

A 75-year-old man presented with pleuritic chest pain and right shoulder pain. A palpable right anterior chest wall mass was noted on physical examination. His past medical history was significant for testicular cancer treated with radiation therapy in 1974.
A computed tomography (CT) scan of the chest, performed without intravenous contrast due to renal insufficiency, revealed an 8-cm lobulated soft tissue mass arising from the right third anterior rib and involving the costal cartilage (Figure 1). Scattered internal calcific deposits were present. It was uncertain if these represented chondroid matrix or destruction of calcified costal cartilage. The mass extended into the chest wall with elevation of the pectoralis muscles. Medially, the mass abutted the right anterior mediastinum. No additional osseous lesions or enlarged mediastinal or hilar nodes were seen. A few nonspecific 1- to 2-mm noncalcified pulmonary nodules were noted. CT of the abdomen and pelvis showed no evidence of metastatic disease.
Figure 1.
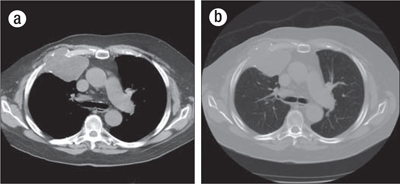
CT images through the chest at the level of the carina in both (a) soft tissue and (b) bone windows demonstrate a lytic mass involving the right anterior third rib, centered at the costochondral junction. A large, lobulated soft tissue component elevates the pectoralis musculature. A few scattered internal calcifications are suggestive of chondroid matrix.
Given the internal calcifications, the leading differential diagnostic consideration was chondrosarcoma; however, the classic “ring and arc” calcifications described in chondroid neoplasms were not definitively present on imaging. Metastasis was another consideration given the patient's history of testicular cancer; however, the location and appearance would be very atypical. The lesion appeared to arise from the chest wall and not the lung itself. Other considerations would include extramedullary plasmacytoma or lymphoma.
An ultrasound-guided needle biopsy of the lesion, prepared for rapid cytologic evaluation using hematoxylin and eosin, showed a myxoid chondroid matrix with chondrocytes present (Figure 2). The subsequently prepared paraffin block from the core biopsy showed a similar chondroid matrix with increasing cellularity at the periphery of the nodule (Figure 3). Though no distinctly atypical chondrocytes were seen, the cellularity was concerning for a possible low-grade chondrosarcoma, and the tumor was excised along with the second, third, and fourth ribs. Grossly, the tumor did not involve the lateral margins but was 12 mm from the inferior resection margin of the fourth rib and 3 mm from the periosteal soft tissue of the superior aspect of the second rib. The neoplasm clearly demonstrated cartilage synthesis, as well as areas of hemorrhage and necrosis (50% of total tumor volume) (Figure 4).
Figure 2.
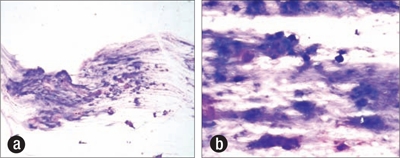
Scrape preparation at time of ultrasound-guided biopsy demonstrating a myxoid chondroid matrix, increased cellularity, and chondrocytes. Diff-Quik stain, (a) ×100, (b) ×400.
Figure 3.
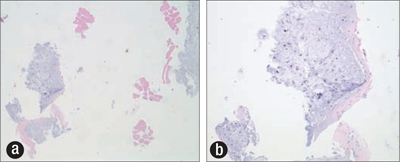
Paraffin cell block prepared from ultrasound-guided biopsy showing focal increased cellularity, chondroid matrix, and chondrocytes. Hematoxylin and eosin stain, (a) ×40, (b) ×100.
Figure 4.
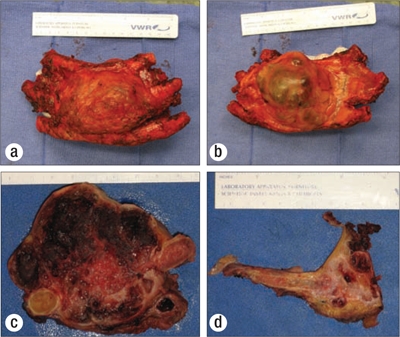
Gross images of the resected tumor, showing the lesion (a) anteriorly, (b) posteriorly, (c) in cross-section, and (d) invading and destroying the third rib.
Microscopically, the tumor showed features suggesting increasing histologic grade, such as areas with distinct marked hypercellularity, increasing nuclear size/atypia, and multinucleated cells (Figure 5). However, the proliferation index, as demonstrated by MiB-1, was low, with most areas demonstrating 1% and focal areas demonstrating 5%. The tumor expanded the medullary cavity and in areas extended beyond the cortex, forming subpleural nodules posteriorly and involving the chest wall tissue anteriorly. The anterior chest wall surgical resection margin was focally involved by tumor. Extension through the pleural surface was not seen. Focal vascular invasion was present, with tumor grossly and microscopically surrounding and invading one blood vessel specifically. The tumor was considered grade 2.
Figure 5.
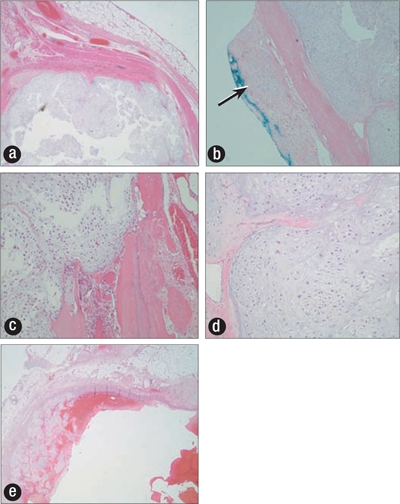
(a) Subpleural tumor nodule (×100). (b) Tumor at anterior resection margin (arrow) (×40). (c, d) Tumor expanding marrow cavity with markedly increased cellularity, nuclear atypia, and occasional multinucleated cells (×400). (e) Tumor expanding blood vessel.
Given the positive margin, radiation therapy was recommended. The patient received 3060 cGy of the planned 5940 cGy of radiation but could not complete the full course due to locoregional side effects. No additional therapy was recommended. A CT scan performed approximately 2 months after radiation therapy showed expected radiation changes and minimal pleural fluid without overt evidence of tumor recurrence. The patient is currently 4 months postradiation.
DISCUSSION
Chondrosarcomas are malignant tumors that produce chondroid matrix. Primary chondrosarcomas arise de novo. Secondary chondrosarcomas occur in preexisting benign cartilaginous neoplasms, such as a complication of a preexisting enchondroma or osteochondroma (1, 2). Tumors that arise in the medullary cavity are considered central, or conventional intramedullary, chondrosarcomas, while those that occur near the surface of bone are referred to as peripheral, or juxtacortical. Primary chondrosarcoma is the third most common primary malignant tumor of bone, but the most common primary neoplasm involving the chest wall (3). Ten percent of chondrosarcomas are radiation-induced (3). A wide variety of primary neoplasms are associated with radiation-induced chondrosarcoma, as follows: Hodgkin's lymphoma, breast adenocarcinoma, squamous cell carcinoma of the larynx, thyroid carcinoma, non–small cell lung carcinoma, and retinoblastoma (4–10). This particular case was felt to represent a primary chondrosarcoma.
Chondrosarcomas tend to be bulky tumors. Most are >4 cm in diameter, and some grow as large as 25 or 35 cm. The most extensive tumors occur in the flat and irregular bones, including the pelvis, ribs, and scapula, where they can grow to a large size before producing symptoms (11). Long bones are involved approximately 45% of the time, with the femur most frequently affected, making up 25% of all cases. In the femur, humerus, tibia, and fibula, the proximal metaphysis is affected more commonly than the distal (11).
Conventional intramedullary chondrosarcomas typically occur in the fourth or fifth decades, with a male to female predominance of 1.5 to 2:1. However, chondrosarcomas involving the ribs and sternum tend to occur in a slightly younger patient population. Within the chest, anterior ribs are most commonly involved, at the costochondral junction (3).
Pain is the most frequent presenting symptom, reported in 95% of patients. The pain is often insidious and progressive, with an average duration of 1 to 2 years prior to presentation. The pain is typically described as aching at rest and worse, sometimes severe, at night. A palpable soft tissue mass or fullness is present in 30% to 80% of patients at presentation. Pathologic fractures are the presenting symptom in 3% to 17% of patients (3).
Two syndromes characterized by multiple enchondromas, Maffucci syndrome and Ollier's disease, have been described. Ollier's disease is more common than Maffucci syndrome and is characterized by multiple enchondromatosis, which may involve a single limb, one half of the body, or the entire skeleton (1, 12, 13). Maffucci syndrome is the constellation of multiple enchondromatosis and soft tissue hemangiomas, with greatest involvement of the hands and feet. In fact, radiographs of the hands and feet are often pathognomonic in Maffucci syndrome, showing multiple soft tissue masses containing phleboliths in addition to the multiple enchondromas (14). Both processes potentially undergo malignant transformation. The range of malignant transformation to chondrosarcoma in Ollier's disease varies widely in the reported literature, ranging from 5% to 50%. In Maffucci syndrome, the incidence of malignant transformation to chondrosarcoma is approximately 15% to 20%. However, the soft tissue hemangiomas can undergo malignant transformation to vascular sarcomas in 3% to 5% of cases. There is also an increased incidence of ovarian and pancreatic malignancies and gliomas in patients with Maffucci syndrome (14).
Imaging considerations
The classic radiographic appearance of chondrosarcoma is a lobulated, mixed lytic and sclerotic lesion. The sclerotic regions on radiographs correspond to chondroid matrix mineralization. In more advanced stages, the mass becomes lobulated with endosteal scalloping and eventual cortical disruption and extension to the adjacent soft tissues. Higher-grade chondrosarcomas contain relatively less extensive chondroid matrix. Well-organized calcific rings within cartilage usually signify a low-grade tumor. High-grade chondrosarcomas may contain more myxoid material and are frequently associated with large areas of noncalcified tumor matrix. When calcification occurs in high-grade chondrosarcomas, it tends to be amorphous, scattered, punctuate, or irregular (11). The degree of endosteal scalloping is considered the best predictor in distinguishing chondrosarcoma from benign enchondroma on radiographs. Deep endosteal scalloping and soft tissue expansion, which are both considered aggressive features, are better evaluated with CT. Endosteal scalloping greater than two thirds the cortical thickness in long bones is strong evidence of chondrosarcoma (3).
CT is optimal for detection and characterization of chondroid matrix, which classically has well formed rings and arcs of mineralization. Both CT and magnetic resonance imaging (MRI) depict high internal water content. MRI is the ideal modality to evaluate the extent of bone marrow involvement and extraosseous extension.
Nuclear imaging also plays a role in the evaluation of chondroid neoplasms. The role of skeletal scintigraphy is largely to determine a monostotic or polyostotic process. Positron emission tomography (PET) can provide more insight into the tumor grade. 18-fluoro-2-deoxy-D-glucose (F18 FDG) uptake has been shown to be proportional to tumor grade, with high standardized uptake values predictive of higher-grade chondrosarcomas. PET cannot differentiate benign cartilaginous tumor from grade 1/low-grade chondrosarcoma; however, PET is highly sensitive in detecting high-grade chondrosarcoma metastasis. PET can be useful in optimizing the biopsy approach and targeting regions of highest measured metabolic activity.
Molecular genetic considerations
There is interobserver variability in the histologic grading of chondrosarcomas, yet significantly different therapeutic approaches are used for grade 1 versus grade 2 lesions. Therefore, definitive molecular markers distinguishing these histologic subtypes are needed. Presently, such markers have not been delineated. However, when considering central and peripheral chondrosarcomas, different molecular genetic profiles have been demonstrated.
Central chondrosarcomas, the most common type, are associated with genetic aberrations involving chromosome 12q13-15 and 9p21. The locus for CDKN2A (p16), a tumor suppressor gene, is located at 9p21. This marker is considered important in progression of chondrosarcoma from low to high grade. Mutations in TP53 are also associated with tumor progression (15).
Peripheral chondrosarcomas are associated with exostosin (EXT) genes, which cause multiple osteochondromas. In the setting of a hereditary syndrome, germline mutations of EXT1 and EXT2 are present, with accompanying loss of the wild-type allele in the cartilage cap of the osteochondroma. When a peripheral chondrosarcoma is solitary, however, a homozygous deletion with decreased EXT expression in the preceding osteochondroma and subsequent peripheral chondrosarcoma is seen (15). The function of EXT is to synthesize heparan sulfate, which stimulates the formation of heparan sulfate proteoglycans, which plays a role in cell signaling. Inactivation of EXT leads to accumulation of heparan sulfate proteoglycans within the cell cytoplasm and Golgi apparatus, and decreased signaling of growth pathways occurs (15). Overexpression of platelet-derived growth factor receptor-alpha and -beta has also been described (15, 16).
Pathology
Permeation of bone, size, and periosteal invasion are specific findings of chondrosarcoma. Cellularity, nuclear atypia (including multinucleated cells) and increased proliferation (>10%) are all indicators of increasing grade and aggressive behavior. Apart from the typical chondrosarcomas, there are some rare types of this tumor including dedifferentiated chondrosarcoma, mesenchymal chondrosarcoma, clear cell chondrosarcoma, and myxoid chondrosarcoma. These rare forms are more aggressive and malignant (15). Chondrosarcomas associated with clinical syndromes were previously discussed.
Treatment
The natural history and prognosis of chondrosarcoma is extremely variable. Overall 5-year survival rates for grade 1 chondrosarcomas are 90% to 94%, while those for grade 3 are 43% to 44%. For grade 1 chondrosarcomas with intact cortex and absence of soft tissue mass, consideration can be given to an intralesional procedure such as curettage with adjunctive ablation. However, if there are aggressive imaging features such as cortical breakthrough or soft tissue mass, or if the tumor is grade 2 or higher, wide surgical excision is required (3).
A Scandinavian study evaluated 106 consecutive chondrosarcomas involving the rib and sternum. The pathologic specimens were reviewed and graded 1 to 4, and the surgical margins were defined as wide, marginal, or intralesional. After wide surgical excision, the 10-year survival rate was 92%, and the local recurrence rate was 4%. In contrast, the 10-year survival rate was 47% and the local recurrence rate 73% in patients treated with an intralesional approach. Prognostic factors for local recurrence included both surgical margins and histologic grade (17).
Due to the slow growth rate of chondrosarcomas—as supported by a low proliferation index with the Mib-1/Ki-67 proliferation marker—chondrosarcomas are known to be relatively radioresistant. Doses exceeding 60 Gy are recommended for effective treatment, which is often not tolerable for adjacent structures (e.g., neurologic tissue)(1, 15).
Other treatment approaches have included particle therapy with protons, which offers the advantage of a minimal exit dose after energy deposits within the tissue, thus effectively sparing adjacent structures (2). Additionally, proton radiotherapy has proven beneficial in the setting of incompletely resected chondrosarcomas present at the skull base and axial skeleton, with reported local control rates of 85% to 100% (18). Another documented approach is the use of radiotherapy with carbon ions, providing the physical advantages of proton therapy combined with higher radiobiologic activity. Local control rates with this approach are 96% and 90% at 3 and 4 years, respectively (2).
Chondrosarcomas present several architectural and structural barriers for systemic chemotherapy. In addition to their low proliferation index, they have poor vascularity and an abundant extracellular matrix that makes tumor perfusion difficult. Further, it is hypothesized that the expression of the multidrug-resistance 1 gene (P-glycoprotein), which confers resistance to doxorubicin, offers another explanation to the relative ineffectiveness of chemotherapeutic regimens (2). There is no suggestion of benefit for low-grade chondrosarcomas with adjuvant chemotherapy, even in the metastatic setting. However, chemotherapy may have some role to play in the treatment of tumors of dedifferentiated and mesenchymal subtypes (especially with increased small blue cells). This latter type of chondrosarcoma is especially chemotherapy sensitive, and tumor responses to conventional treatments have been noted. Specifically, Huvos et al (19) reported a median survival of 37.9 months, with a 50% 3-year survival, 42% 5-year survival, and 28% 10-year survival in a study of 32 patients. Cisplatin and Adriamycin–based regimens seem to have the most efficacy. Investigational classes of drugs including tyrosine kinase inhibitors, hormonal therapies, and angiogenesis inhibitors are also being studied (15). Thus, no standardized proven approach exists for adjuvant chemotherapy in the setting of chondrosarcoma.
References
- 1.Mazanet R, Antman KH. Sarcomas of soft tissue and bone. Cancer. 1991;68(3):463–473. doi: 10.1002/1097-0142(19910801)68:3<463::aid-cncr2820680304>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 2.Cakir O, Topal U, Bayram AS, Tolunay S. Sarcomas: rare primary malignant tumors of the thorax. Diagn Interv Radiol. 2005;11(1):23–27. [PubMed] [Google Scholar]
- 3.Murphey MD, Walker EA, Wilson AJ, Kransdorf MJ, Temple HT, Gannon FH. From the archives of the AFIP: imaging of primary chondrosarcoma: radiologic-pathologic correlation. Radiographics. 2003;23(5):1245–1278. doi: 10.1148/rg.235035134. [DOI] [PubMed] [Google Scholar]
- 4.Weatherby RP, Dahlin DC, Ivins JC. Postradiation sarcoma of bone: review of 78 Mayo Clinic cases. Mayo Clin Proc. 1981;56(5):294–306. [PubMed] [Google Scholar]
- 5.Sauter ER, Keller SM, Curran WJ, Russo J, Langer CJ. Radiation-induced chest-wall chondrosarcoma following surgical resection and radiotherapy for non-small-cell lung cancer. J Natl Cancer Inst. 1993;85(2):162–163. doi: 10.1093/jnci/85.2.162. [DOI] [PubMed] [Google Scholar]
- 6.Schwarz RE, Burt M. Radiation-associated malignant tumors of the chest wall. Ann Surg Oncol. 1996;3(4):387–392. doi: 10.1007/BF02305669. [DOI] [PubMed] [Google Scholar]
- 7.Mohammadianpanah M, Gramizadeh B, Omidvari SH, Mosalaei A. Radiation-induced chondrosarcoma of the maxilla 7-year after combined chemoradiation for tonsillar lymphoma. J Postgrad Med. 2004;50(3):200–201. [PubMed] [Google Scholar]
- 8.Fitzwater JE, Cabaud HE, Farr GH. Irradiation-induced chondrosarcoma. A case report. J Bone Joint Surg Am. 1976;58(7):1037–1039. [PubMed] [Google Scholar]
- 9.Peimer CA, Yuan HA, Sagerman RH. Postradiation chondrosarcoma. A case report. J Bone Joint Surg Am. 1976;58(7):1033–1036. [PubMed] [Google Scholar]
- 10.Sheppard DG, Libshitz HI. Post-radiation sarcomas: a review of the clinical and imaging features in 63 cases. Clin Radiol. 2001;56(1):22–29. doi: 10.1053/crad.2000.0599. [DOI] [PubMed] [Google Scholar]
- 11.Resnick D, Niwayama G, editors. Diagnosis of Bone and Joint Disorders. 2nd ed. Philadelphia: Saunders; 1988. pp. 3897–3919. [Google Scholar]
- 12.Silve C, Jüppner H. Ollier disease. Orphanet J Rare Dis. 2006;37:1–6. doi: 10.1186/1750-1172-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unni K, Inwards C, Bridge J, Kindblom LG, Wold L. AFIP Atlas of Tumor Pathology, 4th series, fascicle 2. Washington, DC: American Registry of Pathology Press; 2005. Tumors of the bones and joints; pp. 52–56. [Google Scholar]
- 14.Zwenneke Flach H, Ginai AZ, Wolter Oosterhuis J, Armed Forces Institutes of Pathology Best cases from the AFIP. Maffucci syndrome: radiologic and pathologic findings. Radiographics. 2001;21(5):1311–1316. doi: 10.1148/radiographics.21.5.g01se301311. [DOI] [PubMed] [Google Scholar]
- 15.Gelderblom H, Hogendoorn PC, Dijkstra SD, van Rijswijk CS, Krol AD, Taminiau AH, Bovée JV. The clinical approach towards chondrosarcoma. Oncologist. 2008;13(3):320–329. doi: 10.1634/theoncologist.2007-0237. [DOI] [PubMed] [Google Scholar]
- 16.Lagonigro MS, Tamborini E, Negri T, Staurengo S, Dagrada GP, Miselli F, Gabanti E, Greco A, Casali PG, Carbone A, Pierotti MA, Pilotti S. PDGFRα, PDGFRβ and KIT expression/activation in conventional chondrosarcoma. J Pathol. 2006;208(5):615–623. doi: 10.1002/path.1945. [DOI] [PubMed] [Google Scholar]
- 17.Widhe B, Bauer HC, Scandinavian Sarcoma Group Surgical treatment is decisive for outcome in chondrosarcoma of the chest wall: a population-based Scandinavian Sarcoma Group study of 106 patients. J Thorac Cardiovasc Surg. 2009;137(3):610–614. doi: 10.1016/j.jtcvs.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 18.Hug EB, Loredo LN, Slater JD, DeVries A, Grove RI, Schaefer RA, Rosenberg AE, Slater JM. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91(3):432–439. doi: 10.3171/jns.1999.91.3.0432. [DOI] [PubMed] [Google Scholar]
- 19.Huvos AG, Rosen G, Dabska M, Marcove RC. Mesenchymal chondrosarcoma. A clinicopathologic analysis of 35 patients with emphasis on treatment. Cancer. 1983;51(7):1230–1237. doi: 10.1002/1097-0142(19830401)51:7<1230::aid-cncr2820510710>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]