

Abstract
Background
Inolimomab, a monoclonal antibody against IL-2Rα (CD25) has shown promising results in the treatment of steroid-resistant acute graft-versus-host disease (aGvHD).
Objective
Our purpose was to characterize its pharmacokinetic (PK) and pharmacodynamic (PD) properties in first line treatment.
Methods
Data arose from 21 patients with aGvHD (8 with IBMTR at score B, 11 at score C and 2 at score D) following Hematopoietic Stem Cell Transplantation after a median delay of 26 days (10 – 127 days). Inolimomab was administrated at 0.1, 0.2, 0.3 or 0.4 mg/kg daily associated with methylprednisolone (2 mg/kg) for 8 or 16 days depending of status at day 9. Then, for responder patients, administrations were continued three times a week until day 28. Inolimomab concentrations and PD data (aGvHD scores) were collected along the study. PD data were assessed in 4 grades according to IBMTR and Glucksberg classification in parallel with Karnofsky scores. Population analysis was developed using NONMEM to define the pharmacokinetic model, to test covariates, and when apparent, to model the exposure-effect relationship by a proportional odds model. Modelling was finally qualified by predictive check.
Results
The best pharmacokinetic model was bi-compartmental. For each score, the most demonstrative exposure-effect graphics linked cumulative AUC to cumulated probabilities to observe a specific score. This relationship was identified as an Emax model for skin (with 2 patient subpopulations: sensitive/less sensitive) and a linear model for intestinal tract and liver. No covariate was identified as influent on any of these parameters.
Conclusion
Inolimomab exposure–effect relationships in first-line treatment of aGvHD have been identified and modeled. The discovered dose effect relationship allows to confirm the treatment response, then to establish the first step towards optimizing the doses of future trials.
Keywords: Acute Disease; Adult; Antibodies, Monoclonal; pharmacokinetics; pharmacology; therapeutic use; Dose-Response Relationship, Drug; Female; Glucocorticoids; therapeutic use; Graft vs Host Disease; prevention & control; Hematopoietic Stem Cell Transplantation; Humans; Immunosuppressive Agents; pharmacokinetics; pharmacology; therapeutic use; Karnofsky Performance Status; Male; Methylprednisolone; therapeutic use; Middle Aged; Models, Biological
BACKGROUND
Allogeneic Haematopoietic Stem Cell Transplantation (HSCT) is an effective and curative treatment for many hematological malignancies [1] related to the existence of a graft-versus-malignancy effect. However, it is frequently associated with graft-versus-host disease (GvHD) which is still responsible for a high rate of treatment-related mortality.[2] Acute GvHD usually involves skin, liver and the intestinal tract, but lymphoid and haematopoietic tissues can also be affected. It is induced by alloreactive T-cells from the donor, which react against the recipient’s tissues and organs. Standard GvHD prophylaxis consists in administering a combination of immunosuppressive drugs with the classical association of cyclosporine A (CsA) and methotrexate.[3] Other therapeutic approaches have been tested including T-cell depletion, that can be performed in vivo or ex vivo. The incidence of GvHD decreases, but this is at the cost of a high risk of rejection and relapse associated to a delayed immune reconstitution.[4] The first line treatment of aGvHD is based on steroids with usually methylprednisolone (MP) at the dose of 2 to 2.5 mg/kg/day.[5] However, steroid resistance is observed in approximately 40 % of the patients and therefore requires alternative treatment.[6,7] No standard therapy really exists in steroid-refractory aGvHD. Therapy could be based on high dose of steroids (10 to 15 mg/kg/day) either alone or in combination with antithymocyte globulins or monoclonal anti-T-cell preparations.[8–10] In some cases, although it cures aGvHD, this treatment is responsible for a strong immunosuppression leading to an increased incidence of severe bacterial, viral infections, and an increased risk of Epstein-Barr Virus (EBV)-related lymphoproliferative disorders.[11,12] Inolimomab (Leukotac®, OPi, Limonest, France) because of its inhibitory effects on activated T-cells, could be useful for the treatment of aGvHD. This murin monoclonal antibody (mAb) specifically targets the α chain (CD25) of the interleukine-2 (IL2) receptor. Activated T-cells express the inducible IL-2Rα chain whereas resting cells and their precursors do not. Consequently, fewer adverse experiences are expected, due to a lower and more targeted immunosuppression activity. Some clinical trials have already been performed in steroid-resistant aGvHD and have shown some promising results in terms of response and survival.[13–17]
A clinical trial of inolimomab given in association with steroids, was conducted as initial therapy of aGvHD. As the compound was well tolerated [15], this clinical trial expected to show a better, longer and less heterogeneous overall response than in steroid-resistant patients. This study presents an original population PK-PD modelling of these clinical data, showing for the first time and in this indication, an exposure-effect relationship of a mAb. The specific aims were: (i) to model the PK of inolimomab given as a repeated dose; (ii) to identify inolimomab exposure-effect relationships on different efficacy markers in aGvHD and (iii) to propose a model in order to help future dose optimization of this treatment.
METHODS
Patients and Treatment
Data were collected from an open label, dose-escalating, non-randomized phase I–II study of inolimomab in association with steroids (MP at 2 mg/kg) as first line therapy for grade II to IV aGvHD following allogeneic HSCT. The main objective of this trial was to establish pharmacokinetics of four dosages of inolimomab. Six French institutions participated in the study (Lyon-Hôpital Edouard Herriot, Clermont-Ferrand-Centre Jean Perrin, Marseille-Institut Paoli Calmettes, Lille-Hôpital Claude Hurriez, Nantes-Hôtel Dieu, Créteil-Hôpital Henri Mondor). The study protocol was approved by the Independent Ethics Committee of Lyon-Centre Léon Bérard. Inclusion criteria were as follows: grade II to IV aGvHD following HSCT using either allogeneic bone marrow or allogeneic peripheral blood progenitor cells, and patients had to be 18 years old with written informed consent to participate in the study. Patients were not included if steroids were part of prophylaxis of aGvHD or in case of aGvHD occurring after the donor lymphocyte infusions. Diagnosis and classification of aGvHD was done according to the Seattle criteria (Glucksberg classification) [18,19] and the IBMTR [20] classification.
After eligibility was confirmed, 21 patients were registered and assigned to 1 of 4 cohorts to receive 30-minute intravenous (IV) inolimomab infusion (0.10, 0.20, 0.30, and 0.40 mg/kg), with 5 patients in each dose group except 0.30 mg/kg group which had 6 patients. The treatment was divided into the induction and maintenance regimen phases. The induction regimen was given from day 1 to day 8 and consisted in a once daily IV infusion of inolimomab at the patient’s assigned dose level. The clinical response assessed at day 9 determined subsequent treatment. Patients with complete response were assigned to receive the maintenance regimen. Patients with Partial Response, Mixed Response, No Response or Progression disease were reassigned to the induction regimen for one week. The maintenance regimen consisted in the administration of IV inolimomab three times a week at the patient’s induction dose level. During both the induction and maintenance regimens, all patients received concomitant IV infusion of MP. Patients received from 6 to 22 administrations of inolimomab depending on their induction and maintenance phase durations. The entire treatment period lasted a maximum of four weeks.
A total of 21 patients (12 men and 9 women; age range, 29–61 years; weight range, 49–93 kg) were enrolled in the trial (Table I). According to their initial disease, they could be classified in 3 groups: 6 patients with good prognosis transplantation of success (for CML in chronic phase, AML, or ALL in first complete remission), 9 with intermediate prognosis (for AML, ALL in second complete remission or above, NHL, myeloma, or hodgkin disease in partial response), and 6 with a poor prognosis (disease refractory or in relapse).
Table I.
Patient’s disease, pretreatment, and donor characteristics.
| Characteristics | Number |
|---|---|
| Initial disease | |
| acute myeloblastic leukemia (AML) | 5 |
| chronic lymphoïd leukemia (CLL) | 2 |
| non hodgkin lymphoma (NHL) | 2 |
| hodgkin disease | 1 |
| myelodisplasia | 2 |
| chronic myeloid leukemia (CML) | 1 |
| acute lymphoblastic leukemia (ALL) | 1 |
| multiple myeloma | 1 |
| myeloid splenomegaly | 1 |
| solid tumors | 5 |
| Transplantation | |
| first | 17 |
| second | 4 |
| Type | |
| peripheral blood stem cell | 15 |
| bone marrow | 6 |
| Status after | |
| complete remission | 8 |
| partial response | 6 |
| stable disease | 0 |
| relapse | 1 |
| progressive disease | 5 |
| chronic phase | 1 |
| Conditionning regimen | |
| non myeloablative | 13 |
| myeloablative | 8 |
| Acute GVHD prophylaxis | |
| Cyclosporine | |
| yes | 20 |
| no | 1 |
| Methotrexate | |
| yes | 8 |
| no | 13 |
| Steroids | |
| yes | 0 |
| no | 21 |
| Other drug | |
| yes | 10 |
| no | 11 |
| Donor and recipient compatibility | |
| Gender | |
| yes | 10 |
| no | 11 |
| ABO | |
| yes | 9 |
| minor incompatibility | 7 |
| major incompatibility | 5 |
| Histocompatibility (HLA) | |
| matched sibling donor | 17 |
| matched unrelated donor | 2 |
| mismatched unrelated donor | 2 |
PK sampling
The median number of PK samples per patient was 12 (range 7–23) and the total number was 318. Blood samples for PK (including peak and trough levels of inolimomab) were collected on day 1 prior to the infusion then 30 minutes, 2, 8 and 16 after completion of the infusion; prior to the infusion and 30 minutes after completion of the administration of study medication on days 2, 3 and 8; and prior to the infusion and 30 minutes after completion of the administration of study medication from day 9 to day 28 for the first three infusions. After collection, they were centrifuged and serums were stored at −20 °C until analysis.
Bioanalysis
Quantification was then carried out in the OPi Research department by a validated inolimomab Enzyme-Linked ImmunoSorbent Assay (ELISA) according to GLP. To trap inolimomab, serum samples were put on a coated plate with goat polyclonal anti-mouse Ig antibodies (GAM). Next, sheep polyclonal anti-mouse IgG1 antibodies used as tracer antibodies were added to the mixture. After incubation with TMB-substrate, the reaction was stopped by the addition of sulfuric acid and absorption was read photometrically to quantify the samples. The range of the immunoassay was from 0.15 to 10 μg/ml, with a sensitivity > 100 ng/ml, a sample intra-assay variation at 8 % and a sample inter-assay variation at 11%.
PD assessments
Acute GvHD grades and performance status were evaluated daily until day 9 then at each administration, from day 10 to day 28, as well as at follow-up (day 60 and day 100) using the Glucksberg–Seattle, IBMTR and Karnofsky classifications [21] (defined in the study as composite scores). Glucksberg criteria determines aGvHD severity (from grades 0, corresponding to no GvHD, to 4, maximum severity) by a combination of different organ scores (skin, intestinal tract and liver) and a decrease in clinical performance. These organs are graded independently from 0 to 4 corresponding respectively to extend of a skin rash, a diarrhea volume, and total bilirubin concentrations. The detailed definition of IBMTR score is given in annex 1. Briefly, it involves the same organs but assigns the score based on maximum involvement in an individual organ system. Therefore, it tends to assign a higher overall grade for aGvHD severity than the Glucksberg.[20] The Karnofsky scale defines overall performance status of a patient from 100% for a normal person with no complaint and no sign of disease to 0 % for a moribund. Independent organs as well as composite scores were considered for pharmacodynamics analysis.
PK analysis
PK and PD analysis were carried out with mixed-effect modeling using NONMEM software version V.[22] Different PK models were tested, including one, two and three compartment models, coupled with linear or nonlinear processes, such as saturable elimination. Inter-individual PK parameter variability was assumed to follow log-normal distribution with non-zero correlations. Residual unexplained variability was modeled as multiplicative. FOCE INTERACTION method was used to fit all PK models. Models were evaluated through goodness of fit plots [23–25] and the parameter precision estimated by asymptotic covariance matrix. Nested models were compared according to likelihood ratio tests (decrease of NONMEM objective function between reduced and full model by Δ=3.84, corresponding to nominal p-value 0.05, for one additional parameter).
PK-PD analysis
For each pharmacodynamic time point assessment, inolimomab exposures were estimated from individual PK profiles predicted from the previously described model. They were defined either as maximal serum concentration (Cmax), Area Under the serum concentration-time Curve (AUC) over last dosing interval, cumulated AUC, or AUC intensity (cumulated AUC/duration) from the first to the last dosing before PD assessment. Cumulated AUC corresponded to the cumulated sum of all AUCs computed for each dosing interval before PD assessment; duration used in AUC intensity calculation corresponded to the treatment duration before PD assessment. The graphical exploration of the exposure-effect relationships was performed with all PD assessments by plotting estimated cumulative probabilities of ordered scores (composite and organ scores) vs. distribution quartiles (25, 50, 75 and 100%) of the above defined drug exposures. Apparent relationships were then quantified by proportional odds models.[26] Cumulative probabilities of the observed score were linked to PK exposure through logit transformation. The nature of this link was tested with different PD models, like Emax, log-linear or linear models. Inter-patient variability of some key parameters was assumed to follow either a normal or a log normal distribution. Parameter estimation was performed using the Laplacian estimation method in NONMEM. The adequacy of the different developed models and selection of the basic model was evaluated by comparing predicted and observed probabilities.
Model Qualification
The model qualification for PK and PK-PD model was conducted in two steps. In regards to the PK model, after inspection of the basic graphics (predictions of a typical patient versus observations, individual predictions versus observations, weighted residuals versus observations, individual predictions and observations versus time), a visual predictive check was conducted.[27] It consisted in simulating (with NONMEM) 200 new datasets with identical patients, dosage regimens, sampling times, and then comparing graphically the simulated concentrations with the observed ones. The qualification of the PK-PD model was also based on a visual predictive check. Here, the purpose was to test the model performance in order to predict probabilities to observe the different grades. Therefore, we compared graphically simulated grades function of exposure, with the observed ones. Then, in order to qualify the PK-PD model for its clinical purpose, a predictive check was more specifically conducted.[28,29] It consisted in simulating (using NONMEM) 1000 new datasets with identical patients, score record time, and then comparing a test statistic deduced from these simulations with observed ones. This test statistic, which is a test quantity that depends only on data was chosen in order to highlight treatment effect.
RESULTS
PK analysis
Amongst all PK models tested, the best results were obtained with a two-compartment model. The goodness of fit plot for this model showed that the mean population and individual predicted concentrations were in good agreement with the observed ones (close to identity) except for a few concentrations over 20μg/ml which were under-predicted. Other tested models included a third compartment or a Michaelis-Menten elimination. The three-compartment model was not identifiable and a non-linear elimination did not show a significant improvement fit, therefore the two-compartment model was eventually retained. The correlation between all PK parameters was then introduced. Parameter estimates are presented on Table II. The half-life of the compound for a typical patient was equal to 44.5h.
Table II.
Population pharmacokinetic parameters obtained from the final model.
| Parameters | Estimate | SE (%) |
|---|---|---|
| Fixed effects | ||
| Clearance (l/hr) | 0.077 | 19 |
| Volume of compartment 1 (Volume 1) (l) | 2.76 | 26 |
| Inter-compartmental clearance (l/hr) | 2.22 | 52 |
| Volume of compartment 2 (Volume 2) (l) | 2.25 | 18 |
| Random effects | ||
| IIV Clearancea | 43% | 87 |
| IIV Volume 1 | 68% | 90 |
| IIV Inter-compartmental clearance | 43% | 425 |
| ρ IIV Clearance, Volume1b | −0.05 | |
| ρ IIV Clearance, Inter-compartmental clearance | −0.76 | |
| ρ IIV Volume 1, Inter-compartmental clearance | −0.62 | |
| Residual error | 0.0913 | 7 |
IIV = inter-individual variability on a fixed effect;
ρ= correlation coefficient between all individual estimations of two parameters.
The most important goodness-of-fit plots of the final PK model are presented in Figure 1. Although it shows a slight under prediction for high concentrations, a visual predictive check (Figure 2) revealed that this has no impact on predicted concentrations: the proportion of concentration points outside the 80 % confidence interval band was in agreement with the expected one. The PK model was consequently considered to be qualified.
Figure 1.
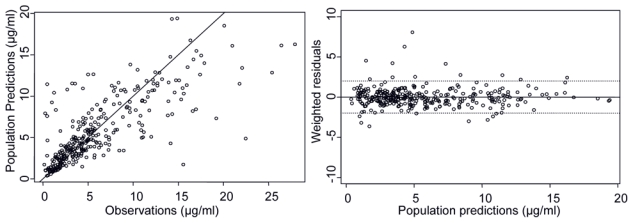
Goodness of fit plots of final PK model. Right plot: solid line represents identity line of population predictions and observation. On weighted residual plots, solid line define zero and dotted lines values for −1.96 and +1.96, the 2.5 and 97.5% quantiles of normal distribution.
Figure 2.
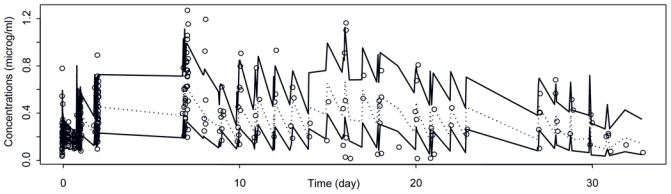
Visual predictive check of final PK model from 200×21 simulated patients. In order to normalize the scale, all concentrations (observed and predicted) are divided by actual received doses. Black lines: 80% confidence interval of predicted concentrations. Dotted line: median of predicted concentrations. Black circles: observed concentrations.
PK-PD analysis
PD assessments included composite scores, with a median number per subject of IBMTR scores at 13, ranging (3–23), a median number per subject of Glucksberg scores at 12, ranging (3–23), and a median number per subject of Karnofsky scores at 18, ranging (7–25). Organ scores (skin, intestinal tract or liver) were recorded with a median number per subject at 18, ranging (7–25). The PK-PD exploratory graphical analysis revealed that composite scores (IBMTR, Glucksberg and Karnofsky) were apparently not related to drug exposure (see Figure 3 with cumulated AUC, other graphics concerning Cmax, AUC and AUC intensity are not shown). On the contrary, organ scores (skin, intestinal tract and liver) revealed patient improvements (i.e. lowest grade probability increase) when plotted versus drug exposure expressed as cumulated AUC or AUC intensity (see cumulated AUC in Figure 4). When AUC or Cmax measured over last dosing interval were used, this relationship was less clear. Consequently, it was chosen to develop the PK-PD model for the 3-organ score cumulated probabilities, conditional on cumulated AUC, and AUC intensity. Based on goodness of fit and parameter uncertainty, only the relationship between cumulated AUC and observed scores was finally considered. In regards to skin, after merging grade 3 and 4 (since only four grades 4 were observed), an Emax model with inter-individual variability on logit and EA50 (cumulated AUC producing 50 % of maximum effect) gave the best results according to equation:
| (1) |
where P(Y≤j) represents the probability to obtain a score Y for skin inferior or equal to the grade j, alphaj represents intercept of the logit for the grade j and γ the coefficient of sigmoïdicity. Individual EA50i distribution across the population revealed two groups of patients (i=1 or 2), one with high EA50, another with low EA50. A mixture probability was then added to EA50i random effect in order to estimate the proportion of those two sub-populations and their respective EA50.[30] For intestinal tract and liver score, the best results were obtained with a linear model (see equation 2) and an inter-individual variability on the logit. All parameter estimates are presented in Table III.
Figure 3.
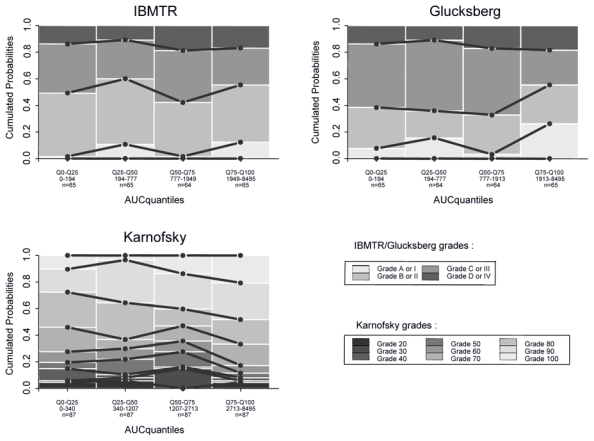
Observed cumulated probabilities of composites scores (IBMTR, Gluscksberg, and Karnofsky) function of predicted cumulated AUC. Data are split in 4 intervals according to quantiles 25, 50 and 75%. It allows for a sufficient number of data (at least n=65), to represent evolution of probability to observe each grade versus different values of predicted cumulated AUC. Solid black line: link between observed cumulated probabilities of a same grade. Black, gray and white bars: observed cumulated probabilities (P(Y≤j).
Figure 4.
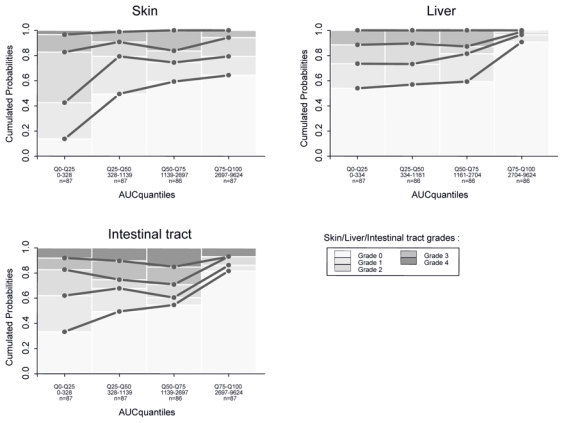
Observed cumulated probabilities of organs scores (Skin, Liver, and Intestinal tract) function of predicted cumulated AUC. For other details, see legend Fig. 4..
Table III.
Population pharmacodynamic parameters obtained from the final models.
| Parameters | Estimate | SE (%) |
|---|---|---|
| Skin model | ||
| Fixed effects | ||
| logit of baseline probability, grade 0 (alpha 0) | −4.79 | 21 |
| logit of baseline probability, grade 1 (alpha 1) | 3.14 | 17 |
| logit of baseline probability, grade 2 (alpha 2) | 2.64 | 25 |
| Emax (maximum effect) | 13.4 | 23 |
| Sigmoïdicity factor | 1 | a |
| EA50 population 1 | 15900 | 35 |
| EA50 population 2 | 609 | 36 |
| Population 1 repartition | 0.35 | 31 |
| Random effects | ||
| IIV EA50 | 0.1 | 45 |
| IIV logit | 7.2 | 57 |
| Gut model | ||
| Fixed effects | ||
| logit of baseline probability, grade 0 (alpha 0) | −0.3 | a |
| logit of baseline probability, grade 1 (alpha 1) | 1.72 | 13 |
| logit of baseline probability, grade 2 (alpha 2) | 1.55 | 25 |
| logit of baseline probability, grade 3 (alpha 3) | 1.75 | 33 |
| slope | 0.000339 | 60 |
| Random effects | ||
| IIV logit | 16.5 | 36 |
| Liver model | ||
| Fixed effects | ||
| logit of baseline probability, grade 0 (alpha 0) | 2 | a |
| logit of baseline probability, grade 1 (alpha 1) | 2.42 | 14 |
| logit of baseline probability, grade 2 (alpha 2) | 3.19 | 19 |
| slope | 0.000429 | 87 |
| Random effects | ||
| IIV logit | 50.2 | 51 |
fixed parameter.
| (2) |
where Emax model of Equation 1 is replaced by a linear model defined by a slope.
A visual predictive check of the PK-PD models for the three organs (Figure 5) revealed an overall good agreement – between 80 % confidence interval band and observed grade probabilities. The next step consisted in global qualification of the PK-PD analysis. For this purpose we evaluated the model by predictive check. We evaluated whether the combination of the PK model and the 3-organ score models (for skin, intestinal tract and liver), correctly predicted global effect therapy expressed by the IBMTR score. In this way, our model could be used to verify a treatment effect. The chosen test statistics was the number of the observed grade at a given time, either calculated from the observed data or predicted conditional on the model. Simulation was performed for the overall treatment duration, but test statistic is only presented at treatment start (top of the Figure 6) and day 28 (bottom of the Figure 6), for which 11 patients remained at treatment end. This graph revealed a good agreement of the 90 % confidence interval band with the observed IBMTR score. The combination of the three models to obtain the IBMTR score is qualified and can be used to define the effect of treatment without modelling the IBMTR score itself. The comparison of the two rows revealed that IBMTR scores decreased with treatment time: IBMTR at day 1 had more probability to be observed at a grade 2 or 3 and eventually 4, whereas at day 28 the highest probability observed and predicted is at grade 0 and then 1 or 2. This observation confirmed the global therapy effect as it was already shown in PKPD profiles of each organ.
Figure 5.
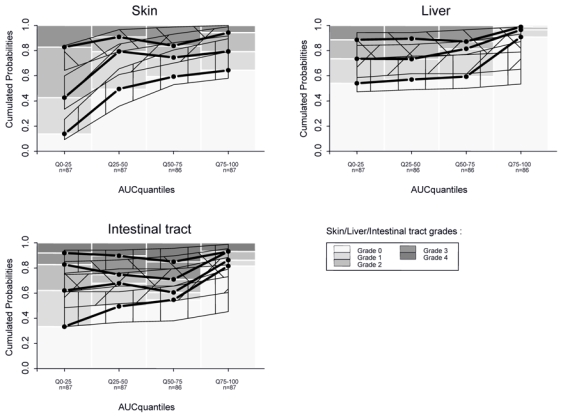
Visual predictive check of final PK-PD models from 200×21 simulated patients. Observations plotted in Figure 4 are compared with model predictions. Thin, solid and black line: prediction and 80% confidence interval of predicted cumulated probabilities.
Figure 6.
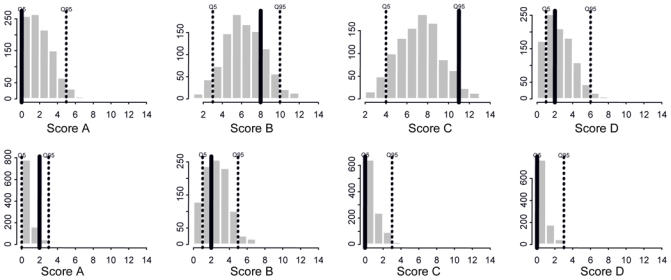
Predictive check of final PK-PD models. Histogram of IBMTR scores from 1000×21 simulated patients at treatment start (top row) and from 1000×11 simulated patients still in the study at day 28 (bottom row). Dotted line: 90% confidence interval of simulated IBMTR. Black line: observed IBMTR in 21 patients at treatment start (top row) and in 11 patients still in the study at day 28 (bottom row).
DISCUSSION
Following promising results observed in 32 steroid-resistant patients who presented aGvHD [31], and more recently, in a retrospective analysis concerning 85 steroid-resistant patients grade II–IV aGvHD [32], inolimomab was proposed as an up-front therapy. As in these clinical trials authors have already observed heterogeneity within organ response with a better and prolonged response for cutaneous aGvHD [32], it therefore clearly appeared that the PD of inolimomab needed to be investigated. Concerning inolimomab and concomitant treatments, patients have received very different exposures in terms of duration as well as dosage. Some authors carried out a multivariate analysis suggesting that a higher total dose of inolimomab might be predictive of a better response.[32] In this context, it also appeared important to identify the PK of this drug [32] in order to take into account in analyses the real inolimomab treatment exposure, and to understand more precisely the PD and its relation to the PK. This type of approach in aGvHD is not very widespread and has not previously involved any type of monoclonal antibody. Only a few authors have already tried to link the PK to the PD for prophylactic treatment. For instance, some noticed a correlation between cyclosporine trough blood concentrations in the early post transplantation period and the probability to observe an aGvHD.[33] By splitting the population into four groups, from no aGvHD, mild aGvHD, moderate aGvHD to severe aGvHD, they observed a decrease in the mean of cyclosporine trough blood concentrations whatever the time periods. Other authors defined binary criteria as the probability to observe at least grade II aGvHD and linked this criteria to busulfan AUC at steady state by a logistic function.[34] Finally, other authors used a threshold value of AUC of unbound mycophenolate in week 1 after transplant to define 2 groups of patients and observed different cumulative proportions of observing a grade II–IV in function of time.[35]
Our study modeled the PK of inolimomab and succeeded to model its exposure-effect relationship. For PK modeling, the population approach taking into account design heterogeneity and individual treatment history, allowed to identify a two-compartment model. Despite some under predictions in the higher concentrations, observations were on the whole well predicted and the model was qualified for calculating individual treatment exposure, which could not be directly computed from the observed data.
To highlight PK-PD relationships, we initially considered aGvHD according to Glucksberg and IBMTR reference scores, and the Karnofsky classification. Since those combined scores are not arranged in order (i.e. grade 0 < grade I < grade II…), the PK-PD relationship is not easy to reveal. For instance, a grade B in IBMTR classification can correspond to grade 2-0-0 or a grade 0-1-2 for skin, liver and intestinal tract, respectively. We found out that the calculation of IBMTR and Glucksberg grades, as well as of the global performance provided by the Karnofsky score were not adapted to highlight an exposure-effect relationship of inolimomab. Actually, some authors have already identified a better efficacy of this compound on a cutaneous form of aGvHD. [32] In the case of targeted efficacy on one organ, one can easily understand that a composite measure of the effect is not relevant for a PK-PD analysis.
Our PK-PD analysis logically focused on each of the three organs and on treatment exposure. It clearly appeared that a relationship was significant in whatever treatment exposure measurement was chosen. The treatment effect was found saturable for skin and was modelled with an Emax model. A mixture model revealed two populations of patients, sensitive and non sensitive.[36–38] For liver as well as for intestinal tract, effect appeared for high cumulated AUC. It is illustrated by a large decrease of patients with severe symptoms (grade 4) and an important increase of patients without symptoms (grade 0). This means that a larger exposure than for skin is required to reach same efficacy for those organs. It also explains why clinically, skin is the first organ to be cured whereas it seems to be more difficult to treat the liver and the intestinal tract. It is illustrated by PK-PD graphs at day 8: from 15 to 20 % of patients still present highest grade for these two organs. Some authors have explained this phenomenon by the difference of bioavailability or pathophysiology of aGvHD depending on organ involvement.[32]
Although the modelling of the IBMTR score could not be performed, this approach allowed us to easily simulate this score by the combination of the three organs on which it is based. It was also possible to use it to define the overall response at the end of treatment and therefore to verify a treatment effect. However, the benefit of this type of approach is that it takes into account the time dependency of the data. In this way, our models can predict the effect for a given patient and a treatment schedule, in function of time throughout the study (see Figure 7) and also give a more precise idea of the disease evolution under treatment. It is also possible to know if one organ presents a rapid or a slow remission. However, these predictions for the time being must be used with caution. As in our models, the effect is related to cumulated AUC and we modelled the PK-PD relationship by positive monotonous functions, predicted effects automatically increase with time. Therefore, no aGvHD relapse could be predicted by the model. Moreover, based on cumulated AUC, we will obtain the same response for a concentration y during x hours or a concentration x during y hours, regardless of the values of x and y.[39] In these conditions, the dosage or regimen optimization based on modelling is not possible. In fact, we used cumulated AUC because we assumed there would be a delay between concentrations and effect. In our case, the nature of the PD data did not allow us to model this delay in another way. However, perspectives of this work, with future data, will be to link scores to serum concentrations using a more physiologic model taking into account drug and effect accumulation. In this case, dosage and regimen optimization of future trial as well as individual therapeutic monitoring of patients could be performed.
Figure 7.
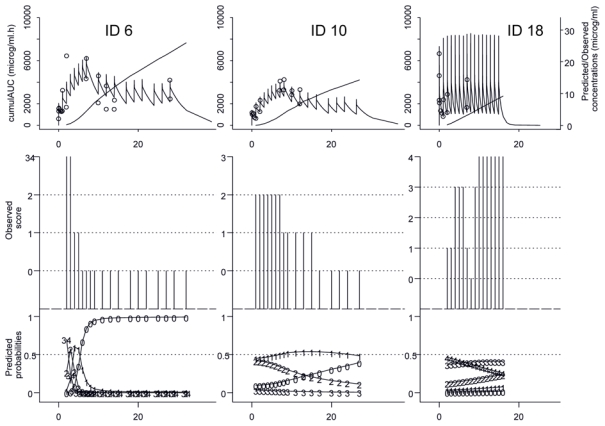
Observations and individual predictions versus time of final PK and PKPD models for 3 patients enrolled in the clinical trial. On the top: Observed concentrations (O) and PK predictions as well as predicted corresponding cumulated AUC (−). In the middle: observed PD grades. In the bottom: predicted probability of a grade observation. For ID 6, PD results for the skin, for ID 10, PD results for the liver, and for ID 18, PD results for intestinal tract.
CONCLUSION
Finally, with this analysis, we highlighted and modelled a PK-PD relationship between cumulated AUC of inolimomab and skin, liver and intestinal tract scores. The modelling of the data allowed us to describe observations as well as to predict an overall response at the end of treatment for this population through scores of IBMTR. This approach, validated for its objective, allowed us to understand better the treatment effect over time and represents the first step towards optimizing the doses of future patients. However, it does still present some limitations, due in particular to the limited number of available patients. They were clearly identified, and future trials are needed to improve clinical use of these models.
ANNEXE 1.
Criteria for IBMTR severity index for acute GvHD.
| Skin involvement | Liver involvement | Gastrointestinal involvement | ||||
|---|---|---|---|---|---|---|
| Indexa | Stage (max) | Extent of rash | Stage (max) | Total bilirubin (μmol/l) | Stage (max) | Volume of diarrhoea (ml/d) |
| A | 1 | < 25% or | 0 | < 34 or | 0 | < 500 |
| B | 2 | 25–50% or | 1–2 | 34–102 or | 1–2 | 550–1500 |
| C | 3 | > 50% or | 3 | 103–255 or | 3 | > 1500 |
| D | 4 | Bullae or | 4 | > 255 or | 4 | Severe pain and ileus |
assign index based on maximum involvement in an individual organ system.
Acknowledgments
Authors wish to thank OPi for providing the pharmacokinetic and pharmacodynamic samples, for reviewing and approving the manuscript.
The funding of this study was provided by a support from OPi and Medicine Faculty of Lyon Sud. I. Darlavoix and C Vermot-Desroches are employees of OPi. C. Dartois’s PhD is supported by Institut de Recherches Internationales Servier. Pascal Girard is supported by INSERM, France. Other authors have no conflict of interests.
REFERENCE LIST
- 1.Storb R. Allogeneic hematopoietic stem cell transplantation--yesterday, today, and tomorrow. Exp Hematol. 2003 Jan;31( 1):1–10. doi: 10.1016/s0301-472x(02)01020-2. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara JL, Deeg HJ. Graft-versus-host disease. N Engl J Med. 1991 Mar 7;324( 10):667–74. doi: 10.1056/NEJM199103073241005. [DOI] [PubMed] [Google Scholar]
- 3.Storb R, Pepe M, Deeg HJ, et al. Long-term follow-up of a controlled trial comparing a combination of methotrexate plus cyclosporine with cyclosporine alone for prophylaxis of graft-versus-host disease in patients administered HLA-identical marrow grafts for leukemia. Blood. 1992 Jul 15;80( 2):560–1. [PubMed] [Google Scholar]
- 4.Keever CA, Small TN, Flomenberg N, et al. Immune reconstitution following bone marrow transplantation: comparison of recipients of T-cell depleted marrow with recipients of conventional marrow grafts. Blood. 1989 Apr;73( 5):1340–50. [PubMed] [Google Scholar]
- 5.Ruutu T, Niederwieser D, Gratwohl A, et al. A survey of the prophylaxis and treatment of acute GVHD in Europe: a report of the European Group for Blood and Marrow, Transplantation (EBMT). Chronic Leukaemia Working Party of the EBMT. Bone Marrow Transplant. 1997 Apr;19( 8):759–64. doi: 10.1038/sj.bmt.1700745. [DOI] [PubMed] [Google Scholar]
- 6.Martin PJ, Schoch G, Fisher L, et al. A retrospective analysis of therapy for acute graft-versus-host disease: secondary treatment. Blood. 1991 Apr 15;77( 8):1821–8. [PubMed] [Google Scholar]
- 7.Weisdorf D, Haake R, Blazar B, et al. Treatment of moderate/severe acute graft-versus-host disease after allogeneic bone marrow transplantation: an analysis of clinical risk features and outcome. Blood. 1990 Feb 15;75( 4):1024–30. [PubMed] [Google Scholar]
- 8.Ross WA. Treatment of Gastrointestinal Acute Graft-Versus-Host Disease. Curr Treat Options Gastroenterol. 2005 Jun;8( 3):249–58. doi: 10.1007/s11938-005-0017-9. [DOI] [PubMed] [Google Scholar]
- 9.Akpek G, Lee SM, Anders V, et al. A high-dose pulse steroid regimen for controlling active chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2001;7( 9):495–502. doi: 10.1053/bbmt.2001.v7.pm11669216. [DOI] [PubMed] [Google Scholar]
- 10.Mollee P, Morton AJ, Irving I, et al. Combination therapy with tacrolimus and anti-thymocyte globulin for the treatment of steroid-resistant acute graft-versus-host disease developing during cyclosporine prophylaxis. Br J Haematol. 2001 Apr;113( 1):217–23. doi: 10.1046/j.1365-2141.2001.02741.x. [DOI] [PubMed] [Google Scholar]
- 11.Curtis RE, Travis LB, Rowlings PA, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi-institutional study. Blood. 1999 Oct 1;94( 7):2208–16. [PubMed] [Google Scholar]
- 12.Micallef IN, Chhanabhai M, Gascoyne RD, et al. Lymphoproliferative disorders following allogeneic bone marrow transplantation: the Vancouver experience. Bone Marrow Transplant. 1998 Nov;22( 10):981–7. doi: 10.1038/sj.bmt.1701468. [DOI] [PubMed] [Google Scholar]
- 13.Hertenstein B, Stefanic M, Sandherr M, et al. Treatment of steroid-resistant acute graft-vs-host disease after allogeneic marrow transplantation with anti-interleukin-2 receptor antibody (BT563) Transplant Proc. 1994 Dec;26( 6):3114–6. [PubMed] [Google Scholar]
- 14.Herbelin C, Stephan JL, Donadieu J, et al. Treatment of steroid-resistant acute graft-versus-host disease with an anti-IL-2-receptor monoclonal antibody (BT 563) in children who received T cell-depleted, partially matched, related bone marrow transplants. Bone Marrow Transplant. 1994 May;13( 5):563–9. [PubMed] [Google Scholar]
- 15.Cuthbert RJ, Phillips GL, Barnett MJ, et al. Anti-interleukin-2 receptor monoclonal antibody (BT 563) in the treatment of severe acute GVHD refractory to systemic corticosteroid therapy. Bone Marrow Transplant. 1992 Nov;10( 5):451–5. [PubMed] [Google Scholar]
- 16.Herve P, Bordigoni P, Cahn JY, et al. Use of monoclonal antibodies in vivo as a therapeutic strategy for acute GvHD in matched and mismatched bone marrow transplantation. Transplant Proc. 1991 Feb;23( 1 Pt 2):1692–4. [PubMed] [Google Scholar]
- 17.Herve P, Wijdenes J, Bergerat JP, et al. Treatment of corticosteroid resistant acute graft-versus-host disease by in vivo administration of anti-interleukin-2 receptor monoclonal antibody (B-B10) Blood. 1990 Feb 15;75( 4):1017–23. [PubMed] [Google Scholar]
- 18.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974 Oct;18( 4):295–304. doi: 10.1097/00007890-197410000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation (second of two parts) N Engl J Med. 1975 Apr 24;292( 17):895–902. doi: 10.1056/NEJM197504242921706. [DOI] [PubMed] [Google Scholar]
- 20.Rowlings PA, Przepiorka D, Klein JP, et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol. 1997 Jun;97( 4):855–64. doi: 10.1046/j.1365-2141.1997.1112925.x. [DOI] [PubMed] [Google Scholar]
- 21.Karnofsky DA, Burchenal JH. The clinical evaluation of chemotherapeutic agents in cancer. In: Macleod CM, editor. Evaluation of Chemotherapeutic Agents. New York: Columbia University Press; 1949. pp. 199–205. [Google Scholar]
- 22.Boeckmann AJ, Sheiner L, Beal SL. NONMEM project group. San Francisco: University of California; 1998. NONMEM User’s Guides. [Google Scholar]
- 23.Ette EI, Ludden TM. Population pharmacokinetic modeling: the importance of informative graphics. Pharm Res. 1995 Dec;12( 12):1845–55. doi: 10.1023/a:1016215116835. [DOI] [PubMed] [Google Scholar]
- 24.Food and Drug Administration. Guidance for industry - Population pharmacokinetics. U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER); 1999. Feb, [Google Scholar]
- 25.Wade JR, Edholm M, Salmonson T. A guide for reporting the results of population pharmacokinetic analyses: a swedish perspective. AAPS PharmSci. 2005 Oct;7( 2):45. doi: 10.1208/aapsj070245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agresti A. Modelling ordered categorical data: recent advances and future challenges. Stat Med. 1999 Sep 15;18( 17–18):2191–207. doi: 10.1002/(sici)1097-0258(19990915/30)18:17/18<2191::aid-sim249>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 27.Duffull SB, Chabaud S, Nony P, et al. A pharmacokinetic simulation model for ivabradine in healthy volunteers. Eur J Pharm Sci. 2000;10( 4):285–94. doi: 10.1016/s0928-0987(00)00086-5. [DOI] [PubMed] [Google Scholar]
- 28.Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001 Apr;28( 2):171–92. doi: 10.1023/a:1011555016423. [DOI] [PubMed] [Google Scholar]
- 29.Rubin DB. Bayesianly justifiable and relevant frequency calculations for the applied statistician. Ann Stat. 1984;12:1151–72. [Google Scholar]
- 30.Shiiki T, Hashimoto Y, Inui K. Simulation for population pharmacodynamic analysis of dose-ranging trials: usefulness of the mixture model analysis for detecting nonresponders. Pharm Res. 2002 Jun;19( 6):909–13. doi: 10.1023/a:1016181505556. [DOI] [PubMed] [Google Scholar]
- 31.Cahn JY, Bordigoni P, Tiberghien P, et al. Treatment of acute graft-versus-host disease with methylprednisolone and cyclosporine with or without an anti-interleukin-2 receptor monoclonal antibody. A multicenter phase III study. Transplantation. 1995 Nov 15;60( 9):939–42. [PubMed] [Google Scholar]
- 32.Bay JO, Dhedin N, Goerner M, et al. Inolimomab in steroid-refractory acute graft-versus-host disease following allogeneic hematopoietic stem cell transplantation: retrospective analysis and comparison with other interleukin-2 receptor antibodies. Transplantation. 2005 Sep 27;80( 6):782–8. doi: 10.1097/01.tp.0000173995.18826.de. [DOI] [PubMed] [Google Scholar]
- 33.Martin P, Bleyzac N, Souillet G, et al. Relationship between CsA trough blood concentration and severity of acute graft-versus-host disease after paediatric stem cell transplantation from matched-sibling or unrelated donors. Bone Marrow Transplant. 2003 Oct;32( 8):777–84. doi: 10.1038/sj.bmt.1704213. [DOI] [PubMed] [Google Scholar]
- 34.Andersson BS, Thall PF, Madden T, et al. Busulfan systemic exposure relative to regimen-related toxicity and acute graft-versus-host disease: defining a therapeutic window for i.v. BuCy2 in chronic myelogenous leukemia. Biol Blood Marrow Transplant. 2002;8( 9):477–85. doi: 10.1053/bbmt.2002.v8.pm12374452. [DOI] [PubMed] [Google Scholar]
- 35.Jacobson P, Rogosheske J, Barker JN, et al. Relationship of mycophenolic acid exposure to clinical outcome after hematopoietic cell transplantation. Clin Pharmacol Ther. 2005 Nov;78( 5):486–500. doi: 10.1016/j.clpt.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Beal SL, Sheiner LB. Conditional Estimation Methods. San Francisco: University of California; 1998. NONMEM User’s Guide - Part VII. [Google Scholar]
- 37.Frey N, Laveille C, Paraire M, et al. Population PKPD modelling of the long-term hypoglycaemic effect of gliclazide given as a once-a-day modified release (MR) formulation. Br J Clin Pharmacol. 2003 Feb;55( 2):147–57. doi: 10.1046/j.1365-2125.2003.01751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zingmark PH, Ekblom M, Odergren T, et al. Population pharmacokinetics of clomethiazole and its effect on the natural course of sedation in acute stroke patients. Br J Clin Pharmacol. 2003 Aug;56( 2):173–83. doi: 10.1046/j.0306-5251.2003.01850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlsson MO, Molnar V, Bergh J, et al. A general model for time-dissociated pharmacokinetic-pharmacodynamic relationship exemplified by paclitaxel myelosuppression. Clin Pharmacol Ther. 1998 Jan;63( 1):11–25. doi: 10.1016/S0009-9236(98)90117-5. [DOI] [PubMed] [Google Scholar]