prepared from Cr(III) bromide and (R)-sulfonamide E, to furnish homopropargyl alcohol 8 in 78% isolated yield with 90% ee. Coupled with the workup based on Amano lipase, this method provides us with a practical route to obtain optically pure (S)-8 in 73% overall yield from 5 in a multi-gram scale. (S)-8 is then transformed to optically pure 1b, the C14-C19 building block of halichondrins and E7389.
Supplementary Material
Acknowledgment
Financial support from the National Institutes of Health (CA 22215) and Eisai Research Institute is gratefully acknowledged.
Footnotes
Supporting Information Available. Experimental details and NMR spectra (47 pages). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Aicher TD, Buszek KR, Fang FG, Forsyth CJ, Jung SH, Kishi Y, Matelich MC, Scola PM, Spero DM, Yoon SK. J. Am. Chem. Soc. 1992;114:3162. [Google Scholar]
- 2.For isolation and synthesis on the halichondrins, see Ref. 1–3 in the preceding Yang Y-R, Kim D-S, Kishi Y. Org. Lett. 2009;11:0000. doi: 10.1021/ol9016589.
- 3.For discovery and development of E7389, see Ref. 5 in the preceding Yang Y-R, Kim D-S, Kishi Y. Org. Lett. 2009;11:0000. doi: 10.1021/ol9016589.
- 4.For reviews on Cr-mediated carbon-carbon bond-forming reactions, see: Saccomano NA. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 1. Oxford: Pergamon; 1991. p. 173. ; (b) Fürstner A. Chem. Rev. 1999;99:991. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]; (c) Wessjohann LA, Scheid G. Synthesis. 1999:1. [Google Scholar]; (d) Takai K, Nozaki H. Proc. Japan Acad. Ser. B. 2000;76:123. [Google Scholar]; (e) Hargaden GC, Guiry PJ. Adv. Synth. Catal. 2007;349:2407. [Google Scholar]
- 5.Kim D-S, Dong C-G, Kim JT, Guo H, Huang J, Tiseni PS, Kishi Y. submitted for publication. [Google Scholar]
- 6.(a) Kurosu M, Lin M-H, Kishi Y. J. Am. Chem. Soc. 2004;126:12248. doi: 10.1021/ja045557j. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z, Huang J, Ma B, Kishi Y. Org. Lett. 2008;10:3073. doi: 10.1021/ol801093p. [DOI] [PubMed] [Google Scholar]
-
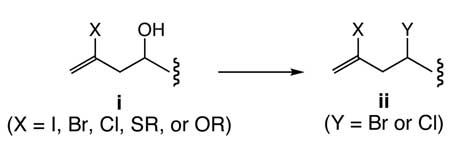
7.No literature example was found for the i → ii transformation where X is I, Br, Cl, SR, and OR. To the contrary, examples are known to demonstrate no racemization where X is H; for example, see:
Clapp CH, Grandizio AM, Yang Y, Kagey M, Turner D, Bicker A, Muskardin D. Biochemistry. 2002;41:11504. doi: 10.1021/bi020229m.
- 8.Carbonyl propargylations with allenylmetal reagents are well established, to synthesize chiral homopropargyl alcohols. For general reviews, see for example: Denmark SD, Fu J. Chem. Rev. 2003;103:2763. doi: 10.1021/cr020050h. Marshall JA, Gung BW, Grachan ML. In: Modern Allene Chemistry Vol 1. Krause N, Hashmi ASK, editors. Weinheim: Wiley-VCH; 2004. p. 493. Gung BW. Org. React. 2004;64:1. Marshall JA. J. Org. Chem. 2007;72:8153. doi: 10.1021/jo070787c. Also see: Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. Int. Ed. 2008;47:5220. doi: 10.1002/anie.200801359. and references cited therein.
- 9.For enantioselective propargylations with a propargyl halide in the presence of low-valent metal, see: Bandini M, Cozzi PG, Umani-Ronchi A. Polydedron. 2000;19:537. ; (b) Bandini M, Cozzi PG, Melchiorre P, Tino R, Umani-Ronchi A. Tetrahedron Asymmetry. 2001;12:1063. [Google Scholar]; (c) Loh T-P, Lin M-J, Tan K-L. Tetrahedron Lett. 2003;44:507. [Google Scholar]
- 10.Inoue M, Nakada M. Org. Lett. 2004;6:2977. doi: 10.1021/ol048826j. [DOI] [PubMed] [Google Scholar]
- 11.Chiral homopropargyl alcohol 8 was synthesized via addition of TMS-acetylene anion to chiral epoxide obtained through Jacobsen’s hydrolytic kinetic resolution of racemic epoxide Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936. doi: 10.1126/science.277.5328.936.
- 12.Guo H, Dong C-G, Kim D-S, Urabe D, Wang J, Kim JT, Liu X, Sasaki T, Kishi Y. submitted for publication. [Google Scholar]
- 13.These included the following sulfonamides: R1=t-Bu/R2=3,5-(CF3)2Ph//R3=(OMe)3 (er=20:1. 65% yield); R1=t-Bu/R2=3,5-(CF3)2Ph/R3=OPh (er=17:1. 68% yield); R1=t-Bu/R2=3,5-(CF3)2Ph/R3=OMe (er=15:1, 65% yield); R1=t-Bu/R2=3,5-(Cl)2Ph/R3=(OMe)3 (er=15:1. 73% yield); R1=t-Bu/R2=3,5-(Cl)2Ph/R3=F (er=11:1. 80% yield); R1=t-Bu/R2=3,5-(CF3)2Ph/R3=F (er=11:1. 80% yield).
- 14.For details, see Supporting Information.
- 15. Hara S, Dojo H, Takinami S, Suzuki A. Tetrahedron Lett. 1983;24:731. For a review, see: Suzuki A. Rev. Heteroatom Chem. 1997;17:271.
- 16.Propargyl alcohol derivatives, including 4-acetylphenyl urethane, were tested.
- 17.Amano lipase PS-800 was found effective to obtain the optically pure (S)-8 (42% isolated yield) from the racemic homopropargyl acetate,14 prepared via propargylation of 5 in the presence of 1 mol % 3,3’-dimethyl-2,2’-dipyridyl-Cr(III) complex, followed by acetylation: Namba K, Wang J, Cui S, Kishi Y. Org. Lett. 2005;7:5421. doi: 10.1021/ol052085k.
- 18.Pluempanupat W, Chavasiri W. Tetrahedron Lett. 2006;47:6821. [Google Scholar]
-
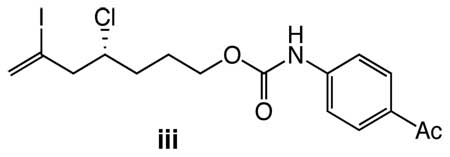
19.The optical purity of 1b was determined via HPLC analysis (OJ-H chiral column) of 4-acetylphenyl urethane iii prepared from 1b. For details, see Supporting Information.


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.