

Abstract
A series of 4-substituted methoxylbenzoyl-aryl-thiazoles (SMART) have been discovered and synthesized as a result of structural modifications of the lead compound 2-arylthiazolidine-4-carboxylic acid amides (ATCAA). The antiproliferative activity of the SMART agents against melanoma and prostate cancer cells was improved from μM to low nM range compared with ATCAA series. The structure-activity relationship was discussed from modifications of “A”, “B” “C” rings and the linker. Preliminary mechanism of action studies indicated that these compounds exert their anticancer activity through inhibition of tubulin polymerization.
Keywords: Thiazolidine, Thiazole, Melanoma, Prostate cancer, Antiproliferative activity, Structure-activity relationship, X-ray Crystal structure, Tubulin polymerization inhibitor
Introduction
In 2008, about 565,650 Americans are expected to die of cancer, more than 1,500 people a day. Cancer is the second most common cause of death in the US, exceeded only by heart disease. In the US, cancer accounts for 1 of every 4 deaths. The 5-year relative survival rate for all cancers patients diagnosed in 1996-2003 is 66%, up from 50% in 1975-1977.1 The improvement in survival reflects progress in diagnosing at an earlier stage and improvements in treatment. Discovering highly effective anticancer agents with low toxicity is a goal of our research.
We have recently discoverd 2-aryl-thiazolidine-4-carboxylic acid amides (ATCAAa, Figure 1) as potent cytotoxic agents for both, prostate cancer and melanoma.2-6 ATCAA was designed from lysophosphatidic acid (LPA) structure with a lipid chain in order to inhibit GPCR (guanine-binding protein-coupled receptor) signaling, which was involved in proliferation and survival of prostate cancer.7-10 The most potent compounds in ATCAA-1 derivatives could inhibit prostate cancer cells with an average IC50 in the range from 0.7 to 1.0 μM and average IC50s against melanoma cells were 1.8~2.6 μM.2 (2RS, 4R)-2-phenyl-thiazolidine-4-carboxylic acid hexadecylamide (ATCAA-1) was sent to the US National Cancer Institute 60 human tumor cell line anticancer drug screen (NCI-60). Results from NCI-60 assay showed that compound ATCAA-1 could inhibit growth of all nine types of cancer cells with IC50 in the range from 0.124 μM (Leukemia, CCRF-CEM) to 3.81 μM (Non-Small Cell Lung Cancer, NCI-H522). SAR studies of ATCAA indicated that replacement of the lipid chain with a bulky aromatic ring in the 4-amide position of ATCAA-2 attached to the thiazolidine ring still kept the anti-proliferative activity.11 This finding afforded us a new point to replace the fatty amide chain with a number of aromatic groups, which would maintain the cytotoxicity. With further investigation of ATCAA-2 analogues, structure modifications were made on thiazolidine ring and 4-carboxylic amide linker. Thus, substituted methoxylbenzoyl-aryl-thiazole (SMART) compounds were discovered and showed highly improved growth inhibition on tested cancer cells in vitro.
Figure 1.

Structures of LPA, ATCAA and SMART
In this paper, we described a series of the SMART agents with general structure as showed in Figure 1. The SMART agents have a structure containing three conjugated aromatic rings (“A”, “B” and “C” rings, respectively) with a ketone linkage between “B” and “C” rings. Thiazole was introduced in “B” ring instead of thiazolidine ring in ATCAA. The linker between “B” and “C” rings was modified from an amide to carbonyl group. The “C” ring was characterized by the presence of differently-substituted phenyl groups, in particular, the 3, 4, 5-trimethoxy substituted phenyl at “C” ring played an important role of antiproliferative activity against melanoma and prostate cancer. Synthesis, SAR studies, biological evaluation and the anticancer mechanism of the SMART analogues was undertaken and reported in this paper.
Chemistry
The general synthesis of the ATCAA and SMART analogues are shown in Scheme 1-3. To prepare ATCAA compounds 2a-b, L-cysteine was allowed to react with appropriate benzaldehydes in ethanol and water at ambient temperature to give cyclized (2RS, 4R)-2-aryl-thiazolidine-4-carboxylic acids 1,12 which were converted to the corresponding BOC-protected derivatives. Reaction of BOC-protected carboxylic acids with 3, 4, 5-trimethoxyaniline using EDCI/HOBt gave corresponding amides, which were treated with TFA to form the target compounds 2a-b.
Scheme 1.

Reagents and conditions: (a) C2H5OH, H2O, r. t.; (b) Boc2O, 1 N NaOH, 1, 4-dioxane, H2O; (c) EDCI, HOBt, TEA, 3, 4, 5-trimethoxyaniline; (d) TFA, CH2Cl2.
Scheme 3.

Reagents and conditions: (a) MeOH/pH=6.4 phosphate buffer, r. t.; (b) EDCI, HOBt, NMM, HNCH3OCH3; (c) CBrCl3, DBU; (d) ArBr/BuLi or ArMgBr, THF; (e) HCl/HOAc; (f) MeOH/CH3COCl; (g) Fe/HOAc; (h) BBr3, CH2Cl2.
To synthesize thiazoline and thiazole series compounds 4a-b and 5, (4R or 4S)-2-Phenyl-4,5-dihydro-thiazole-4-carboxylic acid 3a and 3b were obtained by reacting L- or D-cysteine with benzonitrile in methanol and pH 6.4 phosphate buffer solution at ambient temperature for several days.13 Coupling reactions of 3a-3b with 3, 4, 5-trimethoxyaniline under EDCI/HOBt conditions gave 4R or 4S amide 4a-4b. Oxidation with BrCCl3/DBU of 4a-4b gave the same dehydrogenation thiazole product 5.14
The SMART compounds were synthesized as illustrated in Scheme 3 described. (4R)-2-(substituted phenyl)-4, 5-dihydro-thiazole-4-carboxylic acids 3 were synthesized according the similar preparation of 3a and 3b from L-cysteine with appropriate benzonitriles.13, 15, 16 Compounds 3 can be easily converted to the corresponding Weinreb amides 6a-6p17 using EDCI/HOBt as coupling reagents. Thiazole intermediate 7 can be obtained from BrCCl3/DBU dehydrogenation of 6a-6p. Compound 6 or 7 was reacted with appropriate lithium reagents or Grignard reagents in anhydrous THF to give the final SMART compounds 8a-8z.17 Compound 10 was obtained by the same method. Thiazoline Weinreb amides 6a-6p reacted directly with appropriate lithium reagents or Grignard reagents, after quenching with saturated NH4Cl solution, the mixtures of thiazoline compounds and the corresponding thiazole compounds were afforded. When thiazoline/thiazole mixtures were placed in the solvent and exposed to air under ambient atmosphere for some time (overnight to several days), the thiazoline ring spontaneously dehydrogenated to thiazoles 8a-8z, which were clearly indicated by 1H NMR, mass spectra and elemental analysis. As an example, in solution with deuterated chloroform, mixtures of thiazoline / thiazole compounds can be slowly converted to almost pure thiazole compound 8f after 9 days (see Figure 2). No report of auto-dehydrogenations was found in the literature. Most reports of thiazoline-thiazole dehydrogenations need an oxidant (MnO218), oxidase in biosynthesis19 or catalysts (Hg(OAc)2, K3FeCN6).20 The thiazoline Weinreb amide was reported to undergo dehydrogenation to form thiazole under base/solvent (NaH/MeOH, TMSOK/THF, etc.) conditions.21 The base could abstract the acidic 4-position proton of thiazoline ring, with subsequent intramolecular attack of carbanion on the methoxy amide and release of MeO-, followed by 3-position proton elimination. We did not observe these auto-dehydrogenation phenomena between intermediate thiazoline amides 6a-6p and thiazole amides 7. The triple aromatic ring system formed highly stable conjugated SMART structures 8a-8z which could be a favorable reason for auto-dehydrogenation. X-ray crystallography demonstrated the conjugated structure of compound 8f. Benzoic acid 8r was prepared from the acidic hydrolysis of benzonitrile 8q in HCl/HOAc.22 Methyl ester 8s was obtained by the esterification of 8r in methanol and acetyl chloride. Para-amino compound 8w was synthesized by using iron and acetic acid reduction of para-nitro compound 8p.23 3, 4, 5-trihydroxyl compound 11 was obtained using BBr3 as demethylation reagent.24
Figure 2.
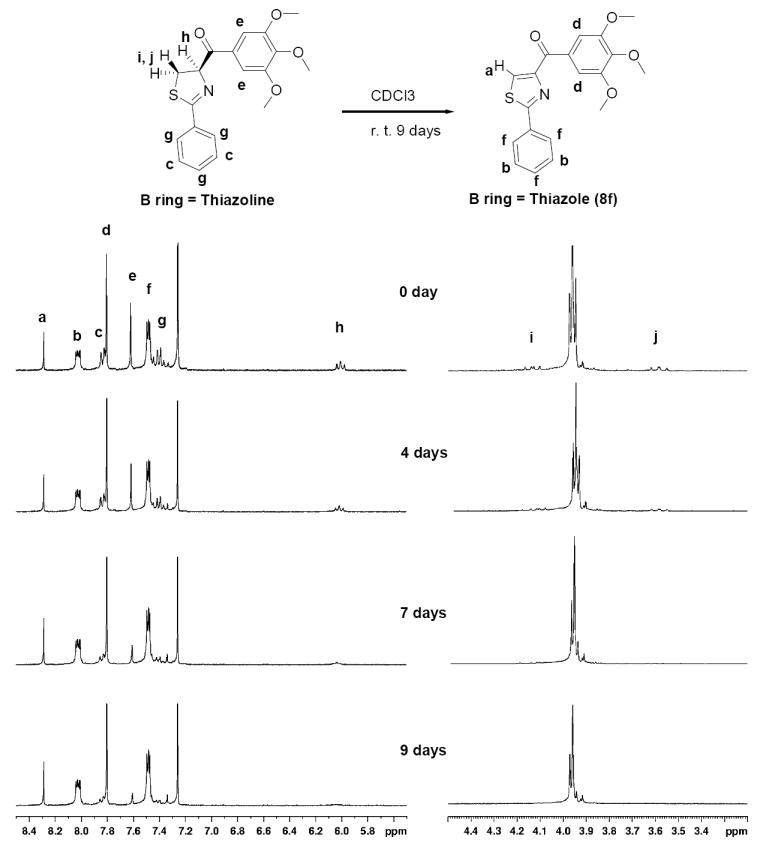
Auto-dehydrogenation from thiazoline to thiazole compound 8f. At 0 day, NMR sample contained thiazoline and thiazole mixtures in CDCl3; ratio is about 3: 2. At 9th day, thiazoline compound was almost converted to thiazole compound 8f
Crystal Structure
The SMART compound 8f was recrystallized from hexane and ethyl acetate, and single colorless crystals suitable for X-ray diffraction were obtained. An ORTEP drawing of 8f with the atom labeling scheme is shown in Figure 3. The X-ray structure showed that 8f molecule contained a conjugated system composed of three aromatic rings and a carbonyl group linker between “B” and “C” ring as expected (“A” ring = phenyl; “B” ring = thiazole; “C” ring = 3, 4, 5-trimethoxyphenyl). As a result, two C-C bonds adjacent to C=O and C-C- bond between “A” phenyl and “B” thiazole ring display (C1-C7 = 1.496(2) Å; C7-C8 = 1.492(2) Å; C10-C11 = 1.471(2) Å) shorter bond lengths than normal C-C single bond (1.54 Å) and longer than normal C=C double bond (1.34 Å) (see Table 1). Thus conjugation of the π system is possible for “A”, “B”, “C” rings and carbonyl group. The carbonyl group is nearly coplanar with the adjacent “B” thiazole ring (O-C7-C1-C6 16.2(2)°, O-C7-C8-C9 9.7(2)°).
Figure 3.
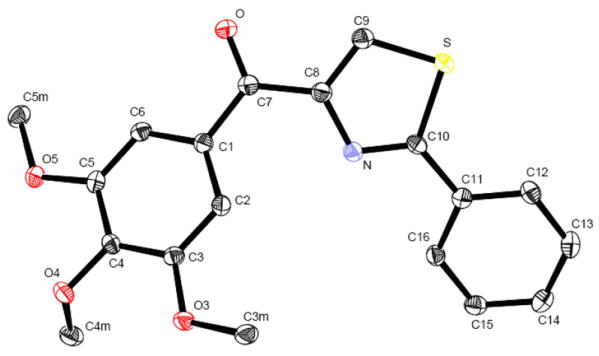
ORTEP drawing of 8f with thermal ellipsoids depicted at 50 % probability level
Table 1.
Selected Geometric Parameters of 8f (Å, °)
| C1—C7 | 1.496(2) | O—C7—C1 | 120.1(2) |
| C7—O | 1.224(2) | C8—C7—C1 | 121.9(2) |
| C7—C8 | 1.492(2) | C9—C8—N | 115.1(2) |
| C8—C9 | 1.371(2) | C9—C8—C7 | 121.7(2) |
| C8—N | 1.380(2) | N—C8—C7 | 123.0(2) |
| C9—S | 1.711(2) | C8—C9—S | 110.0(1) |
| S—C10 | 1.747(2) | C9—S—C10 | 89.6(1) |
| C10—N | 1.303(2) | N—C10—C11 | 123.5(2) |
| C10—C11 | 1.471(2) | N—C10—S | 113.9(1) |
| C2—C1—C6 | 121.2(2) | C11—C10—S | 122.6(1) |
| C2—C1—C7 | 122.3(2) | C10—N—C8 | 111.4(2) |
| C6—C1—C7 | 116.4(2) | C12—C11—C10 | 122.3(2) |
| O—C7—C8 | 118.0(2) | C16—C11—C10 | 118.5(2) |
Biological Results and Discussion
ATCAA to SMART molecules
Modifications of the “B” ring from a thiazolidine to thiazole system and the linker from an amide to a ketone. In the previous ATCAA compounds, we found the thiazolidine ring, which contained a free NH at its 3-position, was important for cytotoxicity. Once the “B” ring thiazolidine moiety was replaced by a thiazoline ring — the antiproliferative activity decreased sharply from 0.6 μM to over 50 μM on WM-164 cell lines.2 The ATCAA-1 fatty amide derivatives that was most effective against melanoma and prostate cancer cell lines were examined and shown to have an IC50 0.4-2.2 μM (Table 2). Replacement of the long fatty chain with a certain aromatic bulky subsistent such as fluorene (ATCAA-2) showed inhibitory activity on both cancer cell lines (IC50 = 1.6 - 3.9 μM)11. The fluorene group in 4-carboxylic amide position was also replaced by 3, 4, 5-trimethoxylphenyl group (2a and 2b), but the potency against both cancer cell lines was lost. The subsequent “B” ring modification from saturated thiazolidine compound 2a to unsaturated thiazole 5 did not show any cytotoxicity against either cancer cell line tested. But thiazoline enantiomers 4a and 4b (R-isomer and S-isomer, showed similar antiproliferative activities) showed improved activity (IC50 = 3.4 - 38.3 μM) compared with 2a, 2b and 5. When the amide CONH linkage between “B” ring and “C” ring was replaced by a carbonyl linker, the mixtures of thiazoline/thiazole ketone 8f were obtained instead of desired thiazoline ketone, because the auto-dehydrogenation between thiazoline and thiazole occurred (the conversion was shown in Figure 2). Surprisingly, introduction of the carbonyl group linker and thiazole led to a significant enhancement of growth inhibition of examined cancer cell lines with a low nanomolar level (8f, IC50 =0.021 - 0.071 μM), which is comparable to the natural anticancer agent Colchicine. Thus a series of the SMART compounds with “B” as a thiazole ring were designed and synthesized based on the discovery of 8f and their anticancer activity was evaluated against melanoma and prostate cancer.
Table 2.
In Vitro Inhibitory Effects of Modificated ATCAA Compounds against the Proliferation of Melanoma (A 375, B16-F1) and Prostate Cancer Cells (DU145, PC-3, LNCaP, PPC-1).
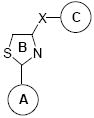 |
A ring | B ringa | C ringb | X | IC50 ± SEM (μM) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | |||||
| ATCAA-1 | p-NHAc-Ph | TZD | C16H33 | CONH | 2.2±0.3 | 2.1±0.2 | 1.7 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | 0.4 ± 0.1 |
| ATCAA-2 | p-NHAc-Ph | TZD | 9H-fluoren-1-yl | CONH | 3.9±0.3 | 2.1±0.1 | 1.9 ± 0.3 | 2.1 ± 0.1 | 3.5 ± 0.7 | 1.6 ± 0.1 |
| 2a | Ph | TZD | 3,4,5-trimethoxyl-Ph | CONH | >100 | >100 | >20 | >20 | >20 | >20 |
| 2b | 3,4,5-trimethoxyPh | TZD | 3,4,5-trimethoxyl-Ph | CONH | >100 | >100 | >20 | >20 | >20 | >20 |
| 4a(4R) | Ph | TZL | 3,4,5-trimethoxyl-Ph | CONH | 38.3± 3.2 | 22.8±1.6 | >20 | >20 | >20 | 5.3± 0.3 |
| 4b(4S) | Ph | TZL | 3,4,5-trimethoxyl-Ph | CONH | 30.4±2.8 | 13.6±1.2 | >20 | 13.2± 2.1 | 16.8± 1.8 | 3.4± 0.2 |
| 5 | Ph | TZ | 3,4,5-trimethoxyl-Ph | CONH | >100 | >100 | >20 | >20 | >20 | >20 |
| 8f | Ph | TZ | 3,4,5-trimethoxyl-Ph | CO | 0.055 ± 0.005 | 0.028 ± 0.005 | 0.071 ± 0.004 | 0.021 ±0.001 | 0.028 ± 0.004 | 0.043 ± 0.005 |
| Colchicine | 0.029 ± 0.005 | 0.020 ± 0.003 | 0.010 ± 0.002 | 0.011 ± 0.001 | 0.016 ± 0.004 | 0.020 ± 0.001 | ||||
TZD=Thiazolidine, TZL=Thiazoline, TZ= Thiazole
For ATCAA-1, “C” position contains a lipid chain.
Modifications of “C” ring of the SMART molecules
We started our investigation of the “C” position of the SMART by introducing different substituted phenyls or alkyl chain. Variation of the phenyl substituents has a remarkable change in effect on potency. The in vitro assay as shown in Table 3 gave us an interesting result but only 3, 4, 5-trimethoxylphenyl in “C” ring (8f) showed excellent inhibition against all cancer cells (IC50= 21 -71 nM, average IC50= 41 nM). Compound 8g, with a 3, 5-dimethoxyphenyl group, showed 6-fold average cytotoxicity lower than 8f against six different cell lines (IC50 = 170-424 nM, calcd. average IC50= 261 nM). Modifications of 8f by removal of one methoxy at meta-position (8e) or two methoxy groups (8b, 8c and 8d) from 8f led to a dramatic loss in activity (IC50 >20 μM). Although ortho- substituted monomethoxy compound 8d exhibited weak activity against a certain cell lines compared with meta-/para-MeO substituted 8c/8b and dimethoxyphenyl compound 8e, none of them showed significant potency in inhibition compared with 8f. Similar trends were also seen in 8h and 8j with 2-fluorophenyl and hexadecyl in “C” ring modifications.
Table 3.
In Vitro Growth Inhibitory Effects of Compounds 8a-8j with Different “C” Rings against the Proliferation of Melanoma (A 375, B16-F1) and Prostate Cancer Cells (DU145, PC-3, LNCaP, PPC-1).
| Compounds 8 | C Ring | IC50 ± SEM (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | ||||
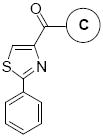 |
8a | Ph | >100 | >100 | >20 | >20 | >20 | >20 | |
| 8b | 4-Methoxy-Ph | >100 | >100 | >20 | >20 | >20 | >20 | ||
| 8c | 3-Methoxy-Ph | >100 | >100 | >20 | >20 | >20 | >20 | ||
| 8d | 2-Methoxy-Ph | 59.4 ± 21.2 | 70.3 ± 32.5 | >20 | >20 | >20 | >20 | ||
| 8e | 3, 4-Dimethoxy-Ph | >100 | >100 | >20 | >20 | >20 | >20 | ||
| 8f | 3,4,5-Trimethoxy-Ph | 0.055 ± 0.005 | 0.028 ± 0.005 | 0.071 ± 0.004 | 0.021 ± 0.001 | 0.028 ± 0.004 | 0.043 ± 0.005 | ||
| 8g | 3, 5-Dimethoxy-Ph | 0.350 ± 0.2 | 0.170 ± 0.1 | 0.424 ± 0.098 | 0.301 ± 0.030 | 0.323 ± 0.041 | 0.242 ± 0.014 | ||
| 8h | 2-Fluoro-Ph | >100 | >100 | >20 | >20 | >20 | >20 | ||
| 8j | Hexadecyla | 18.6±17.5 | 16.0 ± 15.2 | >20 | >20 | >20 | >20 | ||
Compound 8j has a lipid chain at “C” ring position.
Modifications of “A” ring of the SMART molecules
In SAR studies of the ATCAA compounds, we found that the electronic properties of substituents of the phenyl ring in the 2-positioin of the thiazolidine ring strongly affected the anti-cancer activity in ATCAA compounds — electron-withdrawing groups (EWG) on 2-phenyl gave higher activities than those with electron-donating groups (EDG).6 We also introduced different para-substituted EWG and EDG on “A” phenyl ring of the SMART molecules. From the IC50 value against these cancer cell lines, electronic effects of “A” ring phenyl substituents did not show clear influence on antiproliferative activity. Introduction of a weak EWG (4-F in 8n, IC50s: 6 - 43 nM) or weak EDG (4-CH3 in 8k, IC50s: 5 - 21 nM), both increased the potency compared with 8f (see Table 4). The replacement of para- position with strong EWG such as NO2 (8p), CN (8q), CF3 (8t) or introducing strong EDG (3, 4-dimethoxy) to “A” phenyl ring (8o) exhibited comparable antiproliferative activity.
Table 4.
In Vitro Growth Inhibitory Effects of the SMART Compounds with different “A” Rings against the Proliferation of Melanoma (A 375, B16-F1) and Prostate Cancer Cells (DU145, PC-3, LNCaP, PPC-1).
| Compounds 8 | A Ring | IC50 ± SEM (nM) |
||||||
|---|---|---|---|---|---|---|---|---|
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | |||
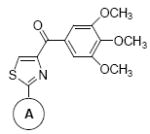 |
8f | Ph | 55 ± 5 | 28 ± 5 | 71 ± 4 | 21 ± 1 | 28 ± 4 | 43 ± 5 |
| 8k | 4-Methyl-Ph | 21 ± 10 | 11 ± 5 | 7 ± 1 | 5± 1 | 6 ± 1 | 6 ± 1 | |
| 8l | 2-Fluoro-Ph | 27 ± 11 | 30 ± 9 | 114 ± 3 | 82 ±9 | 53 ± 4 | 52 ± 3 | |
| 8m | 3-Fluoro-Ph | 287 ± 36 | 304 ± 25 | 35 ± 3 | 24 ± 2 | 11 ± 2 | 21 ± 1 | |
| 8n | 4-Fluoro-Ph | 43 ± 21 | 33 ± 14 | 12 ± 1 | 13 ± 1 | 6 ± 1 | 8 ± 1 | |
| 8o | 3, 4-Dimethoxy-Ph | 161 ± 29 | 34 ± 10 | 102 ± 2 | 69 ± 3 | 38 ± 6 | 56 ± 2 | |
| 8p | 4-Nitro-Ph | 56 ± 12 | 38 ± 9 | 95 ± 5 | 56 ± 1 | 39 ± 4 | 34 ±1 | |
| 8q | 4-Cyano-Ph | 53 ± 16 | 59 ± 24 | 52 ± 2 | 30 ± 7 | 15 ±4 | 19 ± 2 | |
| 8t | 4-Trifluoromethyl-Ph | 92 ± 16 | 23 ± 5 | 50 ± 5 | 58 ± 4 | 94 ± 1 | 76 ± 1 | |
| 8u | 4-Bromo-Ph | 32 ± 5 | 13 ± 2 | 21 ± 4 | 18 ± 3 | 44 ± 3 | 21 ± 5 | |
| 8v | 4-Ethyl-Ph | 70 ± 8 | 17± 2 | 31 ± 4 | 27 ± 4 | 60 ± 5 | 22 ± 3 | |
| 8x | 4-Pyridine | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | |
| 8y | 2-Pyrimidine | 2300 ± 860 | 4100 ± 740 | 2813 ± 92 | 2657 ± 40 | 2370 ± 85 | 1186 ± 22 | |
| 8z | 2-Thienyl | 38 ± 15 | 20 ± 7 | 22 ± 1 | 17 ± 2 | 9 ± 1 | 13 ± 1 | |
| 10 | Ha | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | |
Compound 10 has a proton at “A” ring position.
To compare the effects of ortho-, meta- and para- substitutions, a fluoro atom was introduced to different positions of “A” phenyl ring (8l, 8m and 8n). The various o-, m-, p- substituents did not exhibit equal activities. p-Fluoro substituted 8n has the best activity for examined prostate cancer cells (6-13 nM) while o-fluoro substituted 8l showed the lowest IC50s (27 - 30 nM) against melanoma cells. 8n has similar average IC50s (33 - 43 nM) against melanoma compared with 8l. But o-fluoro substituted 8l has lowest potency (IC50s: 52-114 nM) among the three substituted compounds on prostate cancer cells. Meta-substituted compound 8m showed lowest activity on melanoma cells (IC50s: 287-304 nM) but showed moderate inhibition on prostate cancer cells (IC50s: 23-46 nM).
Turning to the effects of steric hindrance group on the “A” phenyl ring substituents, we found that p-bromo (8u, IC50s: 18-44 nM) caused a decrease in antiproliferative activity relative to p-fluoro position (8n, IC50s: 6-12 nM) only against prostate cancer cells. Reduced activity against both cancer cell lines occurred when p-methyl (8k, IC50s: 5-21 nM) was replaced with a p-ethyl group (8v, IC50s: 17-70 nM).
To investigate if phenyl played an essential role at “A” ring in cytotoxicity, we also removed phenyl at 2-thiazole position and compound 10 was obtained. This modification caused a total loss of activity compared with 8f. The replacement of the “A” ring by pyridine (compound 8x) had the same effect. Moreover, substituting 2-pyrimidine in “A” ring (compound 8y) also caused a significant loss of activity (IC50s: 11.8 – 41.0 μM). However, introducing the thiophene replacement of phenyl (8z) into “A” position improved the potency calcd. 1-3 folds on all examined cell lines (IC50s: 9-38 nM) compared to 8f (IC50s: 21-71 nM).
Addition of pharmaceutically acceptable salt groups to the SMART molecules
Most of the SMART compounds have good solubility in organic solvents such as CHCl3, CH2Cl2 and DMSO. But they show poor water-solubility. We designed and synthesized water-soluble salts of the SMART by introducing a hydrophilic group such as NH2 (8w) and COOH (8r) into “A” ring and generated the HCl or sodium salt. Another modification is replacing “A” / “C” rings in 8a with pyridine (8i, 8x, 8y) or pyrimidine rings, which could also be converted into HCl salts. These modifications reduced the calculated LogP values (LogP = 2.74 - 3.90) compared with 8a and 8f (LogP = 4.46 and 4.08, See Table 5). Introducing p-amino to “A” phenyl (8w) is the only case to increase the antiproliferative activity (HCl salt, IC50s: 11-29 nM) compared with 8f against all cell lines. Although replacing phenyl with pyrimidine (8y) kept partial activity against both cancer cells, the potency range was markedly reduced from nM to μM compared with 8f. Unfortunately introducing COOH to para- phenyl “A” ring and pyridine to “A” or “C” rings (8i, 8r, 8x) all resulted in the total loss of the anti-cancer activity. A total loss of potency was seen in the methyl ester 8s of acid 8r against both cancer cell lines. Demethylation of compound 8f afforded water soluble 3, 4, 5-trihydroxyphenyl at “C” ring compound 11, but this demethylation results in complete loss of antiproliferative activity against all tested cancer cells, which also points out the importance of 3, 4, 5-trimethoxyphenyl at “C” position.
Table 5.
In Vitro Growth Inhibitory Effects of Compounds Contained Hydrophilic Group Against the Proliferation of Melanoma (A 375, B16-F1) and Prostate Cancer Cells (DU145, PC-3, LNCaP, PPC-1).
| Compd | IC50± SEM (nM) |
CLogPa | |||||
|---|---|---|---|---|---|---|---|
| B16-F1 | A375 | DU145 | PC-3 | LNCaP | PPC-1 | ||
| 8i | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | 3.55 |
| 8i·HCl | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | - |
| 8r | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | 3.64 |
| 8s | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | 3.90 |
| 8x | >100000 | >100000 | >20000 | >20000 | 16630 | 18000 | 2.74 |
| 8x·HCl | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | - |
| 8y | 2300±860 | 4100±740 | 2813 ± 92 | 2657 ± 40 | 2370 ± 85 | 1186 ± | 3.04 |
| 8w·HCl | 29±10 | 11±2 | 20 ± 2 | 12 ± 1 | 13 ± 1 | 15 ± 1 | - |
| 11 | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | 3.29 |
| 8a | >100000 | >100000 | >20000 | >20000 | >20000 | >20000 | 4.46 |
| 8f | 55±5 | 28±5 | 71 ± 4 | 21 ± 1 | 28 ± 4 | 43 ± 5 | 4.08 |
Calculated LogP data using Chemoffice 2005, Chemdraw Ultra 9.0 software.
LogP value were calculated based on free base.
Mechanism of action studies: The SMART compounds inhibit tubulin polymerization
By observing cell cycle progression in response to 8f, a significant increase in G2/M phase arrest was detected (unpublished data). This is similar to the induction by an mitotic inhibitor, such as Cochicine.25 To investigate whether the antiproliferative activities of these compounds were related to interaction with tubulin, the SMART compound 8f was evaluated for inhibition of polymerization of purified tubulin in a cell-free system. The results are shown in Figure 4. Compared with non-treated control, 8f (“A” ring = phenyl, “C” ring = 3, 4, 5-trimethoxyphenyl) inhibits tubulin polymerization. The effect of 8f on tubulin assembly was examined at concentrations from 0.625 μM to 20 μM. We observed that compound 8f inhibited tubulin polymerization in a dose-dependent manner (Figure 4), with an IC50 value of 4.23 μM.
Figure 4.

Effect of 8f on tubulin assembly
Conclusions
We have discovered a new class of simple synthetic inhibitors of tubulin polymerization, based on a 2-aryl-4-(3, 4, 5-trimethoxylbenzoyl)-thiazole molecular skeleton, which was derived from thiazolidine ring modification of ATCAA structures. A series of the SMART compounds were synthesized. Chemical modification of different substituted aryl in “A” and “C” rings and structure-activity relationship of the SMART were investigated (Figure 5) based on biological evaluation against melanoma and prostate cancer cells in vitro. Present SAR studies revealed that 3, 4, 5- trimethoxyphenyl was the essential group in the “C” ring to keep excellent antitumor potency. p-Fluoro, p-NH2 and p-CH3 substituents in “A” ring will increase the activity, with no clear difference in effect on activity between EWG and EDG when “A” are substituted phenyl rings. The carbonyl linkage between “B” ring and “C” ring played an important role for the high potency. Further modification of “B” and “C” rings, carbonyl linkage, tubulin binding site studies, mechanism of action of the SMART compounds and in vivo animal testing are currently underway.
Figure 5.

SAR relationship of the SMART molecules
Experimental Section
General
All reagents were purchased from Sigma-Aldrich Chemical Co., Fisher Scientific (Pittsburgh, PA), AK Scientific (Mountain View, CA), Oakwood Products (West Columbia, SC), etc. and were used without further purification. Moisture-sensitive reactions were carried under an argon atmosphere. Routine thin layer chromatography (TLC) was performed on aluminum backed Uniplates. (Analtech, Newark, DE). Melting points were measured with Fisher-Johns melting point apparatus (uncorrected). NMR spectra were obtained on a Bruker AX 300 (Billerica, MA) spectrometer or Varian Inova-500 spectrometer. Chemical shifts are reported as parts per million (ppm) relative to TMS in CDCl3. Mass spectral data was collected on a Bruker ESQUIRE electrospray/ion trap instrument in positive and negative ion modes. Elemental analyses were performed by Atlantic Microlab Inc., (Norcross, GA).
General Procedure for the preparation of (2RS, 4R)-2-Aryl-thiazolidine-4-carboxylic 1
A mixture of L-cysteine (3.16 g, 26.11 mmol) and appropriate aldehyde (26.15 mmol) in ethanol (300 mL) and water (30 mL) was stirred at room temperature for 6-15h, and the solid precipitated out was collected, washed with diethyl ether and dried to afford according (2RS, 4R)-2-aryl-thiazolidine-4-carboxylic acid 1 with yields of 70-99%. At 0 °C, 1 (5.95 mmol) was dissolved in 1N NaOH (6 mL) and 1, 4-dioxane (15 mL), then di-tert-butyldicarbonate (2.80 g, 12.80 mmol) was added slowly and stirred at room temperature for 1 h. The reaction mixture was concentrated in vacuum and washed with ethyl acetate (20 mL). The aqueous phase was adjusted to pH=4 by adding 1N HCl or 5% KHSO4, then extracted with ethyl acetate, dried with magnesium sulfate, filtered and concentrated on vacuum to give corresponding BOC protected acids as white foam-solids, which were used for next step without further purification.
General Procedure for the preparation of (2RS, 4R)-2-Aryl-N-(3,4,5-trimethoxyphenyl)thiazolidine-4-carboxamide 2a-2b
A mixture of appropriate BOC protected carboxylic acids (0.3-0.5g), EDCI (1.2 equiv) and HOBT (1.05 equiv) in CH2Cl2 (20 mL) was stirred at room temperature for 10 min. To this solution, 3,4,5-trimethoxyaniline (1.05 equiv) and Et3N (1.2 equiv) were added and stirring continued at room temperature for 6-8 h. The reaction mixture was diluted with CH2Cl2 (30 mL) and sequentially washed with water, satd. NaHCO3, brine and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude oil, which were stirred with TFA (0.6-1 mL) in 20 mL CH2Cl2 at r. t for 1-8 h to cleave the BOC group. The reaction mixture was concentrated, washed with satd. NaHCO3 and dried over MgSO4. The solvent was removed to yield a crude solid, 2a-2b were purified by column chromatography. Yield was reported as 2 steps yield.
(2RS, 4R)-2-Phenyl-N-(3,4,5-trimethoxyphenyl)thiazolidine-4-carboxamide (2a)
Yield: 69.5 %. M. p. 158-159 °C. 1H NMR (300MHz, CDCl3) δ 9.14 (s, 0.8 H), 8.61 (s, 0.2 H), 7.58-7.32 (m, 5 H), 6.90 (s, 1.6 H), 6.71 (s, 0.4H), 5.71 (dd, 0.2 H, J = 9.0 Hz), 5.42 (dd, 0.8 H, J = 11.7 Hz), 4.53 (dt, 0.8 H), 4.19 (m, 0.2 H), 3.87, 3.80 (s, s, 6 H), 3.82, 3.78 (s, s, 3 H), 3.80-3.78 (m, 0.4 H), 3.62-3.42 (m, 1.6 H), 2.96 (t, 0.2 H, J = 9.0 Hz), 2.74 (dd, 0.8 H, J = 11.7 Hz). MS (ESI) m/z 375.1 [M + H]+, 397.1 [M + Na]+. Anal. (C19H22N2O4S) C, H, N.
(2RS, 4R)-N,2-bis(3,4,5-trimethoxyphenyl)thiazolidine-4-carboxamide (2b)
Yield: 34.5 %. M. p. 147-149 °C. 1H NMR (300MHz, CDCl3) δ 9.10 (s, 0.7 H), 8.59 (s, 0.3 H), 6.90 (s, 1.4 H), 6.80 (s, 0.6 H), 6.74 (s, 1.4H), 6.71 (s, 0.6 H), 5.66 (br, 0.3 H), 5.35 (d, br, 0.7 H, J = 7.5 Hz), 4.52 (br, 0.7 H), 4.21 (br, 0.3 H), 3.90, 3.87, 3.86, 3.84, 3.82, 3.81, 3.79, 3.78 (all s, 18 H), 3.66-3.61, 3.54-3.38 (m, 1.6 H), 2.98, 2.72 (br, 1 H). MS (ESI) m/z 465.1 [M + H]+, 487.1 [M + Na]+. Anal. (C22H28N2O7S) C, H, N.
2-(Substituted-phenyl)-4,5-dihydrothiazole-4-carboxylic acids 3
Substituted benzonitrile (40 mmol) was combined with L- or D- Cysteine (45mmol) in 100 mL of 1:1 MeOH/pH6.4 phosphate buffer solution. The reaction was stirred at 40 °C for 3 days. The precipitate was removed by filtration, and MeOH was removed using rotary evaporation. The remaining solution was added 1M HCl to adjust pH=4 under 0 °C. The resulting precipitate was extracted into CH2Cl2, dried and concentrated to yield a white to light yellow solids 3, which were used directly to next step without purification.
(4R)-2-Phenyl-4,5-dihydrothiazole-4-carboxylic acid (3a)
Yield: 58.3 %. 1H NMR (300MHz, CDCl3) δ 9.31 (br, 1 H), 7.88-7.85 (m, 2 H), 7.55-7.41 (m, 3 H), 5.38 (t, 1 H, J = 9.6 Hz), 3.75 (dt, 2 H, J = 9.6 Hz, 2.7 Hz). MS (ESI) m/z 162.0 [M - COOH]-.
(4S)-2-Phenyl-4,5-dihydrothiazole-4-carboxylic acid (3b)
Yield: 53.9 %. 1H NMR (300MHz, CDCl3) δ 7.89-7.85 (m, 2 H), 7.55-7.41 (m, 3 H), 5.38 (t, 1 H, J = 9.3 Hz), 3.75 (dt, 2 H, J = 9.3 Hz, 2.7 Hz). MS (ESI) m/z 162.0 [M - COOH]-.
2-Phenyl-N-(3,4,5-trimethoxyphenyl)-4,5-dihydrothiazole-4-carboxamide 4a-4b
The compounds were prepared following the same EDCI/HOBt method as 2a-2b described.
(4R)-2-Phenyl-N-(3,4,5-trimethoxyphenyl)-4,5-dihydrothiazole-4-carboxamide (4a)
Yield: 98.7 %. M. p. 121-122 °C. 1H NMR (300MHz, CDCl3) δ 8.98 (s, 1 H), 8.02-7.94, 7.62-7.48 (m, 5 H), 6.93 (s, 2 H), 5.38 (t, 1 H, J = 9.6 Hz), 3.92-3.85 (m, 2 H), 3.87 (s, 6 H), 3.82 (s, 3 H). MS (ESI) m/z 373.1 [M + H]+. Anal. (C19H20N2O4S) C, H, N.
(4R)-2-Phenyl-N-(3,4,5-trimethoxyphenyl)-4,5-dihydrothiazole-4-carboxamide (4b)
Yield: 70.7 %. M. p. 122-123 °C. 1H NMR (300MHz, CDCl3) δ 8.62 (s, 1 H), 7.93-7.90 (m, 2 H), 7.55-7.45 (m, 3 H), 6.88 (s, 2 H), 5.31 (t, 1 H, J = 9.6 Hz), 3.86 (s, 6 H), 3.79 (s, 3 H), 3.83-3.70 (m, 2 H). MS (ESI) m/z 395.1 [M + Na]+, 370.9 [M − 1]-. Anal. (C19H20N2O4S) C, H, N.
2-Phenyl-N- (3,4,5-trimethoxyphenyl)thiazole-4-carboxamide (5)
Yield: 89.7 %. M. p. 157-158 °C. 1H NMR (300MHz, CDCl3) δ 9.30 (s, 1 H), 8.20 (s, 1 H), 8.04-8.01 (m, 2 H), 7.53-7.51 (m, 3 H), 7.08 (s, 2 H), 3.92 (s, 6 H), 3.86 (s, 3 H). MS (ESI) m/z 393.1 [M + Na]+. Anal. (C19H18N2O4S) C, H, N.
2- (Substituted-phenyl)-4, 5-dihydrothiazole-4-carboxylic acid methoxymethylamides 6a-6p and 9
General procedure: A mixture of appropriate 2-(substituted-phenyl)-4, 5-dihydrothiazole-4-carboxylic acid 3 (5mmol), EDCI (6 mmol) and HOBt (5 mmol) in CH2Cl2 (50mL) was stirred for 10 min. To this solution, NMM (5 mmol) and HNCH3OCH3 (5 mmol) were added and stirring continued at room temperature for 6-8 hours. The reaction mixture was diluted with CH2Cl2 (100mL) and sequentially washed with water, satd. NaHCO3, Brine and dried over MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography.
(R)-N-Methoxy-N-methyl-2-phenyl-4,5-dihydrothiazole-4-carboxamide (6a)
Yield: 92.0 %. 1H NMR (300MHz, CDCl3) δ 7.85-7.83 (m, 2 H), 7.48-7.36 (m, 3 H), 5.66 (t, 1 H, J = 9.0 Hz), 3.90 (s, 3 H), 3.88-3.80 (br, 1 H), 3.55-3.47 (dd, 1 H, J = 10.8 Hz, 9.0 Hz), 3.30 (s, 3 H). MS (ESI) m/z 251.0 [M + H]+, 273.0 [M + Na]+.
(R)-N-methoxy-N-methyl-2-p-tolyl-4,5-dihydrothiazole-4-carboxamide (6b)
Yield: 55.8 %. 1H NMR (300MHz, CDCl3) δ 7.79 (d, 2 H, J = 7.8 Hz), 7.22 (d, 2 H, J = 7.8 Hz), 5.68 (t, 1 H, J = 8.7 Hz), 3.91 (s, 3 H), 3.80 (t, 1 H, J = 9.3 Hz), 3.55 (t, 1 H, J = 9.3 Hz), 3.30 (s, 3 H), 2.93 (s, 3 H). MS (ESI) m/z 265.0 [M + H]+, 287.0 [M + Na]+.
(R)-2-(2-fluorophenyl)-N-methoxy-N-methyl-4,5-dihydrothiazole-4-carboxamide (6c)
Yield: 39.6 %. 1H NMR (300MHz, CDCl3) δ 7.91 (dt, 1 H, J = 7.5 Hz, 1.8 Hz), 7.43 (m, 1 H), 7.19-7.09 (m, 2 H), 5.63 (t, 1 H), 3.88 (s, 3 H), 3.83 (br, 1 H), 3.48 (dd, 1 H, J = 11.1 Hz, 9.6 Hz), 3.30 (s, 3 H). MS (ESI) m/z 291.0 [M + Na]+.
(R)-2-(3-fluorophenyl)-N-methoxy-N-methyl-4,5-dihydrothiazole-4-carboxamide (6d)
Yield: 84.3 %. 1H NMR (300MHz, CDCl3) δ 7.60-7.56 (m, 2 H), 7.38 (dt, 1 H, J = 8.1 Hz, 6.0 Hz), 7.16 (dt, 1 H, J = 8.1 Hz, 2.4 Hz), 5.67 (t, 1 H), 3.90 (s, 3 H), 3.86-3.83 (br, 1 H), 3.52 (dd, 1 H, J = 10.8 Hz, 9.3 Hz), 3.30 (s, 3 H). MS (ESI) m/z 291.0 [M + Na]+.
(R)-2-(4-fluorophenyl)-N-methoxy-N-methyl-4,5-dihydrothiazole-4-carboxamide (6e)
Yield: 66.0 %. 1H NMR (300MHz, CDCl3) δ 7.90 (d, 2 H), 7.13 (d, 2 H), 5.63 (t, 1 H), 3.88 (s, 3 H), 3.83 (br, 1 H), 3.46 (dd, 1 H), 3.31 (s, 3 H). MS (ESI) m/z 269.0 [M + H]+.
(R)-2-(3,4-dimethoxyphenyl)-N-methoxy-N-methyl-4,5-dihydrothiazole-4-carboxamide (6f)
Yield: 36.7 %. 1H NMR (300MHz, CDCl3) δ 8.11 (d, 1 H), 7.93 (s, 1 H), 7.19-7.09 (d, 1H), 5.41 (t, 1 H), 3.97 (s, 6H), 3.89 (s, 3 H), 3.73 (br, 1 H), 3.39 (dd, 1 H), 3.31 (s, 3 H). MS (ESI) m/z 333.1 [M + Na]+.
(R)-N-methoxy-N-methyl-2-(4-nitrophenyl)-4,5-dihydrothiazole-4-carboxamide (6g)
Yield: 53.7 %.1H NMR (300MHz, CDCl3) δ 8.25(d, 2 H, J = 9.0 Hz), 8.01 (d, 2 H, J = 9.0 Hz), 5.73 (t, 1 H), 3.90 (s, 3H), 3.87 (br, 1 H), 3.59 (dd, 1 H, J = 11.1 Hz, 9.3 Hz), 3.31 (s, 3 H). MS (ESI) m/z 318.1 [M + Na]+.
(R)-2-(4-cyanophenyl)-N-methoxy-N-methyl-4,5-dihydrothiazole-4-carboxamide (6h)
Yield: 26.7 %. 1H NMR (300MHz, CDCl3) δ 7.94(d, 2 H, J = 8.1 Hz), 7.69 (d, 2 H, J = 8.1 Hz), 5.71 (t, 1 H, J = 9.3 Hz), 3.89 (s, 3 H), 3.87 (br, 1 H), 3.56 (dd, 1 H, J = 10.8 Hz, 9.3 Hz), 3.30 (s, 3 H). MS (ESI) m/z 298.0 [M + Na]+.
(R)-N-methoxy-N-methyl-2-(4-trifluoromethylphenyl)-4,5-dihydrothiazole-4-carboxamide (6i)
Yield: 62.0 %. 1H NMR (300MHz, CDCl3) δ 7.95 (d, 2 H, J = 8.1 Hz), 7.65 (d, 2 H, J = 8.1 Hz), 5.70 (t, 1 H, J = 9.6 Hz), 3.89 (s, 3 H), 3.85 (br, 1 H), 3.55 (dd, 1 H, J = 10.8 Hz, 9.6 Hz), 3.30 (s, 3 H). MS (ESI) m/z 341.0 [M + Na]+.
(R)-2-(4-bromophenyl)-N-methoxy-N-methyl-4,5-dihydrothiazole-4-carboxamide (6j)
Yield: 20.0 %. 1H NMR (300MHz, CDCl3) δ 7.71, 7.53 (d, d, 4 H, J = 8.4 Hz), 5.63 (t, 1 H, J = 9.6 Hz), 3.88 (s, 3 H), 3.84 (t, 1 H, J = 9.6 Hz), 3.52 (dd, 1 H, J = 10.8 Hz, 9.6 Hz), 3.30 (s, 3 H). MS (ESI) m/z 351.0 [M + Na]+.
(R)-N-methoxy-N-methyl-2-(4-ethyl)-4,5-dihydrothiazole-4-carboxamide (6k)
Yield: 77.7 %. 1H NMR (300MHz, CDCl3) δ 7.75(d, 2 H, J = 8.4 Hz), 7.21 (d, 2 H, J = 8.4 Hz), 5.64 (t, 1 H), 3.89 (s, 3 H), 3.81 (m, 1 H), 3.48 (dd, 1 H, J = 10.8 Hz, 9.3 Hz), 3.29 (s, 3 H), 2.67 (q, 2 H), 1.24 (t, 3 H). MS (ESI) m/z 301.0 [M + Na]+.
(R)-N-methoxy-N-methyl-2-(pyridin-4-yl)-4,5-dihydrothiazole-4-carboxamide (6l)
Yield: 66.6 %. 1H NMR (300MHz, CDCl3) δ 8.70 (d, 2 H, J = 9.0 Hz), 7.67 (d, 2 H, J = 9.0 Hz), 5.71 (t, 1 H, J = 9.6 Hz), 3.90 (s, 3 H), 3.73 (t, 1 H), 3.55 (dd, 1 H, J = 10.8 Hz, 9.6 Hz), 3.30 (s, 3 H). MS (ESI) m/z 252.1 [M + H]+, 274.0 [M + Na]+.
(R)-N-methoxy-N-methyl-2-(pyrimidin-2-yl)-4,5-dihydrothiazole-4-carboxamide (6m)
Yield: 32.5 %. 1H NMR (300MHz, CDCl3) δ 8.88 (d, 2 H, J = 4.8 Hz), 7.38 (t, 1 H, J = 4.8 Hz), 5.83 (t, 1 H, J = 9.0 Hz), 3.87 (s, 3 H), 3.56 (dd, 2 H, J = 9.0 Hz), 3.30 (s, 3 H). MS (ESI) m/z 275.0 [M + Na]+.
(R)-N-methoxy-N-methyl-2-(thiophen-2-yl)-4,5-dihydrothiazole-4-carboxamide (6p)
Yield: 58.5 %. 1H NMR (300MHz, CDCl3) δ 7.57 (br, 1 H), 7.49 (d, 1 H, J = 4.8 Hz), 7.09 (dd, 1 H, J = 3.6 Hz, 4.8 Hz), 5.64 (t, 1 H, J = 9.0 Hz), 3.90 (s, 3 H), 3.85 (br, 1 H), 3.57 (dd, 1 H, J = 9.9, 9.0Hz), 3.29 (s, 3 H). MS (ESI) m/z 279.0 [M + Na]+.
N-methoxy-N-methylthiazole-4-carboxamide (9)
Yield: 58.7 %. 1H NMR (300MHz, CDCl3) δ 8.82 (d, 1 H, J = 2.1 Hz), 8.10 (d, 1 H, J = 2.1 Hz), 3.79 (s, 3 H), 3.45 (s, 3 H). MS (ESI) m/z 194.9 [M + Na]+.
2-(Substituted-phenyl)-thiazole-4-carboxylic acid methoxymethylamide 7
General procedure: A solution of 6a-6p (1 equiv) in CH2Cl2 was cooled to 0°C, and distilled DBU (2 equiv) was added. Bromotrichloromethane (1.7 equiv) was then introduced dropwise via syringe over 10 min. The reaction mixtures were allowed to warm to room temperature and stirred overnight. Upon washing with satd. aqueous NH4Cl (2 × 50 mL), the aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic layers were dried on MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography as needed providing compounds 7.
2-Phenyl-thiazole-4-carboxylic acid methoxymethylamide
Yield: 73.6 %. 1H NMR (300MHz, CDCl3) δ 8.01 (s, 1 H), 7.99-7.96 (m, 2 H), 7.47-7.44 (m, 3 H), 3.88 (s, 3 H), 3.49 (s, 3 H). MS (ESI) m/z 271.0 [M + Na]+.
[2-(substituted-phenyl)-thiazol-4-yl]-(3, 4, 5-trimethoxy-phenyl)-methanone 8a-8z and 10. Method 1
To a solution of n-BuLi (1.6M, 0.713 mL) in 8 mL THF was added a solution of 3, 4, 5-Trimethoxybromobenzene (1.09 mmol) in 3 mL THF under -78°C. The mixture was stirred for 2h and a solution of amides 6 or 7 (1.14 mmol) in 3 mL THF was charged. The mixture was allowed to warm to room temperature and stirred for overnight. The reaction mixture was quenched with satd. NH4Cl, extracted with ethyl ether, dried with MgSO4 and exposed in air atmosphere for overnight. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 8a-8z. Method 2: To a solution of corresponding Grignard reagents (0.5M, 3 mL) in 2 mL THF was charged a solution of amides 6 or 7 (1 mmol) in 3 mL THF at 0 °C. The mixtures were stirred for 30 min to 2 hours until amides disappeared on TLC plates. The reaction mixture was quenched with satd. NH4Cl, extracted with ethyl ether, dried with MgSO4 and to set in air atmosphere overnight to yield 6 as starting material. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound 8a-8z. According hydrochloride salt was prepared as following: At 0 °C, to a solution of 10 mL HCl in ethyl ether (2 M) solution was added 8i, 8x or 8w (100 mg) in 5 mL CH2Cl2 (5 mL) and stirred overnight. The hydrochloride precipitate was filtered and washed with ethyl ether. Dying under high vacuum yielded the corresponding salts.
Phenyl (2-phenylthiazol-4-yl)-methanone (8a)
Yield: 76.3 %. M. p. 65-66 °C. 1H NMR (300MHz, CDCl3) δ 8.32-8.29 (m, 2 H), 8.24 (s, 1 H), 8.04-8.00 (m, 2 H), 7.64-7.52 (m, 3 H), 7.50-7.46 (m, 3 H). MS (ESI) m/z 288.0 [M + Na]+. Anal. (C16H11NOS) C, H, N.
(4-Methoxyphenyl)(2-phenylthiazol-4-yl)-methanone (8b)
Yield: 74.8 %. M. p. 105-106 °C. 1H NMR (300MHz, CDCl3) δ 8.41 (d, 2 H), 8.22 (s, 1 H), 8.02 (dd, 2 H), 7.47 (m, 3 H), 7.01 (d, 2 H), 3.80 (s, 3 H). MS (ESI) m/z 318.1 [M + Na]+. Anal. (C17H13NO2S) C, H, N.
(3-Methoxyphenyl)(2-phenylthiazol-4-yl)-methanone (8c)
Yield: 58.8 %. M. p. 43-44 °C. 1H NMR (300MHz, CDCl3) δ 8.23 (s, 1 H), 8.05-8.01 (m, 2 H), 7.93 (d, 1 H), 7.84 (m, 1 H), 7.49-7.40 (m, 4 H), 7.16-7.15 (m, 1 H), 3.89 (s, 3 H). MS (ESI) m/z 318.1 [M + Na]+. Anal. (C17H13NO2S) C, H, N.
(2-Methoxyphenyl)(2-phenylthiazol-4-yl)-methanone (8d)
Yield: 57.4 %. Colorless oil. 1H NMR (300MHz, CDCl3) δ 8.03 (s, 1 H), 7.98-7.95 (m, 2 H), 7.57-7.47 (m, 2 H), 7.47-7.42 (m, 3 H), 7.08-7.01 (m, 2 H), 3.78 (s, 3 H). MS (ESI) m/z 318.1 [M + Na]+. Anal. (C17H13NO2S) C, H, N.
(3, 4-Dimethoxyphenyl)(2-phenylthiazol-4-yl)-methanone (8e)
Yield: 15.3 %. M. p. 89-91 °C. 1H NMR (500MHz, CDCl3) δ 8.24 (s, 1 H), 8.22 (dd, 1 H, J = 8.5 Hz, 2.0 Hz), 8.04-8.02 (m, 2 H), 7.99 (d, 1 H, J = 2.0 Hz), 7.49-7.47 (m, 3 H), 6.98 (d, 1 H, J = 8.5 Hz), 3.99 (s, 6 H). MS (ESI) m/z 348.0 [M + Na]+. Anal. (C18H15NO3S) C, H, N.
(2-Phenyl-thiazol-4-yl)-(3,4,5-trimethoxy-phenyl)-methanone (8f)
Yield: 27.3 %. M. p. 133-135 °C. 1H NMR (300MHz, CDCl3) δ 8.29 (s, 1 H), 8.03 (q, 2 H), 7.80 (s, 2 H), 7.49-7.47 (m, 3 H), 3.96 (s, 6 H), 3.97 (s, 3 H). MS (ESI) m/z 378.1 [M + Na]+. Anal. (C19H17NO4S) C, H, N.
(3, 5-Dimethoxyphenyl)(2-phenylthiazol-4-yl)-methanone (8g)
Yield: 41.5 %. M. p. 84-85 °C. 1H NMR (300MHz, CDCl3) δ 8.23 (s, 1 H), 8.04-8.01 (m, 2 H), 7.99 (d, 2 H, J = 2.4 Hz), 7.49-7.43 (m, 3 H), 6.72 (t, 1 H, J = 2.4 Hz), 3.87 (s, 6 H). MS (ESI) m/z 348.3 [M + Na]+. Anal. (C18H15NO3S) C, H, N.
(2-Fluorophenyl)(2-phenylthiazol-4-yl)-methanone (8h)
Yield: 66.4 %. M. p. 77-79°C. 1H NMR (300MHz, CDCl3) δ 8.48-8.41 (m, 2 H), 8.28 (s, 2 H), 8.04-7.98 (m, 2 H), 7.50-7.46 (m, 3 H), 7.26-7.16 (m, 2 H). MS (ESI) m/z 306.0 [M + Na]+, 283.9 [M - H]-. Anal. (C16H10FNOS) C, H, N.
(2-Phenylthiazol-4-yl)-(pyridin-2-yl)-methanone (8i)
Yield: 20.7 %. M. p. 95-97°C. 1H NMR (300MHz, CDCl3) δ 9.01 (s, 1 H), 8.77 (d, 1 H, J = 4.8 Hz), 8.28 (d, 1 H, J = 7.8 Hz), 8.08-8.05 (m, 2 H), 7.92 (dt, 1 H, J = 7.8 Hz, 1.2 Hz), 7.52 (ddd, 1 H, J = 7.8 Hz, 4.8 Hz, 1.2 Hz), 7.48-7.46 (m, 3 H). (8i·HCl): Yield: 70.6 %. M. p. 105-107°C. 1H NMR (300MHz, DMSO-d6) δ 9.03 (s, 1 H), 8.79 (d, 1 H, J = 4.8 Hz), 8.10 (br, 1 H), 8.08 (br, 1 H), 8.03-8.00 (m, 2 H), 7.73-7.69 (m, 1 H), 7.56-7.54 (m, 3 H). MS (ESI) m/z 267.0 [M + H]+. Anal. (C15H10N2OS, C15H10N2OS·HCl) C, H, N.
1-(2-phenylthiazol-4-yl)-heptadecan-1-one (8j)
Yield: 66.4 %. M. p. 63-64 °C. 1H NMR (300MHz, CDCl3) δ 8.12 (s, 1 H), 8.02-7.99 (m, 2 H), 7.49-7.47 (m, 3 H), 3.16 (t, 2 H, J = 7.5 Hz), 1.82-1.72 (m, 2 H), 1.26 (s, 26 H), 0.88 (t, 3 H, J = 6.9 Hz). MS (ESI) m/z 414.4 [M + H]+. Anal. (C26H39NOS) C, H, N.
(2-p-Tolylthiazol-4-yl)-(3,4,5-trimethoxyphenyl)-methanone (8k)
Yield: 53.2 %. M. p. 116-119 °C. 1H NMR (300MHz, CDCl3) δ 8.25 (s, 1 H), 7.91 (d, 2 H, J = 8.1 Hz), 7.80 (s, 2 H), 7.28 (d, 2 H, J= 8.1 Hz), 3.96 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 392.1 [M + Na]+. Anal. (C20H19NO4S) C, H, N.
[2-(2-Fluorophenyl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8l)
Yield: 39.6 %. M. p. 90-102 °C. 1H NMR (500MHz, CDCl3) δ 8.40 (s, 1 H), 8.33 (dt, 1 H, J = 1.5 Hz, 8.0 Hz), 7.78 (s, 2 H), 7.49-7.44 (m, 1 H), 7.30-7.23 (m, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 396.1 [M + Na]+. Anal. (C19H16FNO4S) C, H, N.
[2-(3-Fluorophenyl)-thiazol-4-yl](3,4,5-trimethoxyphenyl)-methanone (8m)
Yield: 14.1 %. M. p. 122-124 °C. 1H NMR (300MHz, CDCl3) δ 8.31 (s, 1 H), 7.79 (s, 2 H), 7.76-7.74 (m, 2 H), 7.45 (dt, 1 H, J = 6.0 Hz, 8.4 Hz), 7.18 (dt, 1 H, J = 1.8 Hz, 8.4 Hz), 3.97 (s, 3 H), 3.96 (s, 6 H). MS (ESI) m/z 396.1 [M + Na]+. Anal. (C19H16FNO4S) C, H, N.
[2-(4-Fluorophenyl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8n)
Yield: 40.2 %. M. p. 153-155 °C. 1H NMR (300MHz, CDCl3) δ 8.27 (s, 1 H), 8.04-8.00 (dd, 2 H, J = 8.4 Hz, 5.7 Hz), 7.75 (s, 2 H), 7.21-7.15 (t, 3 H, J = 8.4 Hz), 3.97 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 396.1 [M + Na]+. Anal. (C19H16FNO4S) C, H, N.
[2-(3,4-Dimethoxyphenyl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8o)
Yield: 46.6 %. M. p. 145-147 °C. 1H NMR (300MHz, CDCl3) δ 8.20 (s, 1 H), 7.76 (s, 2 H), 7.58-7.54 (m, 2 H), 6.94 (d, 2 H, J = 8.1 Hz), 3.96 (s, 6 H), 3.95 (s, s, 9H). MS (ESI) m/z 438.1 [M + Na]+. Anal. (C21H21NO6S·1/4H2O) C, H, N.
[2-(4-Nitrophenyl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8p)
Yield: 46.4 %. M. p. 199-200 °C. 1H NMR (300MHz, CDCl3) δ 8.38 (d, 2 H, J = 8.7 Hz), 8.34 (s, 1 H), 8.20 (d, 2 H, J = 8.7 Hz), 7.73 (s, 2 H), 3.98 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 423.1 [M + Na]+. Anal. (C19H16N2O6S) C, H, N.
4-[4-(3,4,5-Trimethoxybenzoyl)-thiazol-2-yl]-benzonitrile (8q)
Yield: 45.9 %. M. p. 181-182 °C. 1H NMR (300MHz, CDCl3) δ 8.37 (s, 1 H), 8.13 (d, 2 H, J = 8.4 Hz), 7.78 (d, 2 H, J = 8.4 Hz), 7.72 (s, 2 H), 3.97 (s, 3 H), 3.94 (s, 6 H). MS (ESI) m/z 403.1 [M + Na]+. Anal. (C20H16N2O4S) C, H, N.
4-[4-(3,4,5-Trimethoxybenzoyl)-thiazol-2-yl]-benzoic acid (8r)
Yield: 61.9 %. M. p. >220 °C (dec.). 1H NMR (300MHz, CDCl3) δ 8.65 (s, 1 H), 8.00 (d, d, 4 H), 7.65 (s, 2 H), 3.88 (s, 6 H), 3.80 (s, 3 H). MS (ESI) m/z 397.9 [M - H]-, 353.9 [M - COOH]-,. Anal. (C20H17NO6S) C, H, N.
Methyl-4-[4-(3,4,5-trimethoxybenzoyl)-thiazol-2-yl]-benzoate (8s)
Yield: 72.5 %. M. p. 172-174 °C. 1H NMR (300MHz, CDCl3) δ 8.35 (s, 1 H), 8.12 (dd, 4 H, J = 8.4 Hz), 7.78 (s, 2 H), 3.97 (s, 3 H), 3.96 (s, 3H), 3.95 (s, 6 H). MS (ESI) m/z 436.1 [M + Na]+. Anal. (C21H19NO6S) C, H, N.
(2-(4-(Trifluoromethyl)-phenyl)-thiazol-4-yl)(3,4,5-trimethoxyphenyl)-methanone (8t)
Yield: 45.5 %. M. p. 144-145 °C. 1H NMR (300MHz, CDCl3) δ 8.35 (s, 1 H), 8.14, 7.65 (d, d, 4 H, J = 8.1 Hz), 7.76 (s, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 446.1 [M + Na]+. Anal. (C20H16F3NO4S) C, H, N.
[2-(4-Bromophenyl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8u)
Yield: 51.8 %. M. p. 149-150 °C. 1H NMR (300MHz, CDCl3) δ 8.28 (s, 1 H), 7.89, 7.62 (d, d, 4 H, J = 8.1 Hz), 7.75 (s, 2 H), 3.97 (s, 3 H), 3.94 (s, 6 H). MS (ESI) m/z 456.0, 458.0 [M + Na]+. Anal. (C19H16BrNO4S) C, H, N.
[2-(4-Ethyl-phenyl)-thiazol-4-yl]-(3,4,5-trimethoxy-phenyl)-methanone (8v)
Yield: 40.0 %. M. p. 86-87 °C. 1H NMR (300MHz, CDCl3) δ 8.25 (s, 1 H), 7.93, 7.31 (d, d, 4 H, J = 8.4 Hz), 7.81 (s, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H). MS (ESI) m/z 406.1 [M + Na]+. Anal. (C21H21NO4S) C, H, N.
[2-(4-Amino-phenyl)-thiazol-4-yl]-(3,4,5-trimethoxy-phenyl)-methanone (8w)
Yield: 61.8 %. M. p. 177-179 °C. 1H NMR (300MHz, CDCl3) δ 8.14 (s, 1 H), 7.82, 7.65 (d, d, 4 H, J = 8.4 Hz), 7.78 (s, 2 H), 3.96 (s, 3 H), 3.94 (s, 6 H). (8w·HCl): Yield: 50.1 %. M. p. 166-169 °C. 1H NMR (300MHz, DMSO-d6) δ 8.49 (s, 1 H), 7.84, 6.94 (d, d, 4 H, J = 8.4 Hz), 7.62 (s, 2 H), 3.86 (s, 3 H), 3.79 (s, 6 H). MS (ESI) m/z 393.1 [M + Na]+. Anal. (C19H18N2O4S, C19H18N2O4S·HCl) C, H, N.
[2-(Pyridin-4-yl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8x)
Yield: 29.3 %. M. p. 178-180 °C. 1H NMR (300MHz, CDCl3) δ 8.77 (dd, 2 H, J = 6.0 Hz, 1.5 Hz), 8.40 (s, 1 H), 7.87 (dd, 2 H, J = 6.0 Hz, 1.8 Hz), 7.75 (s, 2 H), 3.98 (s, 3 H), 3.95 (s, 6 H). (8x·HCl): Yield: 92.7 %. M. p. 182-184 °C. 1H NMR (300MHz, CDCl3) δ 8.85 (br, 2 H), 8.52 (s, 1 H), 8.22 (br, 2 H), 7.66 (s, 2 H), 3.98 (s, 3 H), 3.94 (s, 6 H). MS (ESI) m/z 379.1 [M + Na]+. Anal. (C18H16N2O4S, C18H16N2O4S·HCl) C, H, N.
[2-(Pyrimidin-2-yl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8y)
Yield: 51.9 %. M. p. 190-191 °C. 1H NMR (300MHz, CDCl3) δ 8.88 (d, 2 H, J = 4.8 Hz), 8.44 (s, 1 H), 7.73 (s, 2 H), 7.37 (t, 1 H, J = 4.8 Hz), 3.95 (s, 3 H), 3.94 (s, 6 H). MS (ESI) m/z 380.1 [M + Na]+. Anal. (C17H15N3O4S) C, H, N.
[2-(Thiophen-2-yl)-thiazol-4-yl]-(3,4,5-trimethoxyphenyl)-methanone (8z)
Yield: 30.5 %. M. p. 111-113 °C. 1H NMR (300MHz, CDCl3) δ 8.25 (s, 1 H), 7.90 (s, 2 H), 7.58 (dd, 1 H, J = 3.6, 0.9 Hz), 7.46 (dd, 1 H, J = 5.4, 0.9 Hz), 7.12 (dd, 1 H, J = 5.4, 3.6 Hz), 3.98 (s, 6 H), 3.97 (s, 3 H). MS (ESI) m/z 384.1 [M + Na]+. Anal. (C17H15NO4S2) C, H, N.
Thiazol-4-yl-(3,4,5-trimethoxy-phenyl)-methanone (10)
Yield: 49.4 %. M. p. 106-108 °C. 1H NMR (300MHz, CDCl3) δ 8.92 (d, 1 H, J = 2.1 Hz), 8.34 (d, 1 H, J = 2.1 Hz), 7.61 (s, 2 H), 3.94 (s, 3 H), 3.93 (s, 6 H). MS (ESI) m/z 302.0 [M + Na]+. Anal. (C13H13NO4S) C, H, N.
(2-Phenyl-thiazol-4-yl)-(3,4,5-trihydroxy-phenyl)-methanone (11)
To a solution of 8f (123 mg, 0.35 mmol) in 5 mL anh. CH2Cl2 was added BBr3 (1M solution in CH2Cl2, 1.75 mL, 5 mmol) under -78°C. The mixture was stirred for 2h and a solution of amide 7 (1.14 mmol) in 3 mL THF was charged. The mixture was allowed to warm to room temperature slowly and stirred overnight. The reaction mixture was quenched with satd. NH4Cl, extracted with ethyl acetate, dried with MgSO4. The solvent was removed under reduced pressure to yield a crude product, which was purified by column chromatography to obtain pure compound as red crystalline solid. Yield: 50.9 %. M. p. 175-176 °C. 1H NMR (300MHz, DMSO-d6) δ 8.44 (d, 1 H), 8.07-8.04 (m, 2 H), 7.57-7.55 (m, 3 H), 7.33 (s, 2 H). MS (ESI) m/z 336.1 [M + Na]+. Anal. (C16H11NO4S) C, H, N.
X-ray crystallography structure determination
X-ray crystallographic data for 8f were collected from a single crystal mounted with paratone oil on a nylon cryoloop. Data were collected at 100K on a Bruker Proteum CCD area detector, controlled by Proteum2 software,26 using a rotating-anode generator and Osmic mirrors to generate Cu radiation (λ=1.54178Å). The data were reduced using SAINT,27 with an absorption correction applied using SADABS28 based on redundant reflections; this correction included a spherical component. The structure was solved using direct methods (SHELXSx4), which revealed all of the heavy atoms. Structure refinement with SHELXL29 was carried out using full-matrix methods based on F2, and proceeded smoothly. Hydrogen atoms were added to the structural model assuming ideal C-H distances and isotropic ADPs constrained to be similar to that of the bonded carbon atom. In the final model, anisotropic ADPs were refined for all heavy atoms, and isotropic ADPs for chemically-similar hydrogens (e.g. methyl H) were constrained to be identical. The final refinement parameters are: wR2=0.084 for 228 parameters and 3066 independent observations, R1=0.031, S (goodness-of-fit)=1.057. The final structure has been submitted to the Cambridge Crystallographic Data Center for deposition.
Biology
Cell Culture and Cytotoxicity Assay of Melanoma
We examined the antiproliferative activity of the ATCAA and SMART analogues in one human melanoma cell line (A375) and one mouse melanoma cell line (B16-F1). We used activity on fibroblast cells as a control to determine the selectivity of these compounds against melanoma. A375 cells and B16-F1 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). Human dermal fibroblast cells were purchased from Cascade Biologics, Inc., Portland, OR, USA. All cell lines were cultured in DMEM (Cellgro Mediatech, Inc., Herndon, VA, USA), supplemented with 5% FBS (Cellgro Mediatech), 1% antibiotic/antimycotic mixture (Sigma-Aldrich, Inc., St. Louis, MO, USA) and bovine insulin (5 μg/ml; Sigma-Aldrich). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. Standard sulforhodamine B assay was used. Cells were exposed to a wide range of concentrations for 48 h in round-bottomed 96-well plates. Cells were fixed with 10% trichloroacetic acid and washed five times with water. After cells were air-dried overnight and stained with SRB solution, total proteins were measured at 560 nm with a plate reader. IC50 (i.e., concentration which inhibited cell growth by 50% of no treatment controls) values were obtained by nonlinear regression analysis with GraphPad Prism (GraphPad Software, San Diego, CA).
Cell Culture and Cytotoxicity Assay of prostate cancer
We examined the antiproliferative activity of the ATCAA and SMART analogues in four human prostate cancer cell lines (LNCaP, DU 145, PC-3, and PPC-1). LNCaP, PC-3 and DU 145 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). Dr. Mitchell Steiner at University of Tennessee Health Science Center kindly provided PPC-1cells. All prostate cancer cell lines were cultured in RPMI 1640 (Cellgro Mediatech, Inc., Herndon, VA, USA), supplemented with 10% FBS (Cellgro Mediatech). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. 1000 to 5000 cells were plated into each well of 96-well plates depending on growth rate and exposed to different concentrations of a test compound for 96 h in three to five replicates. Cell numbers at the end of the drug treatment were measured by the SRB assay. Briefly, the cells were fixed with 10% of trichloroacetic acid and stained with 0.4% SRB, and the absorbances at 540 nm were measured using a plate reader (DYNEX Technologies, Chantilly, VA). Percentages of cell survival versus drug concentrations were plotted and the IC50 (concentration that inhibited cell growth by 50% of untreated control) values were obtained by nonlinear regression analysis using WinNonlin (Pharsight Corporation, Mountain View, CA).
In Vitro Microtubule Polymerization Assay
Bovine brain tubulin (0.4 mg) (Cytoskeleton, Denver, CO) was mixed with various concentrations(0.625-20 μM) of test compound and incubated in 120 μl of general tubulin buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, pH 6.9 and 1 mM GTP). The absorbance of wavelength at 340 nm was monitored every 60s for 20 min by the SYNERGY 4 Microplate Reader (Bio-Tek Instruments, Winooski, VT). The spectrophotometer was set at 37 °C for tubulin polymerization. The IC50 value was defined as the concentration which can inhibit 50% of microtubule polymerization.
Supplementary Material
Scheme 2.

Reagents and conditions: (a) MeOH / pH=6.4 phosphate buffer, r. t.; (b) EDCI, HOBt, TEA, 3, 4, 5-trimethoxyaniline; (c) CBrCl3, DBU.
Acknowledgments
This research is supported by funds from Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center, the Van Vleet Endowed Professorship, and Division of Pharmaceutics, The Ohio State University. Zhao Wang is supported by the Alma & Hal Reagan Fellowship. Partial support from USPHS CA-125623 is also acknowledged. Support from the Cancer Center Support (CORE) Grant CA 21765 and that of American Lebanese Syrian Associated Charities (ALSAC) is gratefully acknowledged by C. R. Ross.
Footnotes
Abbreviations: ATCAA, 2-Aryl-thiazolidine-4-carboxylic acid amides; LPA, lysophosphatidic acid; GPCR, guanine-binding protein-coupled receptor; SMART, 4-Substituted Methoxylbenzoyl-Aryl-Thiazoles; SAR, structure-activity relationships.
Supporting Information Available. Additional spectral data for compounds, Crystallographic Information File of compound 8f and NCI data for compound ATCAA-1. These material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Cancer Facts & Figures. American Cancer Society; Atlanta, GA: 2008. [Google Scholar]
- 2.Li W, Lu Y, Wang Z, Dalton JT, Miller DD. Synthesis and antiproliferative activity of thiazolidine analogs for melanoma. Bioorg Med Chem Lett. 2007;17:4113–7. doi: 10.1016/j.bmcl.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 3.Li W, Wang Z, Gududuru V, Zbytek B, Slominski AT, Dalton JT, Miller DD. Structure-activity relationship studies of arylthiazolidine amides as selective cytotoxic agents for melanoma. Anticancer Res. 2007;27:883–888. [PubMed] [Google Scholar]
- 4.Lu Y, Wang Z, Li C-M, Li W, Dalton JT, Miller DD. Synthesis and biological evaluation of 2-arylthiazolidine-4-caboxylic acid amides for melanoma and prostate cancer. Abstracts of Papers, 234th ACS National Meeting, Boston, MA, United States. 2007 August 19-23;:MEDI-304. [Google Scholar]
- 5.Gududuru V, Hurh E, Sullivan J, Dalton JT, Miller DD. SAR studies of 2-arylthiazolidine-4-carboxylic acid amides: a novel class of cytotoxic agents for prostate cancer. Bioorg Med Chem Lett. 2005;15:4010–4013. doi: 10.1016/j.bmcl.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 6.Gududuru V, Hurh E, Dalton JT, Miller DD. Discovery of 2-Arylthiazolidine-4-carboxylic Acid Amides as a New Class of Cytotoxic Agents for Prostate Cancer. J Med Chem. 2005;48:2584–2588. doi: 10.1021/jm049208b. [DOI] [PubMed] [Google Scholar]
- 7.Raj GV, Barki-Harrington L, Kue PF, Daaka Y. Guanosine phosphate binding protein coupled receptors in prostate cancer: A review. J Urol. 2002;167:1458–1463. [PubMed] [Google Scholar]
- 8.Kue PF, Daaka Y. Essential role for G proteins in prostate cancer cell growth and signaling. J Urol. 2000;164:2162–7. [PubMed] [Google Scholar]
- 9.Guo R, Kasbohm EA, Arora P, Sample CJ, Baban B, Sud N, Sivashanmugam P, Moniri NH, Daaka Y. Expression and function of lysophosphatidic acid LPA1 receptor in prostate cancer cells. Endocrinology. 2006;147:4883–4892. doi: 10.1210/en.2005-1635. [DOI] [PubMed] [Google Scholar]
- 10.Qi C, Park J-H, Gibbs TC, Shirley DW, Bradshaw CD, Ella KM, Meier KE. Lysophosphatidic acid stimulates phospholipase D activity and cell proliferation in PC-3 human prostate cancer cells. J Cell Physiol. 1998;174:261–272. doi: 10.1002/(SICI)1097-4652(199802)174:2<261::AID-JCP13>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 11.Lu Y, Wang Z, Li C-M, Chen J, Dalton JT, Li W, Miller DD. Synthesis and SAR Study of 2-Aryl-thiazolidine-4-carboxamide Analogues for Melanoma and Prostate Cancer. unpublished article. [Google Scholar]
- 12.Schubert MP. Compounds of thiol acids with aldehydes. J Biol Chem. 1936;114:341–50. [Google Scholar]
- 13.Bergeron RJ, Wiegand J, Dionis JB, Egli-Karmakka M, Frei J, Huxley-Tencer A, Peter HH. Evaluation of desferrithiocin and its synthetic analogs as orally effective iron chelators. J Med Chem. 1991;34:2072–8. doi: 10.1021/jm00111a023. [DOI] [PubMed] [Google Scholar]
- 14.Williams DR, Lowder PD, Gu Y-G, Brooks DA. Studies of mild dehydrogenations in heterocyclic systems. Tetrahedron Lett. 1997;38:331–334. [Google Scholar]
- 15.Bergeron RJ, Wiegand J, Weimar WR, Vinson JR, Bussenius J, Yao GW, McManis JS. Desazadesmethyldesferrithiocin analogues as orally effective iron chelators. J Med Chem. 1999;42:95–108. doi: 10.1021/jm980340j. [DOI] [PubMed] [Google Scholar]
- 16.Zamri A, Abdallah MA. An improved stereocontrolled synthesis of pyochelin, siderophore of Pseudomonas aeruginosa and Burkholderia cepacia. Tetrahedron. 2000;56:249–256. [Google Scholar]
- 17.Nahm S, Weinreb SM. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981;22:3815–18. [Google Scholar]
- 18.Merino P, Tejero T, Unzurrunzaga FJ, Franco S, Chiacchio U, Saita MG, Iannazzo D, Piperno A, Romeo G. An efficient approach to enantiomeric isoxazolidinyl analogs of tiazofurin based on nitrone cycloadditions. Tetrahedron: Asymmetry. 2005;16:3865–3876. [Google Scholar]
- 19.Schneider TL, Shen B, Walsh CT. Oxidase Domains in Epothilone and Bleomycin Biosynthesis: Thiazoline to Thiazole Oxidation during Chain Elongation. Biochemistry. 2003;42:9722–9730. doi: 10.1021/bi034792w. [DOI] [PubMed] [Google Scholar]
- 20.Fuller NA, Walker J. Experiments on the dehydrogenation of some thiazolines derived from cysteine. J Chem Soc C: Organic. 1968:1526–9. doi: 10.1039/j39680001526. [DOI] [PubMed] [Google Scholar]
- 21.Mislin GL, Burger A, Abdallah MA. Synthesis of new thiazole analogs of pyochelin, a siderophore of Pseudomonas aeruginosa and Burkholderia cepacia. A new conversion of thiazolines into thiazoles. Tetrahedron. 2004;60:12139–12145. [Google Scholar]
- 22.Diana GD, McKinlay MA, Brisson CJ, Zalay ES, Miralles JV, Salvador UJ. Isoxazoles with antipicornavirus activity. J Med Chem. 1985;28:748–52. doi: 10.1021/jm00383a010. [DOI] [PubMed] [Google Scholar]
- 23.Pratt YT, Drake NL. Quinolinequinones. V. 6-Chloro- and 7-chloro-5,8-quinolinequinones. J Am Chem Soc. 1960;82:1155–61. [Google Scholar]
- 24.McOmie JFW, Watts ML, West DE. Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron. 1968;24:2289–92. [Google Scholar]
- 25.Margolis RL, Wilson L. Addition of colchicine--tubulin complex to microtubule ends: the mechanism of substoichiometric colchicine poisoning. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:3466–70. doi: 10.1073/pnas.74.8.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proteum2. Bruker AXS Inc.; Madison, Wisconsin, USA: 2005. [Google Scholar]
- 27.SAINT. Bruker AXS Inc.; Madison, Wisconsin, USA.: 1998. [Google Scholar]
- 28.SADABS. Bruker AXS Inc.; Madison, Wisconsin, USA.: 2000. [Google Scholar]
- 29.Sheldrick GM. SHELXL-97. University of Göttingen; Germany: 1997. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.