

Abstract
Matrix metalloproteinases (MMPs) were discovered because of their role in amphibian metamorphosis, yet they have attracted more attention because of their roles in disease. Despite intensive scrutiny in vitro, in cell culture and in animal models, the normal physiological roles of these extracellular proteases have been elusive. Recent studies in mice and flies point to essential roles of MMPs as mediators of change and physical adaptation in tissues, whether developmentally regulated, environmentally induced or disease associated.
The founding member of the matrix metalloproteinase (MMP) family, collagenase, was identified in 1962 by Gross and Lapiere, who found that tadpole tails during metamorphosis contained an enzyme that could degrade fibrillar collagen1,2. Subsequently, an interstitial collagenase, collagenase-1 or MMP1, was found in diseased skin and synovium3. In vitro, MMP1 initiates degradation of native fibrillar collagens, crucial components of vertebrate extracellular matrix (ECM), by cleaving the peptide bond between Gly775–Ile776 or Gly775–Lys776 in native type I, II or III collagen molecules3,4. Further research led to the discovery of a family of structurally related proteinases (23 in human, 24 in mice), now referred to as the MMP family.
Interest in MMPs increased in the late 1960s and early 1970s following observations that MMPs are upregulated in diverse human diseases including rheumatoid arthritis and cancer. Importantly, high levels of MMPs often correlated with poor prognosis in human patients (reviewed in REF. 5). However, recent clinical data indicate that the relationship between MMPs and disease is not simple; for example, increased MMP activity can enhance tumour progression or can inhibit it (reviewed in REF. 6). This complex relationship between MMP expression and cancer has increased the basic and clinical interest in understanding MMP function in vivo, but it has also focused attention on MMPs and pathology, and relatively less attention has been focused on the normal roles of these enzymes. Surely we do not have 23 MMPs in our bodies just to promote tumours. A main question remains unanswered: what is the normal function of the MMP gene family in development? This Review focuses on what we have learned about the normal in vivo functions of MMPs from genetic analysis.
MMPs in vivo: analysis of MMP mutants
Analysis of genetic knockouts of MMPs offers opportunities to identify essential functions of an MMP and to validate candidate substrates by looking for cleavage products in the control animal and uncleaved proteins in the mutant animal. At least 14 mouse MMP mutants have been generated. The initial characterizations have described surprisingly subtle phenotypes, with all MMP-knockout lines surviving to birth (TABLE 1). Possible explanations for the seeming dispensability of MMPs during embryonic development include enzymatic redundancy, enzymatic compensation and adaptive development, although it is possible that MMPs are not important until after embryonic development. MMPs have many overlapping substrates in vitro5, which indicates possible genetic redundancy in vivo. Indeed, redundancy has now been shown with the recent generation of MMP double mutants7,8. Compensation has also been shown in the MMP family9,10. In the next sections, we will discuss mutant mouse phenotypes that rely on genetic approaches. Researchers can also now take advantage of simpler genetic model systems, such as Drosophila melanogaster, in which it is possible to mutate all MMP genes.
Table 1.
Selected MMP- and TIMP-null mutant phenotypes
| MMP gene | Mutant phenotypes (mouse, unless noted) |
|---|---|
| Mmp2 | Reduced body size152; reduced neovascularization55; decreased primary ductal invasion in the mammary gland54; reduced lung saccular development153 |
| Mmp3 | Altered structure of neuromuscular junctions154; reduced purse stringing during wound healing86; altered secondary branching morphogenesis in the mammary gland54 |
| Mmp7 | Innate immunity defects83; decreased re-epithelialization after lung injury85 |
| Mmp8 | Increased skin tumours104; resistance to tumour necrosis factor (TNF)-induced lethal hepatitis103 |
| Mmp9 | Bone-development defects36; defective neuronal remyelination after nerve injury155; delayed healing of bone fractures39; impaired vascular remodelling156; impaired angiogenesis36 |
| Mmp10 | Increased inflammation and increased mortality in response to infection or wounding (W. C. Parks, personal communication) |
| Mmp11 | Delayed mammary tumorigenesis157 |
| Mmp12 | Diminished recovery from spinal cord crush158; increased angiogenesis due to decreased angiostatin128 |
| Mmp13 | Bone remodelling defects7, 40; reduced hepatic fibrosis159; increased collagen accumulation in atherosclerotic plaques160 |
| Mmp14 | Skeletal remodelling defects42, 43, 45, 161; angiogenesis defects46; inhibition of tooth eruption and root elongation162; defects in lung and submandibular gland46, 163 |
| Mmp19 | Obesity164 |
| Mmp20 | Defects in tooth enamel165 |
| Mmp23 | No phenotype reported |
| Mmp24 | Abnormal response to sciatic nerve injury166 |
| Mmp28 | Increased inflammatory response (W. C. Parks, personal communication) |
| DmMmp1 | Defective tracheal tube growth (see text)75; failure of head eversion at metamorphosis75 |
| DmMmp2 | Failure of tissue histolysis at metamorphosis75 |
| Timp1 | Accelerated endometrial gland formation167; impaired learning and memory168; accelerated hepatocyte cell-cycle progression169; increased resistance to Pseudomonas infection107 |
| Timp2 | Motor defects170 |
| Timp3 | Accelerated apoptosis in mammary glands171; impaired bronchiole branching172; enhanced metastatic dissemination173 |
| Timp4 | No phenotype reported174 |
| DmTimp | Inflated wings175; autodigesting gut175 |
Dm, Drosophila melanogaster; Mmp, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinase.
MMP proteolysis
MMPs are members of the metzincin group of proteases, which are named after the zinc ion and the conserved Met residue at the active site11,12. Recent work has generated a unified peptidase nomenclature13 in which MMPs include the M10A subfamily, the M10 family and the MA clan of metallopeptidases. Mammalian MMPs share a conserved domain structure (FIG. 1) that consists of a catalytic domain and an autoinhibitory pro-domain. The pro-domain contains a conserved Cys residue that coordinates the active-site zinc to inhibit catalysis. When the pro-domain is destabilized or removed, the active site becomes available to cleave substrates. Most MMP-family members also contain a hemopexin domain, attached at their C termini by a flexible hinge. The hemopexin domain encodes a four-bladed β-propeller structure that mediates protein–protein interactions. This domain also contributes to proper substrate recognition, activation of the enzyme, protease localization, internalization and degradation14. The structures of the catalytic and hemopexin domains of several MMPs, including MMP1, MMP2, MMP3 and MMP14 (also known as membrane type 1 MMP (MT1-MMP)), have been solved (reviewed in REFS 13, 15). MMP2 and MMP9 also have fibronectin type II repeats, which mediate binding to collagens, inserted into the catalytic domain. Most MMPs are secreted proteins; however there are six MT-MMPs: MMP14, MMP15, MMP16 and MMP24 have transmembrane domains and short cytoplasmic tails; MMP17 and MMP25 have glycosylphosphatidylinositol (GPI) linkages. Also, MMP23 is a type II transmembrane protein. The activity of MMPs is controlled at many levels and the regulation of MMP activity remains a topic of intense research (BOX 1).
Figure 1. Schematic structure of MMPs.

a | Matrix metalloproteinases (MMPs) are expressed as pro-proteins. A conserved Cys residue in the pro-domain coordinates the zinc ion, which would otherwise be used for catalysis. The pro-domain is removed by a combination of a cleavage in the domain and a cleavage between the pro-domain and the catalytic domain. b | Most MMPs share a conserved domain structure of pro-domain, catalytic domain, hinge region and hemopexin domain (1). All MMPs are synthesized with a signal peptide, which is cleaved during transport through the secretory pathway. MMP2 and MMP9 have three fibronectin type II repeats in their catalytic domains (2). Membrane type MMPs (MT-MMPs) are linked to the plasma membrane either by a transmembrane domain or by a glycosylphosphatidylinositol (GPI) linkage, attached to the hemopexin domain (3). Minimal MMPs lack the hinge and hemopexin domains (4). MMP21 has a truncated hinge domain. Drosophila melanogaster DmMMP2 has an insertion of 214 amino acids into its hinge domain. MMP23 (not shown) has a non-conserved N-terminal domain that consists of an immunoglobulin IgC2 domain and a ShKT domain; it is unclear if MMP23 contains a Cys residue switch.
Box 1. Regulation of extracellular proteolysis.
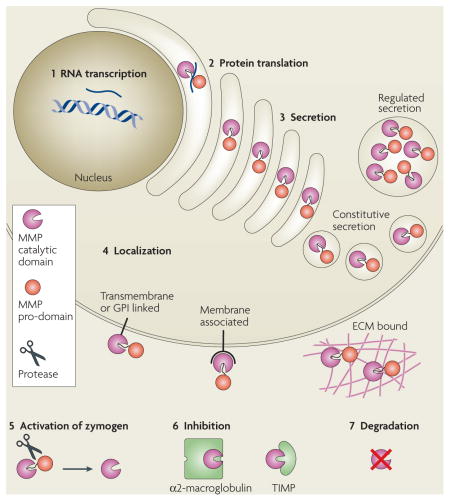
It is easier to assay for the presence or absence of matrix metalloproteinase (MMP) RNA transcripts than for protein abundance or enzyme-activity levels. However, MMP function can be regulated at many levels. In addition to (1) RNA transcription and (2) protein synthesis, MMP function can be regulated at the levels of (3) secretion, intracellular trafficking, (4) subcellular or extracellular localization, (5) activation of the zymogen form, (6) expression of their endogenous protein inhibitors, such as tissue inhibitors of metalloproteinases (TIMPs) and α2-macroglobulin118, and (7) protease degradation. Also, substrate availability and accessibility determine the degree to which MMP activity is used. This regulatory complexity has made it especially difficult to infer the spatial and temporal details of MMP activity from RNA-expression patterns. For details on the regulation of MMP activity, see REFS 2,16,119 and references therein. For a specific example of the regulation of MMP13 activity, see REF. 120. ECM, extracellular matrix; GPI, glycosylphosphatidylinositol.
Functions of MMP proteolysis
Historically, MMPs were thought to function mainly as enzymes that degrade structural components of the ECM. However, MMP proteolysis can create space for cells to migrate, can produce specific substrate-cleavage fragments with independent biological activity, can regulate tissue architecture through effects on the ECM and intercellular junctions, and can activate, deactivate or modify the activity of signalling molecules, both directly and indirectly16 (FIG. 2). Because cells have receptors (for example, integrins) for structural ECM components, MMPs can also affect cellular functions by regulating the ECM proteins with which the cells interact17. In many cases, MMP cleavage of ECM substrates generates fragments that have different biological activities from their precursors. For example, the cleavage of laminin-5 or collagen IV results in the exposure of cryptic sites that promote migration18,19. Type I collagen degradation that is mediated by MMP1 is necessary for epithelial cell migration and wound healing in culture models20. Cleavage of ECM proteins by MMPs can also release ECM-bound growth factors, including insulin growth factors and fibroblast growth factors21,22. Alternative mechanisms of action have also been observed, including functional intermolecular MMP complexes: MMP14 binds to tissue inhibitor of metalloproteinase-2 (TIMP2), which binds to pro-MMP2, thereby positioning it for activation by a second molecule of MMP14 (REF. 23). Furthermore, human MMP11 has an alternative splice isoform that functions as an intracellular proteinase and enters the nucleus24.
Figure 2. Possible modes of MMP action.

Recent work has dramatically expanded our understanding of the possible substrates that can be subject to matrix metalloproteinase (MMP) cleavage, and has similarly expanded our awareness of the means by which MMPs can affect cell behaviour. a | MMPs can cleave components of the extracellular matrix (ECM), resulting in increased space for cell or tissue movement. b | Alternatively, MMP proteolysis can generate specific cleavage products that then signal in an autocrine or paracrine manner (for example, cleavage of collagen IV α3 chain by MMP9 yields tumstatin, an anti-angiogenic peptide that functions by binding to the αvβ3 integrin127). c | MMPs can also directly regulate epithelial tissue architecture through cleavage of intercellular junctions or the basement membrane. d | MMPs can activate or modify the action of latent signalling molecules, resulting in diverse cellular consequences. For example, cleavage of vascular endothelial growth factor (VEGF) by MMPs changes angiogenic outcome by modifying the binding and diffusion properties of VEGF64. e | MMPs can deactivate or modify the action of active signalling molecules, resulting in changes in proliferation, cell death, differentiation or cell motility. For example, MMP2 cleavage of stromal-cell derived factor-1 (SDF1, also known as CXC-motif ligand-12 (CXCL12)) results in its inactivation100.
MMP substrates include peptide growth factors, tyrosine kinase receptors, cell-adhesion molecules, cytokines and chemokines, as well as other MMPs and unrelated proteases (BOX 2). MMPs and the related families of proteinases, the ADAMs (a disintegrin and metalloproteases) and ADAM-TSs (ADAMs with thrombo spondin repeats), are important in shedding plasma-membrane-bound proteins. ADAMs and ADAM-TSs participate in shedding growth factors that are synthesized as cell-membrane-bound precursor forms, including heparin-binding epidermal growth factor (HB-EGF), neuregulin, amphiregulin and transforming growth factor-α (TGFα)25–28. Cleavage of other membrane proteins, such as E-cadherin and CD44, results in the release of specific, biologically active fragments of their extra cellular domains, and in increased invasive behaviour29,30. Cell-surface-adhesion molecules, such as syndecan-1, are also shed by soluble and membrane MMPs31,32. MMP9 and MMP12 contribute to proteolytic shedding of the lipopolysaccharide (LPS) receptor CD14, and therefore influence innate host defense33. TGFβ is another important MMP substrate, the activation of which frequently alters cell migration; for example, MMP9 restrains corneal epithelial migration through the activation of TGFβ34. Both MMP2 and MMP9 can release TGFβ from an inactive extracellular complex that consists of TGFβ, TGFβ latency-associated protein (which is the pro-domain of TGFβ), and latent TGFβ-binding protein35.
Box 2. Biological consequences of MMP proteolysis.
Converting structural matrix proteins to signalling molecules*
Collagen II: fragment is a bone morphogenetic protein (BMP) antagonist125,126
Collagen IV α1–α6 noncollagenous-1 (NC1) domains are anti-angiogenic after cleavage121,124,127
Collagen 18 has an NC1 domain (endostatin), which is anti-angiogenic121,128
Structural changes to matrix proteins
Cleavage of perlecan129
Changes in tissue architecture
Chemoattraction
Increase in chemokine activity following cleavage: interleukin-8 (IL8, also known as CXC-motif ligand-8 (CXCL8); the mouse LIX)93,94
Decrease in chemokine activity following cleavage: monocyte chemotactic protein-1 (MCP1, also known as CCL2 or JE)92,99–101
Change in chemotaxis: gradients form by shedding of syndecan32,141
Proliferation
Cell survival
Neuronal survival factor: stromal-cell derived factor-1 (SDF1, also known as CXCL12); HIV dementia145
Activate latent signalling molecules
Cleavage of insulin-growth-factor (IGF)-binding protein (IGFBP) to release active IGF21
Latent transforming growth factor-β (TGFβ) to active TGFβ35
Tumour necrosis factor-α (TNFα) shedding146
Shedding of Ninjurin A to signal release of cell adhesion78
Change in the range of action of a signalling molecule
Vascular endothelial growth factor (VEGF): change in the range of diffusion through the modulation of heparin binding64
Membrane versus soluble TNFα147
Differentiation
An important ongoing challenge is to link our understanding of MMP substrates that have been identified in vitro or in cell culture with the substrates that are cleaved by MMPs in vivo. A technical limitation of recent approaches is that MMPs are often expressed as isolated catalytic domains without their substrate-recognizing hemopexin domains. These catalytic domains have been proven to be capable of cleaving hundreds of substrates in vitro, including nearly all of the components of the ECM.
MMPs in bone modelling and remodelling
Bone is an important site of ongoing tissue remodelling during development, homeostasis and repair. Bone develops according to one of three distinct processes: intramembranous ossification, endochondral ossification (FIG. 3a) or pseudo-metamorphic ossification. The clavicle and some skull bones develop by intramembranous ossification, in which mesenchymal precursors differentiate directly into osteoblasts. The appendicular and axial skeleton, including the long bones, develops by endochondral ossification, whereby a cartilage template forms first and is then resorbed and replaced by mineralized bone. Cartilage cells, or chondrocytes, differentiate and are replaced at the growth plate, where they follow a stereotyped progression of proliferation, differentiation, hypertrophy, angiogenic invasion and apoptosis. In pseudo-metamorphic ossification, a cartilage template functions as a temporary mould to shape the deposition of bone. Even after bones have formed, they are continually remodelled throughout life.
Figure 3. Skeletal phenotypes of MMP mutants.

a | Long bones in mice and humans develop through the process of endochondral ossification, in which a cartilage template forms first and then is resorbed and replaced by mineralized bone. This process requires extensive matrix remodelling and invasion of new blood vessels. The schematic is adapted from REF. 150 © (2000) Elsevier, and REF. 151 © (1999) Macmillan Magazines Ltd. b | Matrix metalloproteinase-9 (Mmp9)- and Mmp13-null femurs display greatly expanded hypertrophic cartilage zones (HC; red line) and altered trabecular bone (TB; blue line). Despite this expansion, Mmp9- and Mmp13-null phenotypes eventually resolve, resulting in good bone formation. The Mmp9 Mmp13 double mutant has an even greater expansion of hypertrophic cartilage, and significantly and persistently shorter long bones. Images courtesy of D. Stickens, D. Behonick and N. Ortega, University of California, San Francisco, USA.
Loss of MMP9 is associated with growth-plate defects
The first developmental phenotype reported in an MMP-knockout mouse was a defect in endochondral ossification of long bones36. Deletion of Mmp9 results in expansion of the zone of hypertrophic chondrocytes in the growth plate36 because of a failure of apoptosis (FIG. 3b). A relevant MMP9 substrate seems to be galectin-3, a lectin with anti-apoptotic activity that can be secreted and localized to the ECM. MMP9 cleaves and inactivates galectin-3. Indeed, galectin-3-mutant mice display growth-plate defects that are opposite of the defects of Mmp9-deficient mice, with premature apoptosis of the hypertrophic chondrocytes37. Uncleaved galectin-3 is also enriched specifically at the growth plate in Mmp9 mutants. Uncleaved galectin-3, added exogenously to wild-type bone explants, causes an expansion of the growth plates that is similar to the phenotype of the Mmp9 mutants, whereas cleaved galectin-3 has no effect38.
MMP9 is also highly expressed during bone healing after fracture, and bone fractures in Mmp9-mutant mice heal more slowly than in controls. Exogenous vascular endothelial growth factor (VEGF) rescues the repair defects, indicating that MMP9 processing of VEGF might be rate limiting in bone repair39. Intriguingly, the Mmp9-null animals heal from stabilized fractures through endochondral ossification rather than through intramembranous ossification as in wild type39, indicating that a different mechanism is used to heal the fracture in the absence of MMP9.
MMP13 functions in bone formation and remodelling
Mmp13 mutants show an expansion of the zone of hypertrophic chondrocytes and a delay in apoptosis, indicating that MMP13 is required for the transition from cartilage to bone at the growth plates of long bones7,40 (FIG. 3b). MMP13 cleaves type II collagen and aggrecan in vivo, and these cleavage products can be recognized in a tight zone that is just distal to the zone of ossification and angiogenesis. The primary defect in Mmp13 mutants is a failure of chondrocytes to remodel the ECM that is rich in type II collagen and aggrecan. Collagen cleavage does not occur in the absence of MMP13, whereas both MMP13 and MMP9 cleave aggrecan. The cleavage product of aggrecan is absent in the Mmp9 Mmp13 double mutant7. It is worth noting that there are no phenotypic consequences of replacing endogenous aggrecan with aggrecan that is resistant to MMP cleavage, indicating that other mechanisms exist for the removal of aggrecan at the cartilage–bone transition41.
The second function of MMP13 in long-bone development occurs during ossification. The cartilage ECM becomes the scaffold for mineralization, forming bone spicules or trabeculae. Mmp13 mutants exhibit irregularly shaped spicules, demonstrating that MMP13 is required for their initial modelling7. MMP13 also functions in the continual remodelling of spicules; Mmp13 mutants have an abnormal increase in trabecular bone mass that persists into adulthood. Conditional Mmp13 mutants in bone or cartilage show that the regulation of hypertrophic chondrocyte apoptosis by MMP13 and the initial MMP13-mediated spicule-modelling function reside in chondrocytes. The MMP13-mediated remodelling function that regulates bone mass resides in osteoblasts. Therefore, MMP13 functions in bone formation, both at the growth plate and at the spicules, and in bone remodelling.
MMP14 deficiency results in lethality
MMP14 has various roles in skeletal development. Mmp14 knockouts are the only lethal MMP-mutant mice; the mice are normal at birth but develop multiple abnormalities and die by 3–12 weeks42,43. Mmp14 mutants are grossly defective in the remodelling of the connective tissue. Loss of an ECM-degrading enzyme would be expected to result in increased bone deposition; paradoxically, Mmp14 mutants instead show secondary effects of increased bone resorption and defective secondary ossification centres42,43. Osteogenic cells from Mmp14-mutant mice cannot degrade collagen and do not form bone when transplanted subcutaneously into host immunodeficient mice42. Taken together, the phenotypes of Mmp14-mutant mice strongly support the physiological relevance of the in vitro data, which show that collagen types I, II and III are substrates for MMP14 (REF. 44).
Some initial bone modelling is also dependent on MMP14. The pseudo-metamorphic form of bone development was discovered only after aberrant residual cartilaginous tissues were found in Mmp14 mutants45. In wild-type mice, some skull bones and the diaphysis of long bones are formed by osteoblasts that lay down mineralized bone in close apposition to pre-existing cartilage that functions as a mould and later undergoes apoptosis. These bones develop by the replacement of a juvenile tissue with an adult tissue, a process comparable to metamorphosis. In the absence of MMP14, these cartilaginous templates persist.
Mmp2- Mmp14-null mice die at birth8. These double mutants resemble much older Mmp14 single mutants, indicating that MMP2 and MMP14 function redundantly. It is clear that MMP14 activates MMP2, because MMP2 activity is reduced, but not eliminated, in Mmp14-null mice43,46. However, other activators also exist, probably including MMP15 and MMP16. Last, MMP14 has MMP2-independent roles in bone and connective-tissue remodelling and during angiogenesis8,43,46.
Human MMP mutations lead to defects in bone development
Three human skeletal disease conditions are associated with loss-of-function MMP mutations. These diseases are a rare osteolytic syndrome, which is caused by MMP2 mutations47, the Missouri variant of spondyloepimetaphyseal dysplasia (SEMD), which is caused by point mutations in MMP13 (REF. 48), and the tooth enamel defect amelogenesis imperfecta, which is caused by splice-acceptor mutations in MMP20 (REF. 49).
Although the Mmp2-knockout mouse has only a subtle skeletal defect50, human MMP2 mutants have an osteolytic (vanishing bone) syndrome, which involves progressive destruction and resorption of bone, as well as dwarfism and arthritis47. The defects of this syndrome are similar to the phenotype of Mmp14-mutant mice. As with the Mmp14 mice, the paradoxical observation is that a loss-of-function mutation in an ECM-degrading protease causes, rather than inhibits, bone degradation. This phenotype correlates with an excessive number of osteoclasts, which might compensate for the primary failure of another type of ECM degradation.
The similarity between the human disease caused by MMP2 mutation and the Mmp14-knockout mice is particularly interesting in light of the biochemical interaction between these two proteases. Membrane-bound MMP14 activates secreted MMP2 by cleaving its pro-domain51,52. Indeed, active MMP2 is significantly decreased in Mmp14-null mice, strongly indicating that MMP2 is an in vivo substrate of MMP14 (REF. 43). Therefore, it is clear that MMPs have many important roles in bone development.
MMP2 and MMP3 in mammary development
The epithelial ductal network of the mammary gland expands greatly during puberty in response to both local and systemic signals53 (FIG. 4a). This process requires degradation of the basement membrane and ECM, restructuring of the endogenous vascular network and large-scale epithelial morphogenesis — all these processes suggest MMP involvement.
Figure 4. Mammary gland phenotypes of MMP mutants.

a | The length of the ductal network of the mammary gland is formed through the invasion and bifurcation of terminal end buds (TEBs). New branches initiate off of these primary ducts, in a process known as secondary branching. b | Mmp2- and Mmp3-null animals have reciprocal phenotypes; Mmp2-null mammary glands have deficient primary invasion and excess secondary branching, whereas Mmp3-null mammary glands have normal invasion and deficient secondary branching. These findings show that different proteases are used to accomplish these tissue-invasion processes. Images courtesy of A.J.E. and J. Trumbull, University of California, San Francisco, USA.
Combined pharmacological and genetic analysis of MMP2 and MMP3 revealed a role for MMP activity during mammary development54. Branching morphogenesis in the mammary gland occurs by two spatially and temporally distinct mechanisms: terminal end bud (TEB) elongation and secondary branching. The length of the mammary ductal network is established through the invasion of TEBs through the mammary fat pad. Secondary branches initiate laterally from the main ducts. Mmp2- and Mmp3-null mammary glands have reciprocal phenotypes in branching morphogenesis. Mmp2-null mammary glands are deficient in TEB invasion, but have an excess of secondary branches, whereas Mmp3-null mammary glands have normal invasion, but are characterized by deficient secondary branching54 (FIG. 4b). Inhibition of MMP function with a chemical inhibitor (GM6001) results in deficient primary invasion and an excess of secondary branches. Overexpression of the endogenous MMP inhibitor TIMP1 limits TEB invasion, but has a minor effect on secondary branching54. These observations led to a model in which MMP2 promotes ductal invasion and inhibits secondary branching, whereas MMP3 promotes secondary-branch initiation.
MMPs in blood-vessel remodelling
Analysis of a broad range of MMP mutants shows that embryonic vascular development proceeds normally, yet defects are observed in both normal and pathological postnatal vascular remodelling and angiogenesis (TABLE 1). These data imply that MMPs might have a specific role in postnatal refinement, remodelling and neoangiogenesis, but not in the original construction of the embryonic vascular networks.
Mmp9-mutant mice show defects in angiogenesis at the growth plate of long bones36. Similarly, Mmp14-mutant mice lack appropriate vascular invasion at secondary ossification centres, despite normal levels of VEGF and its receptor VEGFR2 (also known as FLK1) (REF. 43). This lack of vascular growth is recapitulated in an experimentally induced corneal angiogenesis assay43, which induces new blood-vessel growth in control mice. Blood-vessel growth is not observed in Mmp14 mutants43, whereas it is reduced but not eliminated in Mmp2-mutant animals55. In a laser-induced injury model of retinal degeneration, neovascularization is reduced in single Mmp2 and Mmp9 mutants and is strongly reduced in the Mmp2 Mmp9 double mutant, indicating that these MMPs function redundantly56. Mmp2 Mmp14 double mutants also have developmental vascular defects, including capillaries with extremely small lumens. These defective blood vessels are sufficient to support embryonic growth and development, but not postnatal survival.
How do MMPs contribute to vascular remodelling? Likely mechanisms include proteolysis of type I collagen, modification of platelet-derived growth factor (PDGF) signalling, regulation of perivascular cells and processing of VEGF. Invading blood vessels in postnatal tissues encounter ECM that is rich in type I collagen, a protein that is not highly expressed in embryos. When control aortic explants are embedded in three-dimensional collagen matrices57, they sprout neovessels and the endothelial cells invade the collagen matrix in a growth-factor-dependent manner. Strikingly, Mmp14-null explants do not sprout neovessels or invade the collagen matrix, whereas explants from Mmp2- and Mmp9-null mice are indistinguishable from control explants. However, when Mmp14-null explants are embedded in a fibrin matrix (similar to the provisional ECM at a wound site) they form capillaries, demonstrating matrix specificity. So, MMP14 contributes to postnatal vascular development by cleaving type I collagen; the relative absence of type I collagen in the embryo might partially explain the lack of embryonic vascular defects.
MMP9 and MMP14 have another effect on the vasculature: the perivascular (or smooth muscle) cells that ensheath the endothelial cells of the blood vessels are missing or their density is significantly decreased in normal vessels and during tumour neoangiogenesis57–59. This defect is especially evident in the smaller arterioles of the brain, and many of the remaining perivascular cells have irregular morphology. This phenotype is similar to that of mice that carry weak PDGF-B alleles. Signalling that occurs downstream of PDGF is attenuated in Mmp14-null mice, and PDGF receptor-β (PDGFRβ) co-immunoprecipitates with MMP14, demonstrating that they form a physical complex60. These data indicate a novel and direct function for MMP14 in PDGF signalling.
MMPs and VEGF signaling
MMP processing of VEGF might have an important role in physiological and tumour angiogenesis. VEGF is stored extracellularly: after secretion, VEGF binds to the ECM, from where it must be released to initiate angiogenesis61. Small insulinomas form constitutively in the RIP1-Tag (rat insulin promoter 1–T-antigen) mouse pancreatic islet cancer model, but only 1–2% of these develop into angiogenic adenomas and carcinomas62. Despite continuous expression of VEGF and its receptor, VEGFR2, VEGF availability is limited and cannot bind to its receptor in pre-angiogenic tumours. MMP9 mobilizes VEGF and initiates angiogenesis. Importantly, when the Mmp9 mutation is crossed into the RIP1-Tag background, fewer tumours become angiogenic, supporting the role of MMP9 in mobilizing VEGF63.
MMPs can also cleave VEGF, separating the matrix-binding domain from the receptor-binding domain. Uncleaved VEGF is enriched in at least one MMP9-mutant postnatal tissue, the hypertrophic chondrocyte zone38, which indicates that VEGF is an important downstream effector, and possibly a substrate, of MMP9. Truncated VEGF has different effects on tumour blood vessels than does uncleavable VEGF; truncated VEGF increases vessel diameter, whereas uncleavable VEGF increases vessel sprouting64. Mmp9-null mice display defective post-embryonic neovasculature, suggesting that in wild-type mice, postnatal blood vessels respond differently to cleaved and uncleaved forms of VEGF. Perhaps the sprouting-initiation function of uncleaved VEGF is the only VEGF function required in embryos; however, this function of VEGF is unaffected in Mmp9-mutant embryos.
MMPs regulate tissue remodelling in flies
The results from mouse mutants indicate that during development individual MMPs are required specifically for tissue-remodelling events — bone remodelling, mammary development and vascular remodelling. Although the mouse double mutants that have been examined survive to birth7,8,56, it is not feasible to knockout all the MMPs in a mouse to test the general role of the MMP family. MMP expression and inhibition have been examined in many other model systems, including Xenopus laevis65,66, hydra67, zebrafish68–70 and Caenorhabditis elegans71,72; in C. elegans, genetic analysis indicates that a worm MMP contributes to a developmentally regulated cell-invasion event71. The fruitfly D. melanogaster has fewer MMPs than any other model system studied, with only two MMPs73,74, and mutants have been generated that lack both genes75. D. melanogaster MMP double mutants eliminate concerns about redundancy and compensation.
The D. melanogaster MMP genes, DmMmp1 and DmMmp2, maintain the conserved MMP domain structure, and DmMmp2 also encodes a predicted GPI membrane anchor73,74 (FIG. 1). Although neither fly gene has a single orthologue among the mammalian MMPs (despite the nomenclature), fly TIMP can inhibit mammalian MMPs76 and mammalian TIMPs can inhibit fly MMPs74, indicating conservation between the systems. Double mutants made from null alleles can complete embryonic development and can progress partway through the larval stages, a time of tremendous growth. Even when the maternal contribution of both MMP genes is removed from the double-mutant embryo, MMPs are not required for viability or morphogenesis until the middle of larval development75. Therefore, fly experiments confirm and extend the mammalian MMP-mutant results, which indicate that the MMP family specializes in remodelling, rather than in the regulation of the initial cell and tissue migrations that are required during embryogenesis. It is worth noting, however, that morpholino-inhibition experiments indicate that MMPs are essential for embryonic axis formation in zebrafish70.
Loss of DmMmp1 results in defects in tracheal remodeling
What are the fundamental roles of MMPs if an organism that lacks MMPs can complete embryo genesis? Although the single and double fly mutants can grow to the larval stages, their phenotypes indicate that even in insects, MMPs participate in tissue remodelling. DmMmp1 mutants are unable to remodel the breathing tubes known as tracheae. The tracheae are a system of branched interconnected tubes that allow the diffusion of oxygen to all internal tissues; during normal larval growth, tracheal tubes grow 14-fold in length and 7–40-fold in diameter77. In DmMmp1 mutants, as the larva grows, the tracheal tubes cannot grow concomitantly, they stretch inappropriately, and within a few days, they break from the stretching tension (FIG. 5a). These tubes also cannot dilate properly, and instead display inappropriate constrictions. At a cellular level, it seems that the epithelial cells that comprise the tubes cannot release their attachments from the ECM that lines the tubes75.
Figure 5. Phenotypes of Drosophila melanogaster DmMmp1 and DmMmp2 mutant flies.

a | Drosophila melanogaster tracheal development requires the dorsal tracheal trunks to extend with the growth of the larva. In DmMmp1-mutant flies, the tracheal trunks stretch tight and finally break, resulting in larval lethality. b | During D. melanogaster metamorphosis, larval tissue is histolysed and adult dorsal epithelium migrates to the midline and fuses. In DmMmp2 mutants, the midline closure of the epithelium fails. Images adapted from REF. 75 © (2003) Elsevier.
Recently, a new signalling protein that is likely to participate in the normal remodelling of the tracheal tubes was identified78. This signalling protein, Ninjurin A, physically associates with DmMMP1 and colocalizes with it at tracheal cell surfaces. In cell culture, the Ninjurin A ectodomain is shed by DmMMP1 protein, and the liberated ectodomain signals cells to release adhesion. It is likely that in larvae, DmMMP1 releases the Ninjurin A ectodomain, which signals a release of cell adhesion from the tracheal cuticle78. The Ninjurin-family proteins were first identified because of their upregulation in injured rat neurons. Therefore, it seems that this new MMP substrate, the ectodomain of which functions as a previously unsuspected cell–cell signal, is conserved between insects and mammals.
DmMmp2 is required for tissue histolysis
The requirement for DmMmp2 in D. melanogaster is reminiscent of the original MMP identification in metamorphic tadpole-tail histolysis. Like frogs, insect metamorphosis requires extensive tissue histolysis, and this histolysis requires an MMP: in the DmMmp2 mutants, tissues that are normally slated for histolysis persist inappropriately. DmMmp2 is also required for the fusion of two epithelial sheets that meet at the midline of the fly during metamorphosis, as the mutants display cleft thoraxes75 (FIG. 5b). Further work on identifying the cellular defect in these mutants might shed light on the roles of MMPs in other epithelial fusion events, such as palate formation, which goes awry in cleft palate birth defects.
That embryonic development can be successfully completed without MMP function does not preclude the possibility that MMPs normally participate in embryogenesis. Further analysis might elucidate specific MMP functions that are non-essential or that might be obscured by adaptive development. For example, DmMMP2 is expressed in the central nervous system during development, and during motor-neuron outgrowth, DmMmp2-mutant axons inappropriately lose adhesion to one another. These data indicate that MMPs cleave a substrate that is important for axon guidance (A.P.-M. and H. Broihier, unpublished results).
Whereas the study of mammalian MMPs began many decades ago, the study of fly MMPs is still new and the topics of research are still expanding. Recent studies in flies have focused on the role of fly MMPs in tumour metastasis, and DmMmp1 was found to contribute significantly to the invasiveness of tumours79,80. Additionally, the fruitfly offers opportunities for forward genetic screening as a method of identifying new substrates and pathways, methods not available in mammalian systems. Yet even in these early days, fly experiments have genetically identified a limited role for the MMP family in embryogenesis, they have highlighted a crucial role for MMPs in tissue remodelling and they have uncovered a new conserved substrate, the Ninjurin signalling protein.
MMPs in inflammation and wounding
The role of MMPs is not limited to developmental processes, and mouse-mutant experiments are beginning to show that MMPs are also required to maintain homeostasis in response to environmental challenges, such as wounding and infection. This topic has been reviewed recently81, but here we will discuss a few of the most important findings.
MMP7 is involved in innate immunity and wound healing
MMP7 (or matrilysin) has no known role in development and is not expressed at high levels in embryonic development. In healthy adult tissue, MMP7 is expressed only in mucosal epithelia and it is upregulated and proteolytically activated in response to bacterial exposure in epithelia82. Consistent with this expression pattern, Mmp7-mutant mice are more easily infected with intestinal bacteria. This susceptibility is partly caused by the inability of Mmp7 mutants to proteolytically activate an endogenous antibiotic peptide, pro-cryptdin. In vivo, the precursor, but not the mature forms, of cryptdin is found in samples of intestinal epithelia from Mmp7-mutant mice83. These experiments firmly establish a role for MMP7 in innate immunity.
MMP7 is also required for wound healing. Epithelial cells at the edges of wounded tracheal explants migrate towards each other and fuse. MMP7 seems to mediate wound-induced epithelial migration by cleaving E-cadherin84. MMP7 colocalizes with E-cadherin and cleaves its extracellular domain in wounded epithelia, which results in a loosening of cell–cell attachments. In wounded Mmp7-mutant mice, epithelial cells do not migrate and E-cadherin cleavage does not occur85. MMP3 also functions in epidermal wound healing, as skin wounds of Mmp3-mutant mice heal more slowly than those of control mice, owing to a deficit in actin purse-string formation86.
Pro-inflammatory and anti-inflammatory MMP functions
Both injury and infection induce inflammation, another physiological response to environmental challenge that requires MMPs. Lung epithelium controls the recruitment of inflammatory cells to alveoli, through the production of chemoattractants, following injury, infection or exposure to allergens or chemicals. Injury induces leukocytes to migrate across the epithelium into the alveolar air spaces in response to a chemoattractant gradient. The transmembrane heparan sulphate proteoglycan syndecan-1 sequesters many chemokines. The formation of the chemoattractant gradient requires MMP-mediated shedding of the syndecan-1 ectodomain. Subsequent chemokine release results in the migration of leukocytes into the air spaces and epithelial repair. In the absence of MMP7, neutrophils remain stuck outside the epithelia in injured lungs, and subsequent inflammation-mediated lung damage is attenuated32. Neutrophil migration is defective in Mmp7 mutants owing to the absence of the neutrophil attractant CXC-motif ligand-1 (CXCL1; also known as keratinocyte-derived chemokine (KC); or GROα in humans) in the fluid of the alveolar lumens. In summary, injury promotes MMP7 expression, MMP7 cleaves syndecan-1 and syndecan-1 shedding releases CXCL1, which recruits neutrophils.
MMPs can be both pro-inflammatory and anti-inflammatory. MMPs facilitate inflammatory cell recruitment87,88 and clearance of inflammatory cells89–91 by cleaving inflammatory mediators, resulting in a tightly regulated inflammatory response92. Several chemokines, including C–C motif ligand-7 (CCL7) and CXCL12 (REF. 92), are substrates for MMP2. MMP9 cleaves and activates CXCL6 and CXCL8 (also known as interleukin-8 (IL8)), whereas it inactivates CXCL1 and CXCL4 (REFS 93,94). In asthma models, eosinophils from Mmp2- or Mmp9-deficient mice fail to migrate into the airways and accumulate in the interstitium, thereby predisposing the animals to asphyxiation95,96. MMP2 and MMP9 participate in a regulatory loop that dampens allergic inflammation. The interstitial eosinophil accumulation might be explained by the disruption of transepithelial chemokine gradients, which affect CCL7 (also known as MCP3 or MARC), CCL11 (also known as eotaxin) and CCL17 (also known as TARC). MMP2 and MMP9 also process S100A8 and S100A9, two neutrophil- and macrophage-specific chemoattractant proteins that are found in the bronchioalveolar fluid of asthmatic mice97. These MMPs establish the chemotactic gradient that is required for clearance of lung inflammatory cells and the prevention of lethal asphyxiation. In a mouse model of the autoimmune skin blistering disease, bullous pemphigoid, Mmp9-null mice have decreased recruitment of neutrophils and fail to develop epidermal blistering. This defect is caused by a lack of inactivation of α1-proteinase inhibitor by MMP9, which does not allow neutrophil elastase to function98.
Truncation of macrophage-derived CCL7 by MMP2 and MMP14 results in the formation of peptides that can bind to the CC chemokine receptor and function as antagonists99–101. Limited N-terminal proteolytic processing of CXCL8 and the CXC chemokine LIX (the mouse equivalent of CXCL5 and CXCL6) by MMP9 and MMP8, respectively, results in the generation of chemoattractants that are more potent than the full-length molecules102,103. Accordingly, Mmp8-null animals104 are protected from tumour necrosis factor-α (TNFα)-induced lethal hepatitis because of impaired LIX release and impaired leukocyte influx into the liver103. A tripeptide neutrophil chemo-attractant N-acetyl Pro-Gly-Pro, derived from the breakdown of ECM, shares sequence and structural homology with an important domain on α-chemokines and causes chemotaxis through CXCR2 (REF. 105).
MMPs in human inflammatory disease models
Most human pathologies have an inflammatory component, as do their mouse models. The same MMP can have different roles in different conditions. For example, MMP9 contributes to inflammation in mouse models of stroke, heart attack, Alzheimer’s disease, some aspects of asthma and other lung inflammatory conditions, aortic aneurysms and autoimmune encephalomyelitis9,106–110; however, it functions as an anti-inflammatory agent in models of inflammatory skin and kidney diseases89,111,112. Current data indicate that MMP12 contributes to emphysema113,114, whereas MMP3 and MMP9 contribute to skin inflammatory conditions89,115. By contrast, MMP8 protects against skin inflammatory responses104 and MMP2 protects against inflammation of the brain and spinal cord116. Collectively, these observations indicate that MMPs function non-redundantly to regulate inflammation, which is potentially of great medical significance. Indeed, tumour progression also triggers inflammation, and the clinical observations that MMPs are upregulated in cancer5 might reflect the roles of MMPs in triggering and controlling inflammation.
Conclusions and future directions
The MMP family of extracellular proteinases is present in most multicellular organisms, including plants and animals13. MMP function can be most simply analysed in D. melanogaster, which has only two MMP genes. The fly mutants demonstrate that MMPs are dispensable, both individually and together, for embryonic fly development, but are crucial for tissue growth and tissue–ECM remodelling in the larvae and during larval development. In mammals, the 24 MMP genes seem to share redundant functions. Analysis of single-MMP-mutant mice (TABLE 1) has identified developmental phenotypes in postnatal mammary, skeletal and circulatory development, three prominent sites of postnatal tissue and ECM remodelling. A particularly striking aspect of these phenotypes is that MMPs are not required to build blood vessels or bones in the embryo, but are required for their postnatal development and tissue remodelling. Examination of mouse mutants also reveals an important function of MMPs in regulating tissue response to environmental challenges, such as wounding, infection and inflammation. The genetic analysis from humans and model systems argues for a crucial role for MMPs as active regulators of post-natal tissue development, tissue remodelling and tissue repair in response to injury, infection or disease.
Current efforts to delete the remaining mouse MMPs and continuing analyses of existing mutants will reveal how MMPs are deployed in development and disease. Further studies with compound knockouts might yield lethal phenotypes, thereby revealing essential roles in development. Complementing the analysis of compound mouse mutants will be experiments that use pharmacological manipulations of both individual and classes of MMPs, as well as analyses in other genetically tractable model systems. Biochemical challenges include the visualization of MMP activity in living cells and tissues, the development of non-cleavable substrates and the determination of the complete in vitro MMP degradome117. An important question is the extent to which MMPs have a structural role in remodelling tissues versus a role in regulating access to signalling molecules.
Another challenge is to determine the relative importance of extracellular proteolysis in general, and MMP-mediated proteolysis in particular, as a post-translational regulatory mechanism in development and disease. Which stages in normal development, which cellular processes and which signalling pathways are strongly influenced by MMPs? A key to answering these questions will be to increase our understanding of the relevant in vivo substrates for specific MMPs. The challenge is to link our in vitro understanding of potential functions of MMPs with an in vivo understanding of how MMPs function in a given cellular process. Joining together biochemical approaches and MMP genetics with mutant substrates should make it possible to make progress on this important problem.
Conceptually, one of the main advances in MMP biology has been the realization that extracellular proteolysis need not simply be a mechanism of destroying structure or information. Instead, diverse studies demonstrate that MMPs can release growth factors from the ECM and cell surface, activating latent proteins and generating new bioactive molecules through proteolysis (BOX 2). We view extracellular proteolysis as another form of post-translational modification. From this perspective, proteolysis can function to destroy a protein, but it can equally provide a mechanism to grant cells conditional access to a particular signalling molecule. Significantly, the products of MMP cleavage can function locally (for example, cleavage of E-cadherin) or at a long distance from the site of proteolysis (for example, chemokine activation). Given that many MMP phenotypes have been observed in the remodelling of tissues that do not require MMPs for their initial formation, it is tempting to speculate that MMPs might function specifically as regulators of post-embryonic cell motility and tissue architecture.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (NIH) to Z.W. and to A.P.-M and a March of Dimes Basil O’Connor award to A.P.-M. A.J.E. was supported by an NIH National Research Service Award Institutional Fellowship and by the California Breast Cancer Research Program. We apologize to the many scientists whose papers we were unable to cite owing to space constraints.
- Fibrillar collagen
Polymerized, supramolecular collagen that has been organized into fibrils
- collagen types I
II and III form fibrils
- Extracellular matrix
Complex, ordered mixture of structural and signalling molecules that surrounds cells
- Enzymatic redundancy
Two enzymes that are expressed in the same time and place that can fully substitute for each other’s essential functions
- Enzymatic compensation
Upregulation of an enzyme, which is normally not expressed (or is expressed at a low level) to substitute for the absence of a mutated enzyme
- Adaptive development
Alternative developmental trajectory whereby an organism compensates for the loss of a gene by doing some essential function in a reproducibly different manner
- Catalytic domain
A domain that cleaves other proteins
- Pro-domain
An autoinhibitory domain that prevents the catalytic domain from functioning
- Hemopexin domain
A four-bladed β-propeller domain that mediates protein–protein interactions
- Tissue inhibitor of metalloproteinases
(TIMPs). Endogenous protein inhibitors of matrix metalloproteinase function
- Intramembranous ossification
Direct differentiation of mesenchymal precursors into osteoblasts, as in the clavicle and in some skull bones
- Endochondral ossification
Bone development in which a cartilage template forms first and is then replaced by mineralized bone in the growth plate, as in the long bones
- Pseudo-metamorphic ossification
Bone development in which a cartilage template functions as a temporary mould to shape the deposition of bone, as in the mandible
- Osteoblast
A cell that secretes unmineralized type-I-collagen-rich extracellular matrix and builds bone
- Growth plate
The region of developing appendicular and axial skeleton that is responsible for growth in length of bones
- Aggrecan
The main chondroitin sulphate proteoglycan in cartilage
- Trabeculae
A fine network of bony spicules in trabecular bone
- Osteoclast
A cell that resorbs the mineralized matrix of the bone
- Terminal end bud
A highly proliferative epithelial structure at the end of invading mammary ductal epithelium during puberty
- Secondary branching
Also known as side branching. A process during mammary development whereby secondary ducts initiate laterally off of main ducts during puberty
- Corneal angiogenesis assay
Stimulated neovascularization assay in which test substances are implanted in the cornea of a rabbit or mouse and the cornea is monitored for the development of new blood vessels
- Perivascular cells
Also known as pericytes, mural cells or smooth muscle cells. These cells are tightly associated with endothelial cells and aid in the function and regulation of vascular networks
- Endothelial cells
Cells that constitute the lining of the blood vessels
- MMP degradome
The complete list of matrix metalloproteinase proteolytic substrates
Footnotes
Competing interests statement
The authors declare no competing financial interests.
DATABASES
The following terms in this article are linked online to:
Entrez Genome: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
DmMmp1 | DmMmp2
UniProtKB: http://ca.expasy.org/sprot
MMP1 | MMP2 | MMP3 | MMP9 | MMP14 | MMP15 | MMP16 | MMP17 | MMP23 | MMP24 | MMP25
FURTHER INFORMATION
Andrea Page-McCaw's homepage: www.rpi.edu/research/biotech/researchers/page-mccaw.html
Zena Werb's homepage: http://anatomy.ucsf.edu/Pages/werb.html
International Proteolysis Society: hhttp://www.protease.org
MEROPS: http://merops.sanger.ac.uk
Contributor Information
Andrea Page-McCaw, Email: pagema@rpi.edu.
Andrew J. Ewald, Email: andrew.ewald@ucsf.edu.
Zena Werb, Email: zena.werb@ucsf.edu.
References
- 1.Gross J, Lapiere CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci USA. 1962;47:1014–1022. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nature Rev Mol Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 3.Birkedal-Hansen H, et al. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- 4.Welgus HG, Kobayashi DK, Jeffrey JJ. The collagen substrate specificity of rat uterus collagenase. J Biol Chem. 1983;258:14162–14165. [PubMed] [Google Scholar]
- 5.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 6.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. A critical analysis of why MMP inhibitors failed as cancer therapeutics, despite great promise. [DOI] [PubMed] [Google Scholar]
- 7.Stickens D, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh J, et al. Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene. 2004;23:5041–5048. doi: 10.1038/sj.onc.1207688. [DOI] [PubMed] [Google Scholar]
- 9.Ducharme A, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudolph-Owen LA, Hulboy DL, Wilson CL, Mudgett J, Matrisian LM. Coordinate expression of matrix metalloproteinase family members in the uterus of normal, matrilysin-deficient, and stromelysin-1-deficient mice. Endocrinology. 1997;138:4902–4911. doi: 10.1210/endo.138.11.5478. [DOI] [PubMed] [Google Scholar]
- 11.Bode W, Gomis-Ruth FX, Stockler W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 1993;331:134–140. doi: 10.1016/0014-5793(93)80312-i. [DOI] [PubMed] [Google Scholar]
- 12.Stocker W, et al. The metzincins--topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Science. 1995;4:823–840. doi: 10.1002/pro.5560040502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rawlings ND, Morton FR, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2006;34:D270–D272. doi: 10.1093/nar/gkj089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Overall CM. Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol Biotechnol. 2002;22:51–86. doi: 10.1385/MB:22:1:051. [DOI] [PubMed] [Google Scholar]
- 15.Maskos K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie. 2005;87:249–263. doi: 10.1016/j.biochi.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Streuli C. Extracellular matrix remodelling and cellular differentiation. Curr Opin Cell Biol. 1999;11:634–640. doi: 10.1016/s0955-0674(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 18.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. Demonstration that cleavage of an ECM protein reveals a cryptic function, in this case chemokinesis. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, et al. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154:1069–1080. doi: 10.1083/jcb.200103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pilcher BK, et al. The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J Cell Biol. 1997;137:1445–1457. doi: 10.1083/jcb.137.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fowlkes JL, Thrailkill KM, Serra DM, Suzuki K, Nagase H. Matrix metalloproteinases as insulin-like growth factor binding protein-degrading proteinases. Prog Growth Factor Res. 1995;6:255–263. doi: 10.1016/0955-2235(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 22.Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem. 1996;271:10079–10086. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Juttermann R, Soloway PD. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem. 2000;275:26411–26415. doi: 10.1074/jbc.M001270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo D, Mari B, Stoll I, Anglard P. Alternative splicing and promoter usage generates an intracellular stromelysin 3 isoform directly translated as an active matrix metalloproteinase. J Biol Chem. 2002;277:25527–25536. doi: 10.1074/jbc.M202494200. [DOI] [PubMed] [Google Scholar]
- 25.Peschon JJ, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki M, Raab G, Moses MA, Fernandez CA, Klagsbrun M. Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J Biol Chem. 1997;272:31730–31737. doi: 10.1074/jbc.272.50.31730. [DOI] [PubMed] [Google Scholar]
- 27.Montero JC, Yuste L, Diaz-Rodriguez E, Esparis-Ogando A, Pandiella A. Differential shedding of transmembrane neuregulin isoforms by the tumor necrosis factor-α-converting enzyme. Mol Cell Neurosci. 2000;16:631–648. doi: 10.1006/mcne.2000.0896. [DOI] [PubMed] [Google Scholar]
- 28.Sternlicht MD, et al. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noe V, et al. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 2001;114:111–118. doi: 10.1242/jcs.114.1.111. [DOI] [PubMed] [Google Scholar]
- 30.Kajita M, et al. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. 2001;153:893–904. doi: 10.1083/jcb.153.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Endo K, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278:40764–40770. doi: 10.1074/jbc.M306736200. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 33.Senft AP, Korfhagen TR, Whitsett JA, Shapiro SD, LeVine AM. Surfactant protein-D regulates soluble CD14 through matrix metalloproteinase-12. J Immunol. 2005;174:4953–4959. doi: 10.4049/jimmunol.174.8.4953. [DOI] [PubMed] [Google Scholar]
- 34.Mohan R, et al. Matrix metalloproteinase gelatinase B (MMP-9) coordinates and effects epithelial regeneration. J Biol Chem. 2002;277:2065–2072. doi: 10.1074/jbc.M107611200. [DOI] [PubMed] [Google Scholar]
- 35.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 36.Vu TH, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. First MMP-null mutant with a tissue-remodelling phenotype and defects in development and angiogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colnot C, Sidhu SS, Balmain N, Poirier F. Uncoupling of chondrocyte death and vascular invasion in mouse galectin 3 null mutant bones. Dev Biol. 2001;229:203–214. doi: 10.1006/dbio.2000.9933. [DOI] [PubMed] [Google Scholar]
- 38.Ortega N, Behonick DJ, Colnot C, Cooper DN, Werb Z. Galectin-3 is a downstream regulator of matrix metalloproteinase-9 function during endochondral bone formation. Mol Biol Cell. 2005;16:3028–3039. doi: 10.1091/mbc.E04-12-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–4133. doi: 10.1242/dev.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inada M, et al. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci USA. 2004;101:17192–17197. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Little CB, et al. Matrix metalloproteinases are not essential for aggrecan turnover during normal skeletal growth and development. Mol Cell Biol. 2005;25:3388–3399. doi: 10.1128/MCB.25.8.3388-3399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmbeck K, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. Demonstration that loss of an MMP decreases ECM remodelling with the surprising consequence of increased bone loss and arthritis; previously these phenotypes were thought to be the consequence of excess proteolysis. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Z, et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohuchi E, et al. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997;272:2446–2451. doi: 10.1074/jbc.272.4.2446. [DOI] [PubMed] [Google Scholar]
- 45.Holmbeck K, Bianco P, Chrysovergis K, Yamada S, Birkedal-Hansen H. MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: a critical process in skeletal growth. J Cell Biol. 2003;163:661–671. doi: 10.1083/jcb.200307061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oblander SA, et al. Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev Biol. 2005;277:255–269. doi: 10.1016/j.ydbio.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 47.Martignetti JA, et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nature Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- 48.Kennedy AM, et al. MMP13 mutation causes spondyloepimetaphyseal dysplasia, Missouri type 115, 2832–2842 (SEMDMO) . J Clin Invest. 2005 doi: 10.1172/JCI22900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JW, et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005;42:271–275. doi: 10.1136/jmg.2004.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoue K, et al. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 2006;281:33814–33824. doi: 10.1074/jbc.M607290200. [DOI] [PubMed] [Google Scholar]
- 51.Strongin AY, et al. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 52.Sato H, et al. A matrix metalloproteinase expressed on the surface of invasive tumor cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. Discovery of the first transmembrane MMP, MMP14 (MT1-MMP). This discovery changed the concept of pericellular proteolysis. [DOI] [PubMed] [Google Scholar]
- 53.Sternlicht MD, Kouros-Mehr H, Lu P, Werb Z. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74:365–381. doi: 10.1111/j.1432-0436.2006.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiseman BS, et al. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162:1123–1133. doi: 10.1083/jcb.200302090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kato T, et al. Diminished corneal angiogenesis in gelatinase A-deficient mice. FEBS Lett. 2001;508:187–190. doi: 10.1016/s0014-5793(01)02897-6. [DOI] [PubMed] [Google Scholar]
- 56.Lambert V, et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17:2290–2292. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- 57.Chun TH, et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167:757–767. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Filippov S, et al. MT1-matrix metalloproteinase directs arterial wall invasion and neointima formation by vascular smooth muscle cells. J Exp Med. 2005;202:663–671. doi: 10.1084/jem.20050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chantrain CF, et al. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 2004;64:1675–1686. doi: 10.1158/0008-5472.can-03-0160. [DOI] [PubMed] [Google Scholar]
- 60.Lehti K, et al. An MT1-MMP–PDGF receptor-β axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19:979–991. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–1326. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergers G, Hanahan D, Coussens LM. Angiogenesis and apoptosis are cellular parameters of neoplastic progression in transgenic mouse models of tumorigenesis. Int J Dev Biol. 1998;42:995–1002. Discovery that MMP-regulated bioavailability of VEGF regulated the angiogenic switch early in tumour progression. [PubMed] [Google Scholar]
- 63.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169:681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hasebe T, Hartman R, Fu L, Amano T, Shi YB. Evidence for a cooperative role of gelatinase A and membrane type-1 matrix metalloproteinase during Xenopus laevis development. Mech Dev. 2007;124:11–22. doi: 10.1016/j.mod.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harrison M, et al. Matrix metalloproteinase genes in Xenopus development. Dev Dyn. 2004;231:214–220. doi: 10.1002/dvdy.20113. [DOI] [PubMed] [Google Scholar]
- 67.Leontovich AA, Zhang J, Shimokawa K, Nagase H, Sarras MP., Jr A novel hydra matrix metalloproteinase (HMMP) functions in extracellular matrix degradation, morphogenesis and the maintenance of differentiated cells in the foot process. Development. 2000;127:907–920. doi: 10.1242/dev.127.4.907. [DOI] [PubMed] [Google Scholar]
- 68.Yoong S, et al. Characterization of the zebrafish matrix metalloproteinase 9 gene and its developmental expression pattern. Gene Expr Patterns. 2007;7:39–46. doi: 10.1016/j.modgep.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 69.Bai S, et al. Matrix metalloproteinase expression and function during fin regeneration in zebrafish: analysis of MT1-MMP, MMP2 and TIMP2. Matrix Biol. 2005;24:247–260. doi: 10.1016/j.matbio.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 70.Zhang J, Bai S, Zhang X, Nagase H, Sarras MP., Jr The expression of novel membrane-type matrix metalloproteinase isoforms is required for normal development of zebrafish embryos. Matrix Biol. 2003;22:279–293. doi: 10.1016/s0945-053x(03)00020-9. [DOI] [PubMed] [Google Scholar]
- 71.Sherwood DR, Butler JA, Kramer JM, Sternberg PW. FOS-1 promotes basement-membrane removal during anchor-cell invasion in C. elegans. Cell. 2005;121:951–962. doi: 10.1016/j.cell.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 72.Wada K, et al. Cloning of three Caenorhabditis elegans genes potentially encoding novel matrix metalloproteinases. Gene. 1998;211:57–62. doi: 10.1016/s0378-1119(98)00076-6. [DOI] [PubMed] [Google Scholar]
- 73.Llano E, et al. Structural and enzymatic characterization of Drosophila Dm2-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. J Biol Chem. 2002;277:23321–23329. doi: 10.1074/jbc.M200121200. [DOI] [PubMed] [Google Scholar]
- 74.Llano E, Pendas AM, Aza-Blanc P, Kornberg TB, Lopez-Otin C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J Biol Chem. 2000;275:35978–35985. doi: 10.1074/jbc.M006045200. [DOI] [PubMed] [Google Scholar]
- 75.Page-McCaw A, Serano J, Sante JM, Rubin GM. Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev Cell. 2003;4:95–106. doi: 10.1016/s1534-5807(02)00400-8. A clear demonstration of the important role of MMPs in tissue remodelling. [DOI] [PubMed] [Google Scholar]
- 76.Wei S, Xie Z, Filenova E, Brew K. Drosophila TIMP is a potent inhibitor of MMPs and TACE: similarities in structure and function to TIMP-3. Biochemistry. 2003;42:12200–12207. doi: 10.1021/bi035358x. [DOI] [PubMed] [Google Scholar]
- 77.Beitel GJ, Krasnow MA. Genetic control of epithelial tube size in the Drosophila tracheal system. Development. 2000;127:3271–3282. doi: 10.1242/dev.127.15.3271. [DOI] [PubMed] [Google Scholar]
- 78.Zhang S, et al. An MMP liberates the Ninjurin A ectodomain to signal a loss of cell adhesion. Genes Dev. 2006;20:1899–1910. doi: 10.1101/gad.1426906. Demonstration that MMP cleavage regulates adhesion and signalling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uhlirova M, Bohmann D. JNK- and Fos-regulated MMP1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006;25:5294–5304. doi: 10.1038/sj.emboj.7601401. Following loss of MMPs, mutant tissue overproliferates, resists apoptosis, leaves its site of origin and invades other organs, ultimately causing lethality. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beaucher M, Herspberger E, Page-McCaw A, Shearn A. Metastatic ability of Drosophila tumors depends on MMP activity. Dev Biol. 2006 Dec 5; doi: 10.1016/j.ydbio.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 81.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature Rev Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 82.Lopez-Boado YS, et al. Bacterial exposure induces and activates matrilysin in mucosal epithelial cells. J Cell Biol. 2000;148:1305–1315. doi: 10.1083/jcb.148.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilson CL, et al. Regulation of intestinal-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 84.McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol. 2003;162:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dunsmore SE, et al. Matrilysin expression and function in airway epithelium. J Clin Invest. 1998;102:1321–1331. doi: 10.1172/JCI1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bullard KM, et al. Impaired wound contraction in stromelysin-1-deficient mice. Ann Surg. 1999;230:260–265. doi: 10.1097/00000658-199908000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haas TL, Madri JA. Extracellular matrix-driven matrix metalloproteinase production in endothelial cells: implications for angiogenesis. Trends Cardiovasc Med. 1999;9:70–77. doi: 10.1016/s1050-1738(99)00014-6. [DOI] [PubMed] [Google Scholar]
- 88.Haro H, et al. Matrix metalloproteinase-3-dependent generation of a macrophage chemoattractant in a model of herniated disc resorption. J Clin Invest. 2000;105:133–141. doi: 10.1172/JCI7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang M, et al. Matrix metalloproteinase deficiencies affect contact hypersensitivity: stromelysin-1 deficiency prevents the response and gelatinase B deficiency prolongs the response. Proc Natl Acad Sci USA. 1999;96:6885–6889. doi: 10.1073/pnas.96.12.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zheng T, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kumagai K, et al. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J Immunol. 1999;162:4212–4219. [PubMed] [Google Scholar]
- 92.Overall CM, McQuibban GA, Clark-Lewis I. Discovery of chemokine substrates for matrix metalloproteinases by exosite scanning: a new tool for degradomics. Biol Chem. 2002;383:1059–1066. doi: 10.1515/BC.2002.114. [DOI] [PubMed] [Google Scholar]
- 93.Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood. 2000;96:2673–2681. [PubMed] [Google Scholar]
- 94.D’Haese A, et al. In vivo neutrophil recruitment by granulocyte chemotactic protein-2 is assisted by gelatinase B/MMP-9 in the mouse. J Interferon Cytokine Res. 2000;20:667–674. doi: 10.1089/107999000414853. [DOI] [PubMed] [Google Scholar]
- 95.Corry DB, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nature Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Corry DB, et al. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Greenlee KJ, et al. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol. 2006;177:7312–7321. doi: 10.4049/jimmunol.177.10.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu Z, et al. The serpin α1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 99.McQuibban GA, et al. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. First demonstration that a main role of MMPs is to regulate chemokine activity and therefore the chemoattraction of immune cells. [DOI] [PubMed] [Google Scholar]
- 100.McQuibban GA, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276:43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 101.McQuibban GA, et al. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- 102.Van Den Steen PE, et al. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur J Biochem. 2003;270:3739–3749. doi: 10.1046/j.1432-1033.2003.03760.x. [DOI] [PubMed] [Google Scholar]
- 103.Van Lint P, et al. Resistance of collagenase-2 (matrix metalloproteinase-8)-deficient mice to TNF-induced lethal hepatitis. J Immunol. 2005;175:7642–7649. doi: 10.4049/jimmunol.175.11.7642. [DOI] [PubMed] [Google Scholar]
- 104.Balbin M, et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nature Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 105.Weathington NM, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nature Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 106.Asahi M, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee MM, et al. Tissue inhibitor of metalloproteinase 1 regulates resistance to infection. Infect Immun. 2005;73:661–665. doi: 10.1128/IAI.73.1.661-665.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vermaelen KY, et al. Matrix metalloproteinase-9-mediated dendritic cell recruitment into the airways is a critical step in a mouse model of asthma. J Immunol. 2003;171:1016–1022. doi: 10.4049/jimmunol.171.2.1016. [DOI] [PubMed] [Google Scholar]
- 109.Lanone S, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and-12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;110:463–474. doi: 10.1172/JCI14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pyo R, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lelongt B, et al. Matrix metalloproteinase 9 protects mice from anti-glomerular basement membrane nephritis through its fibrinolytic activity. J Exp Med. 2001;193:793–802. doi: 10.1084/jem.193.7.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McMillan SJ, et al. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol. 2004;172:2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- 113.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 114.Morris DG, et al. Loss of integrin α(v)β6-mediated TGF-β activation causes Mmp12-dependent emphysema. Nature. 2003;422:169–173. doi: 10.1038/nature01413. [DOI] [PubMed] [Google Scholar]
- 115.Liu Z, et al. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest. 2005;115:879–887. doi: 10.1172/JCI23977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Esparza J, Kruse M, Lee J, Michaud M, Madri JA. MMP-2 null mice exhibit an early onset and severe experimental autoimmune encephalomyelitis due to an increase in MMP-9 expression and activity. FASEB J. 2004;18:1682–1691. doi: 10.1096/fj.04-2445com. [DOI] [PubMed] [Google Scholar]
- 117.Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nature Rev Mol Cell Biol. 2002;3:509–519. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 118.Sottrup-Jensen L, Birkedal-Hansen H. Human fibroblast collagenase-α-macroglobulin interactions. Localization of cleavage sites in the bait regions of five mammalian α-macroglobulins. J Biol Chem. 1989;264:393–401. [PubMed] [Google Scholar]
- 119.Chase AJ, Newby AC. Regulation of matrix metalloproteinase (matrixin) genes in blood vessels: a multi-step recruitment model for pathological remodelling. J Vasc Res. 2003;40:329–343. doi: 10.1159/000072697. [DOI] [PubMed] [Google Scholar]
- 120.Leeman MF, Curran S, Murray GI. The structure, regulation, and function of human matrix metalloproteinase-13. Crit Rev Biochem Mol Biol. 2002;37:149–166. doi: 10.1080/10409230290771483. [DOI] [PubMed] [Google Scholar]
- 121.Ortega N, Werb Z. New functional roles for non-collagenous domains of basement membrane collagens. J Cell Sci. 2002;115:4201–4214. doi: 10.1242/jcs.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–3979. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 123.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nature Rev Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 124.Sund M, et al. Function of endogenous inhibitors of angiogenesis as endothelium-specific tumor suppressors. Proc Natl Acad Sci USA. 2005;102:2934–2939. doi: 10.1073/pnas.0500180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Larrain J, et al. BMP-binding modules in chordin: a model for signalling regulation in the extracellular space. Development. 2000;127:821–830. doi: 10.1242/dev.127.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Robertis EM. Spemann’s organizer and self-regulation in amphibian embryos. Nature Rev Mol Cell Biol. 2006;7:296–302. doi: 10.1038/nrm1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hamano Y, et al. Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via αV β3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. Shows the molecular mechanisms that underlie the function of new bioactive molecules generated from extracellular matrix by MMPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Houghton AM, et al. Macrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res. 2006;66:6149–6155. doi: 10.1158/0008-5472.CAN-04-0297. [DOI] [PubMed] [Google Scholar]
- 129.Fjeldstad K, Kolset SO. Decreasing the metastatic potential in cancers--targeting the heparan sulfate proteoglycans. Curr Drug Targets. 2005;6:665–682. doi: 10.2174/1389450054863662. [DOI] [PubMed] [Google Scholar]
- 130.Gu Z, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Trackman PC. Diverse biological functions of extracellular collagen processing enzymes. J Cell Biochem. 2005;96:927–937. doi: 10.1002/jcb.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fukui N, et al. Processing of type II procollagen amino propeptide by matrix metalloproteinases. J Biol Chem. 2002;277:2193–2201. doi: 10.1074/jbc.M105485200. [DOI] [PubMed] [Google Scholar]
- 133.Dusek RL, et al. The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J Biol Chem. 2006;281:3614–3624. doi: 10.1074/jbc.M508258200. [DOI] [PubMed] [Google Scholar]
- 134.Illman SA, Lehti K, Keski-Oja J, Lohi J. Epilysin (MMP-28) induces TGF-β mediated epithelial to mesenchymal transition in lung carcinoma cells. J Cell Sci. 2006;119:3856–3865. doi: 10.1242/jcs.03157. [DOI] [PubMed] [Google Scholar]
- 135.Radisky DC, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. Works out the molecular mechanisms downstream of MMP cleavage that lead to tumour development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Duong TD, Erickson CA. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn. 2004;229:42–53. doi: 10.1002/dvdy.10465. [DOI] [PubMed] [Google Scholar]
- 137.Song W, Jackson K, McGuire PG. Degradation of type IV collagen by matrix metalloproteinases is an important step in the epithelial-mesenchymal transformation of the endocardial cushions. Dev Biol. 2000;227:606–617. doi: 10.1006/dbio.2000.9919. [DOI] [PubMed] [Google Scholar]
- 138.Sternlicht MD, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. Demonstrates that MMPs not only facilitate cell invasion, but can also initiate tumour progression directly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Takahashi C, et al. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad Sci USA. 1998;95:13221–13226. doi: 10.1073/pnas.95.22.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fernandez-Patron C, Radomski MW, Davidge ST. Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor. Circ Res. 1999;85:906–911. doi: 10.1161/01.res.85.10.906. [DOI] [PubMed] [Google Scholar]
- 141.Xu J, Park PW, Kheradmand F, Corry DB. Endogenous attenuation of allergic lung inflammation by syndecan-1. J Immunol. 2005;174:5758–5765. doi: 10.4049/jimmunol.174.9.5758. [DOI] [PubMed] [Google Scholar]
- 142.Hinkle CL, et al. Multiple metalloproteinases process protransforming growth factor-α (proTGF-α) Biochemistry. 2003;42:2127–2136. doi: 10.1021/bi026709v. [DOI] [PubMed] [Google Scholar]
- 143.Sahin U, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ii M, Yamamoto H, Adachi Y, Maruyama Y, Shinomura Y. Role of matrix metalloproteinase-7 (matrilysin) in human cancer invasion, apoptosis, growth, and angiogenesis. Exp Biol Med. 2006;231:20–27. doi: 10.1177/153537020623100103. [DOI] [PubMed] [Google Scholar]
- 145.Zhang K, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nature Neurosci. 2003;6:1064–1071. doi: 10.1038/nn1127. Shows that MMPs regulate survival factors and excess activity can lead to cell death. [DOI] [PubMed] [Google Scholar]
- 146.Mizgerd JP, Spieker MR, Doerschuk CM. Early response cytokines and innate immunity: essential roles for TNF receptor 1 and type I IL-1 receptor during Escherichia coli pneumonia in mice. J Immunol. 2001;166:4042–4048. doi: 10.4049/jimmunol.166.6.4042. [DOI] [PubMed] [Google Scholar]
- 147.Xu H, Uysal KT, Becherer JD, Arner P, Hotamisligil GS. Altered tumor necrosis factor-α (TNF-α) processing in adipocytes and increased expression of transmembrane TNF-α in obesity. Diabetes. 2002;51:1876–1883. doi: 10.2337/diabetes.51.6.1876. [DOI] [PubMed] [Google Scholar]
- 148.Chun TH, et al. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125:577–591. doi: 10.1016/j.cell.2006.02.050. This study shows that MMPs contribute to the architectural context of cells and argues that this might be necessary for the conversion of pre-adipocytes to adipocytes. [DOI] [PubMed] [Google Scholar]
- 149.Alexander CM, Selvarajan S, Mudgett J, Werb Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. J Cell Biol. 2001;152:693–703. doi: 10.1083/jcb.152.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gerber HP, Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000;10:223–228. doi: 10.1016/s1050-1738(00)00074-8. [DOI] [PubMed] [Google Scholar]
- 151.Harper J, Klagsbrun M. Cartilage to bone — angiogenesis leads the way. Nature Med. 1999;5:617–618. doi: 10.1038/9460. [DOI] [PubMed] [Google Scholar]
- 152.Itoh T, et al. Unaltered secretion of β-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- 153.Kheradmand F, Rishi K, Werb Z. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J Cell Sci. 2002;115:839–848. doi: 10.1242/jcs.115.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.VanSaun M, Herrera AA, Werle MJ. Structural alterations at the neuromuscular junctions of matrix metalloproteinase 3 null mutant mice. J Neurocytol. 2003;32:1129–1142. doi: 10.1023/B:NEUR.0000021907.68461.9c. [DOI] [PubMed] [Google Scholar]
- 155.Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NG2 proteoglycan. J Neurosci. 2003;23:11127–11135. doi: 10.1523/JNEUROSCI.23-35-11127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Galis ZS, et al. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ Res. 2002;91:852–859. doi: 10.1161/01.res.0000041036.86977.14. [DOI] [PubMed] [Google Scholar]
- 157.Masson R, et al. In vivo evidence that the stromelysin-3 metalloproteinase contributes in a paracrine manner to epithelial cell malignancy. J Cell Biol. 1998;140:1535–1541. doi: 10.1083/jcb.140.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wells JE, et al. An adverse role for matrix metalloproteinase 12 after spinal cord injury in mice. J Neurosci. 2003;23:10107–10115. doi: 10.1523/JNEUROSCI.23-31-10107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Uchinami H, Seki E, Brenner DA, D’Armiento J. Loss of MMP 13 attenuates murine hepatic injury and fibrosis during cholestasis. Hepatology. 2006;44:420–429. doi: 10.1002/hep.21268. [DOI] [PubMed] [Google Scholar]
- 160.Deguchi JO, et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 161.Holmbeck K, et al. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J Cell Sci. 2005;118:147–156. doi: 10.1242/jcs.01581. [DOI] [PubMed] [Google Scholar]
- 162.Beertsen W, et al. On the role of MT1-MMP, a matrix metalloproteinase essential to collagen remodeling, in murine molar eruption and root growth. Eur J Oral Sci. 2002;110:445–451. doi: 10.1034/j.1600-0722.2002.21384.x. [DOI] [PubMed] [Google Scholar]
- 163.Atkinson JJ, et al. Membrane-type 1 matrix metalloproteinase is required for normal alveolar development. Dev Dyn. 2005;232:1079–1090. doi: 10.1002/dvdy.20267. [DOI] [PubMed] [Google Scholar]
- 164.Pendas AM, et al. Diet-induced obesity and reduced skin cancer susceptibility in matrix metalloproteinase 19-deficient mice. Mol Cell Biol. 2004;24:5304–5313. doi: 10.1128/MCB.24.12.5304-5313.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Caterina JJ, et al. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2002;277:49598–49604. doi: 10.1074/jbc.M209100200. [DOI] [PubMed] [Google Scholar]
- 166.Komori K, et al. Absence of mechanical allodynia and Aβ-fiber sprouting after sciatic nerve injury in mice lacking membrane-type 5 matrix metalloproteinase. FEBS Lett. 2004;557:125–128. doi: 10.1016/s0014-5793(03)01458-3. [DOI] [PubMed] [Google Scholar]
- 167.Zhou HE, Zhang X, Nothnick WB. Disruption of the TIMP-1 gene product is associated with accelerated endometrial gland formation during early postnatal uterine development. Biol Reprod. 2004;71:534–539. doi: 10.1095/biolreprod.104.029181. [DOI] [PubMed] [Google Scholar]
- 168.Chaillan FA, et al. Involvement of tissue inhibition of metalloproteinases-1 in learning and memory in mice. Behav Brain Res. 2006;173:191–198. doi: 10.1016/j.bbr.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mohammed FF, et al. Metalloproteinase inhibitor TIMP-1 affects hepatocyte cell cycle via HGF activation in murine liver regeneration. Hepatology. 2005;41:857–867. doi: 10.1002/hep.20618. [DOI] [PubMed] [Google Scholar]
- 170.Jaworski DM, Soloway P, Caterina J, Falls WA. Tissue inhibitor of metalloproteinase-2 (TIMP-2)-deficient mice display motor deficits. J Neurobiol. 2006;66:82–94. doi: 10.1002/neu.20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fata JE, et al. Accelerated apoptosis in the TIMP-3-deficient mammary gland. J Clin Invest. 2001;108:831–841. doi: 10.1172/JCI13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gill SE, Pape MC, Khokha R, Watson AJ, Leco KJ. A null mutation for tissue inhibitor of metalloproteinases-3 (TIMP-3) impairs murine bronchiole branching morphogenesis. Dev Biol. 2003;261:313–323. doi: 10.1016/s0012-1606(03)00318-x. [DOI] [PubMed] [Google Scholar]
- 173.Cruz-Munoz W, et al. Enhanced metastatic dissemination to multiple organs by melanoma and lymphoma cells in Timp-3−/− mice. Oncogene. 2006;25:6489–6496. doi: 10.1038/sj.onc.1209663. [DOI] [PubMed] [Google Scholar]
- 174.English JL, et al. Individual Timp deficiencies differentially impact pro-MMP-2 activation. J Biol Chem. 2006;281:10337–10346. doi: 10.1074/jbc.M512009200. [DOI] [PubMed] [Google Scholar]
- 175.Godenschwege TA, Pohar N, Buchner S, Buchner E. Inflated wings, tissue autolysis and early death in tissue inhibitor of metalloproteinases mutants of Drosophila. Eur J Cell Biol. 2000;79:495–501. doi: 10.1078/0171-9335-00072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.