

Abstract
Activation of the peroxisome proliferator-activated receptor α (PPARα) is associated with increased fatty acid catabolism and is commonly targeted for the treatment of hyperlipidemia. To identify latent, endogenous biomarkers of PPARα activation and hence increased fatty acid β-oxidation, healthy human volunteers were given fenofibrate orally for 2 weeks and their urine profiled by UPLC-QTOFMS. Biomarkers identified by the machine learning algorithm random forests included significant depletion by day 14 of both pantothenic acid (>5-fold) and acetylcarnitine (>20-fold), observations that are consistent with known targets of PPARα including pantothenate kinase and genes encoding proteins involved in the transport and synthesis of acylcarnitines. It was also concluded that serum cholesterol (-12.7%), triglycerides (-25.6%), uric acid (-34.7%), together with urinary propylcarnitine (>10-fold), isobutyrylcarnitine (>2.5-fold), (S)-(+)-2-methylbutyrylcarnitine (5-fold), and isovalerylcarnitine (>5-fold) were all reduced by day 14. Specificity of these biomarkers as indicators of PPARα activation was demonstrated using the Ppara-null mouse. Urinary pantothenic acid and acylcarnitines may prove useful indicators of PPARα-induced fatty acid β-oxidation in humans. This study illustrates the utility of a pharmacometabolomic approach to understand drug effects on lipid metabolism in both human populations and in inbred mouse models.
Introduction
Peroxisome proliferator-activated receptors (PPAR) belong to the nuclear hormone receptor superfamily and are involved in the transcriptional activation of many target genes regulating energy metabolism, adipogenesis, angiogenesis, cell proliferation and inflammation.1 In the liver PPARα activates fatty acid and cholesterol catabolism, gluconeogenesis, heme synthesis and partly regulates amino acid metabolism and the urea cycle.1-6 The fibrate group of drugs, potent agonists of PPARα, is widely prescribed to treat hyperlipidemias.7 In particular, these drugs mediate an increase in fatty acid β-oxidation which in turn lowers serum triglyceride levels and reduces insulin resistance.8 It is anticipated that an in-depth study of the metabolic changes caused by PPARα agonist administration to humans should reveal the specificity of action for this class of drugs.
Metabolomics, the global qualitative and quantitative analysis of small molecules present in a biofluid, shows great promise as a means to identify biomarkers of drug efficacy.9, 10 1H Nuclear magnetic resonance spectroscopy and liquid chromatography mass spectrometry are two commonly deployed metabolomic platforms.9, 11 When coupled with multivariate data analysis techniques or machine learning algorithms unique chemical fingerprints of drug efficacy can be identified thus directing investigators to specific biochemical pathways.12-16
Hypolipidemic fibrate drugs have been studied extensively in rodent models to identify both biomarkers of PPARα activation and early indicators of peroxisome proliferation and hepatotoxicity.13-16 For example, non-invasive urinary biomarkers of the PPARα synthetic agonist, Wy-14,643, were identified and found to be specific for PPARα activation by comparing wild-type and Ppara-null mice.13 Similarly, nicotinamide and related metabolites were found to be elevated in treated rats and were shown to correlate with PPARα activation.14-16 These historical biomarkers of PPARα activation are summarized in Table 1. However, such biomarkers identified in rodent models have yet to be extended to and validated in humans.
Table 1.
Historical Biomarkers of PPARα Activation
| Chemical Name | Empirical Formula | Mono-isotopic Mass | [M+H]+ | [M-H] - | Ligand | Animal Model | Refs. |
|---|---|---|---|---|---|---|---|
| N-Methyl-2-pyridone-3-carboxamide (2PY) | C7H8N2O2 | 152.0586 | 153.0664 | 151.0508 | Fenofibrate GWα | Rat | 14-16 |
| N-Methyl-4-pyridone-3-carboxamide (4PY) | C7H8N2O2 | 152.0586 | 153.0664 | 151.0508 | Fenofibrate GWα | Rat | 14-16 |
| 11β-Hydroxy-3,20-dioxopregn-4-en-21-oic acid (HDOPA) | C21H28O5 | 360.1937 | 361.2015 | 359.1858 | Wy-14,643 | Mouse | 13 |
| 11β,20-Dihydroxy-3-oxopregn-4-en-21-oic acid (DHOPA) | C21H30O5 | 362.2093 | 363.2171 | 361.2015 | Wy-14,643 | Mouse | 13 |
| N-Methylnicotinamide | C7H9N2O | 137.0715 | 138.0793 | 136.0637 | Fenofibrate GWα Wy-14,643 | Rat Mouse | 13-16 |
| Nicotinamide N-oxide | C6H6N2O2 | 138.0429 | 139.0508 | 137.0351 | Wy-14,643 | Mouse | 13 |
| Nicotinamide | C6H6N2O | 122.0480 | 123.0558 | 121.0402 | Wy-14,643 | Mouse | 13 |
In the present investigation, urine samples from 10 healthy, unrelated human volunteers taking fenofibrate orally (200 mg/day for 14 days) were compared using metabolomic analysis to understand the urinary pharmacometabolomic properties of PPARα activation. Ultra-performance liquid chromatography (UPLC) coupled with electrospray ionization quadrupole time-of-flight mass spectrometry (ESI-QTOFMS) was used to profile each volunteer's urinary metabolome. To identify latent, endogenous biomarkers of PPARα activation, random forests, a machine learning algorithm, was used to model the day 0, day 7, and day 14 mass spectrometric data purged of all fenofibrate and related drug metabolite ions. Metabolomic analysis identified urinary metabolites associated with the fenofibrate-induced increase in β-oxidation. The present investigation in human volunteers provides compelling evidence for the utility of metabolomics in evaluating drug efficacy and demonstrates that the burden of metabolic effects of fenofibrate is consistent with its action on PPARα.
Materials and Methods
Authentic Compounds
Pantothenic acid, debrisoquine hemisulfate, and 4-nitrobenzoic acid were purchased from Sigma (St. Louis, MO). Acylcarnitines were obtained from the Metabolic Laboratory, Vrije Universiteit Medical Center (Amsterdam, The Netherlands). Fenofibric acid was obtained from AK Scientific, Inc. (Mountain View, CA). All other reagents were HPLC grade.
Human Volunteers
The study protocol was approved by the Ethics Committee of the General Teaching Hospital in Prague, Czech Republic. Ten healthy unrelated volunteers (3 males, 7 females) were enrolled after obtaining their written informed consent. The volunteer demographics are shown in Table 2. Personal history was taken from each volunteer, who also underwent clinical examination and received a general clinical chemistry screen. Each volunteer refrained from taking any medication 28 days prior to enrollment in the study and no drugs except the study medication were permitted.
Table 2.
Demographics and Clinical Biochemistry of Healthy Volunteers from Day 0, and Days7 and 14 After Fenofibrate Administration.
| Volunteer | Day | Uric Acid#@ (μmol/l) |
Bilirubin (μmol/l) |
ALT (μkat/l) |
AST (μkat/l) |
Cholesterol#@ (mmol/l) |
Triglycerides@ (mmol/l) |
Creatinine Clearance (ml/s) |
GFR (ml/s) |
|---|---|---|---|---|---|---|---|---|---|
|
F1 Female Age 23 |
0 | 349 | 23.2 | 0.19 | 0.23 | 4.08 | 0.81 | 1.675 | 2.56 |
| 7 | 250 | 11.8 | 0.19 | 0.28 | 3.49 | 0.32 | 1.698 | 2.21 | |
| 14 | 300 | 12.8 | 0.28 | 0.37 | 3.5 | 0.5 | 1.463 | 1.88 | |
|
F2 Male Age 31 |
0 | 278 | 12.7 | 0.33 | 0.39 | 4.75 | 0.6 | 2.107 | 2.16 |
| 7 | 205 | 10 | 0.34 | 0.4 | 4.36 | 0.82 | 2.006 | 1.91 | |
| 14 | 166 | 13.4 | 0.32 | 0.43 | 3.99 | 0.57 | 1.69 | 2.13 | |
|
F3 Female Age 25 |
0 | 235 | 9.9 | 0.4 | 0.4 | 4.26 | 0.78 | 1.832 | 1.91 |
| 7 | 161 | 7.3 | 0.37 | 0.39 | 3.49 | 0.34 | 1.541 | 1.78 | |
| 14 | 153 | 6.1 | 0.34 | 0.36 | 3.63 | 0.45 | 1.245 | 1.83 | |
|
F4 Female Age 27 |
0 | 265 | 7.5 | 0.26 | 0.37 | 4.52 | 0.9 | 1.994 | 1.93 |
| 7 | 195 | 7.6 | 0.27 | 0.43 | 4.8 | 0.85 | 1.345 | 7.72 | |
| 14 | 186 | 13 | 0.35 | 0.51 | 3.96 | 0.77 | 1.742 | 1.93 | |
|
F5 Female Age 23 |
0 | 235 | 7.4 | 0.18 | 0.33 | 3.99 | 0.95 | 1.426 | 1.63 |
| 7 | 123 | 8.2 | 0.22 | 0.35 | 3.62 | 0.5 | 1.877 | 1.52 | |
| 14 | 143 | 7 | 0.19 | 0.32 | 2.93 | 0.77 | 1.949 | 1.34 | |
|
F6 Female Age 23 |
0 | 212 | 8.4 | 0.19 | 0.46 | 4.31 | 0.74 | 1.485 | 1.23 |
| 7 | 153 | 6.6 | 0.24 | 0.42 | 3.79 | 0.9 | 1.269 | 1.09 | |
| 14 | 140 | 6.7 | 0.21 | 0.36 | 3.61 | 0.45 | 1.165 | 1.23 | |
|
F7 Female Age 23 |
0 | 288 | 9.1 | 0.4 | 0.38 | 4.33 | 1.02 | 1.446 | 1.35 |
| 7 | 168 | 4.7 | 0.29 | 0.32 | 3.6 | 0.72 | 0.994 | 1.3 | |
| 14 | 179 | 10.8 | 0.26 | 0.23 | 3.85 | 0.74 | 1.281 | 1.26 | |
|
F8 Female Age 24 |
0 | 267 | 10.1 | 0.32 | 0.34 | 4.18 | 0.72 | 0.804 | 1.59 |
| 7 | 154 | 8.7 | 0.25 | 0.35 | 4.1 | 0.41 | 0.973 | 1.58 | |
| 14 | 172 | 6.2 | 0.24 | 0.22 | 3.72 | 0.79 | 0.75 | 1.5 | |
|
F9 Male Age 27 |
0 | 280 | 10.6 | 0.5 | 0.51 | 4.63 | 1.7 | 1.034 | 2.01 |
| 7 | 200 | 8.5 | 0.52 | 0.43 | 4.81 | 1.54 | 1.09 | 1.94 | |
| 14 | 165 | 7.5 | 0.47 | 0.5 | 4.8 | 1.07 | 1.912 | 2.21 | |
|
F10 Male Age 23 |
0 | 395 | 6 | 0.52 | 0.41 | 4.01 | 1.02 | 1.948 | 2.13 |
| 7 | 224 | 12.6 | 0.54 | 0.48 | 3.66 | 0.96 | 1.844 | 1.96 | |
| 14 | 227 | 8.2 | 0.53 | 0.48 | 3.58 | 0.76 | 1.71 | 2.1 | |
means significantly (p<0.05 by repeated measures ANOVA with Bonferroni's Test for Multiple Comparisons) different when comparing day 0 and day 7
means significantly (p<0.05 by repeated measures ANOVA with Bonferroni's Test for Multiple Comparisons) different when comparing day 0 and day 14
Animal Studies
All animal studies were approved by the National Cancer Institute Animal Care and Use Committee prior to initiation of the study. Male C57Bl/6 wild-type (n=3) and Ppara-null (n=3) were fed either standard NIH31 diet or an NIH31 modified diet containing 0.1% fenofibrate ad libitum (Bioserv, Frenchtown, NJ). Control 24-h urines were collected in metabolic cages (Tecniplast USA, Exton, PA) prior to the start of the 0.1% fenofibrate diet. After 7 days on the diet, mice were placed in metabolic cages for 24-h urine collection. Urines were frozen at -80°C until use.
Fenofibrate Dosing
Each volunteer received a 200 mg capsule containing fenofibrate (Lipanthyl 200 mg, Laboratoires Fournier S.A., Dijon/Fontaine les Dijon, France) daily for 14 days. The drug was taken orally with lunch under the supervision of a study nurse. Urine for metabolomic analysis was collected over a 24 h period prior to treatment (day 0) and on days 7 and 14 of the treatment. Blood for clinical chemistry was collected between 07.30 and 08.00 h on days 0, 7, and 14. Creatinine clearance from the timed 24-h urine collection was computed. Urines were frozen at -80°C until use.
Sample Preparation for UPLC-ESI-QTOFMS Analysis
Urine samples were diluted with an equal volume of solvent containing 50% acetonitrile in HPLC grade water, 1 μM debrisoquine hemisulfate (ESI+ internal standard), and 40 μM 4-nitrobenzoic acid (ESI- internal standard). The samples were vortexed briefly and centrifuged at maximum speed for 20 min at 4°C to remove particulates and precipitated protein. The supernatant was transferred to an autosampler vial and sealed.
Ultra-Performance Liquid Chromatography coupled Quadrupole Time-of-Flight Mass Spectroscopy (UPLC-QTOFMS) of Urine
Five μl of deproteinated urine was chromatographed on a 50 × 2.1 mm Acquity 1.7 μm C18 column (Waters Corp., Milford, MA) using an Acquity UPLC system (Waters) as described.17, 18 Sample injection order was as follows: Volunteer 1 Day 0, Volunteer 1 Day 7, Volunteer 1 Day 14, to Volunteer 10 Day 14. For quantitation of urinary acylcarnitines, authentic compounds and urine samples were chromatographed on a 100 × 2.1 mm Acquity 1.7 μm C18 column. QuanLynx (Waters) was used to calculate the urinary acylcarnitine concentrations.
Chromatogram Deconvolution
Centroided and integrated mass chromatographic data were aligned using MarkerLynx software (Waters) to generate a data matrix consisting of peak areas corresponding to a unique m/z and retention time. The following parameters were used: mass tolerance = 0.02 Da, mass window = 0.05 Da, and retention time window = 0.20 min. The total ion current (TIC) for each sample was first normalized by summing the TIC to 10,000. The peak area corresponding to protonated creatinine (m/z = 114.0670+, retention time = 0.33 min) was used to normalize all the peak areas in a sample. Fenofibrate and its metabolites were excluded from the data matrix by excluding m/z measurements that ran later than 5.00 min.
Random Forests Analysis
ESI+ and ESI- data matrices were merged along with the clinical data from Table 2. The merged matrix was constructed such that each m/z and retention time pair was represented by a unique variable. Merging the datasets permitted the simultaneous evaluation of standard clinical indices and the urinary metabolomics data. In the R software environment (version 2.4.1), the machine learning algorithm, random forests,19 was used to compare day 0 and day 7, day 0 and day 14, or day 7 and day 14, and important variables were identified as those highly ranked on the variable importance list. Twenty-five independent random forests models were constructed (ntrees=10000) and the variable importance ranks were averaged across all 25 models. Bootstrapping the results from the 25 independent random forests was used to determine the 95% confidence intervals of the variable importance ranks. Random forests models were optimized by constructing 25 independent models based on a subset (e.g., the top 10, 20, 50, 75, 150, 250, 350, 500, 750, or 1000 most important variables) of the top ranked variables. The model having the lowest classification error using the fewest number of variables was assumed to contain the most robust variables. Bootstrapping was used to monitor the model variation over the set of 25 random forests models. The common significant variables spanning the two week study were defined using the ABarray package.
Molecular Ion Concentration Measurements
For pantothenic acid quantitation, both mouse and human urine samples were diluted in an equal volume of HPLC grade water containing debrisoquine hemisulfate (final concentration 1 μM) and separated on a Phenomenex Luna C18(2) column (Torrence, CA). The following multiple reaction monitoring (MRM) transitions were monitored in ESI+ mode using an API2000 SCIEX triple-quadrupole tandem mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA): pantothenic acid (219.9→90.1 m/z) and the internal standard, debrisoquine hemisulfate (176.1→134.2 m/z). Fenofibric acid was chromatographed on Phenomenex Luna Phenyl-Hexyl column and was monitored in ESI-mode using the following transitions: fenofibric acid (316.9→221.5 m/z) and the internal standard, 4-nitrobenzoic acid (166.0→109.6 m/z). Absolute peak areas (normalized by the internal standard peak area value) were used for quantitation and values estimated by comparison with a standard curve. Results were corrected for the dilution, multiplied by the 24-h urine volume, and expressed as μmol per 24 h.
Acylcarnitines were best resolved by UPLC. Human urines were diluted in HPLC water 1:10 for propionyl-, (S)-(+)-2-methylbutyryl-, and isovalerylcarnitine measurements, and 1:100 for acetyl- and isobutyrylcarnitine measurements. Mouse urines were diluted 1:5 for acetylcarnitine measurements. Other acylcarnitines including butyryl-, pivaloyl-, valeryl-, hexanoyl-, octanoyl-, decanoyl-, dodecanoyl-, tetradecanoyl-, hexadecanoyl-, and octadecanoyl-carnitine were resolved as standards, but fell below the limit of detection in the urine samples. Five μl of diluted urine as well as standards ranging from 0 to 50 μM were chromatographed on a 100 × 2.1 mm Acquity 1.7 μm C18 column as above. QuanLynx software (Waters) was used to quantify the urine metabolites based on peak area. Debrisoquine hemisulfate (1 μM) was used as the internal standard. Absolute peak areas (acylcarnitine/debrisoquine) were used for quantitation. Results were corrected for the dilution, multiplied by the 24-h urine volume, and expressed as μmol per 24 h.
Statistical Analysis
Statistical analysis of differences in ion concentration was performed using GraphPad Prism (San Diego, CA). The significance of metabolite concentration differences was determined using repeated measures ANOVA with Bonferroni correction for multiple comparisons. An unpaired t test was used when comparing the means of day 0 and day 7 in the mouse studies. Outliers were detected using Grubb's test (QuickCalcs, GraphPad Software, San Diego, CA) P-values less than p = 0.05 were considered significant.
Results
Clinical Chemistry
Prior to dosing on day 0, and on days 7 and 14 after daily oral administration of 200 mg fenofibrate, blood was drawn for routine clinical chemistry to monitor liver function and to assess cholesterol and triglyceride levels. Results of the clinical assays are summarized in Table 2. No significant changes in bilirubin, aspartate transaminase (AST) or alanine transaminase (ALT) levels were observed. Uric acid levels were significantly reduced on day 7 (mean ± standard deviation; 280 ± 55 μmol/l on day 0 to 183 ± 38 on day 7, p<0.001) and remained reduced on day 14 (280 ± 55 μmol/l on day 0 to 183 ± 48, p<0.001). Cholesterol levels were also reduced on day 7 (4.3 ± 0.26 mmol/l on day 0 to 3.97 ± 0.52 on day 7, p<0.05) and remained reduced on day 14 (4.3 ± 0.26 mmol/l on day 0 to 3.76 ± 0.47, p<0.001). No significant change was observed with high-density lipoprotein cholesterol (HDLc) serum levels (1.5 ± 0.41 mmol/l on day 0 to 1.5 ± 0.37 in day 7 and 1.5 ± 0.43 on day 14) while low-density lipoprotein cholesterol (LDLc) was slightly but significantly reduced (2.4 ± 0.47 mmol/l on day 0 to 2.1 ± 0.48 on day 7, p<0.05; and 2.4 ± 0.47 mmol/l on day 0 to 1.9 ± 0.65 on day 14, p<0.001). Triglycerides were reduced on day 7 but failed significance testing (0.92 ± 0.30 mmol/l on day 0 to 0.74 ± 0.37 on day 7, p>0.05) but were significantly reduced on day 14 (0.92 ± 0.30 mmol/l on day 0 to 0.69 ± 0.19 on day 14, p<0.05). No significant differences were observed with creatinine clearance or glomerular filtration rate (GFR).
Random Forests Modeling
The machine learning algorithm random forests19 was used to model the urinary metabolome and clinical indices of samples obtained on day 0, day 7, and day 14 after fenofibrate administration. Random forests has been demonstrated to handle metabolomics datasets well, it may be less susceptible to over-parameterization, and it can provide convenient estimates of the most important variables used in classification.12, 20, 21 Twenty-five independent random forests models were used to obtain an average ranked list of those variables contributing most significantly and consistently to group classification. Optimized models of day 0 and day 7 (classification error less than 3%) as well as day 0 and day 14 (classification error approximately 10%) are shown in Figure 1A-B. Two samples that did not cluster with the other day 14 samples (Volunteer 6 and 9) are discernible in Figure 1B. No meaningful difference was found when comparing day 7 and day 14 (data not shown). The subsets of variables used to generate the optimized models were then compared to define a conservative, yet robust, set of common variables that spanned the 2-week study (Figure 1C). Detailed results of the common metabolomic profile are described in Table 3. Exclusion of the male human volunteer data from the random forests analysis did not influence the results suggesting gender did not influence the classifications (data not shown).
Figure 1.
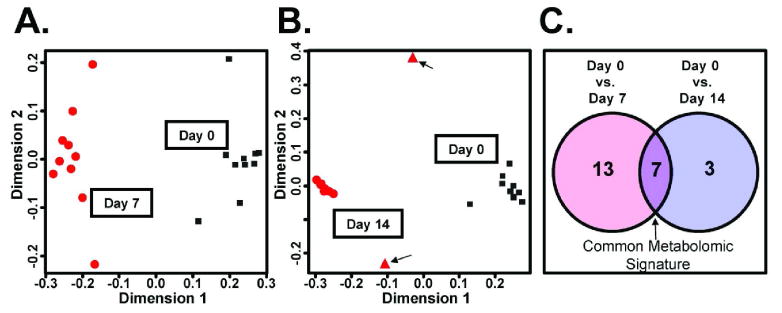
Modeling of day 0, day 7, and day 14 urine metabolomics data from optimized random forests models using the proximity matrix. (A) A multidimensional scaling (MDS) plot of day 0 and day 7 using the random forests proximity matrix. Black squares indicate day 0 and red circles indicate day 7. (B) An MDS plot of day 0 and day 14. Black squares indicate day 0 and red circles indicate day 14. Two potential outliers are indicated as red triangles and with arrows. (C) A Venn diagram describing the number of specific (day 0 and day 7, 13 highly ranked biomarkers; day 0 and day 14, 3 highly ranked biomarkers) that generated the optimal model and the number of common important variables, 7.
Table 3. Summary of Common Important Variables from Random Forests Analysis of Day 0, Day 7, and Day 14.
| Identity | m/z (ESI+) | Retention Time (min) | Empirical Formula | Mass Error (ppm) | Day 0 vs. Day 7 Mean Rank (95% CI) |
Day 0 vs. Day 14 Mean Rank (95% CI) |
|---|---|---|---|---|---|---|
| Common Biomarkers (and adducts) Spanning 2 Week Study | ||||||
| Pantothenic Acid† a | 242.1003+ | 2.07 | C9H17NO5Na+ | -0.4 | 1.32 (1.16-1.52) | 1.56 (1.2-1.9) |
| b | 220.1179+ | 2.07 | C9H18NO5+ | -2.7 | 1.68 (1.48-1.84) | 3.24 (2.5-4.0) |
| c | 202.1063+ | 2.06 | C9H16NO4+ | -7.9 | 4.28 (3.95-4.56) | 7.6 (6.8-8.2) |
| Acetylcarnitine | 204.1233+ | 0.37 | C9H18NO4+ | -1.5 | 4.36 (4.12-4.6) | 3.04 (2.4-3.7) |
| Serum Uric Acid* | na | na | na | na | 6.6 (6.4-6.8) | 11.4 (10.8-12.0) |
| unknown | 308.1831+ | 4.73 | na | na | 10.4 (9.92-10.9) | 5.8 (5.0-6.5) |
| unknown | 319.1658+ | 3.45 | na | na | 13.5 (12.7-14.2) | 5.2 (4.4-5.9) |
| Other Notable Biomarkers | ||||||
| Serum Cholesterol* | na | na | na | na | 26.2 (24.8-27.8) | 15.1 (13.9-16.4) |
| Carnitine | 162.1133+ | 0.32 | C7H16NO3+ | 1.9 | 28.2 (26.2-30.9) | 11.8 (10.6-13.1) |
| Isovalerylcarnitine | 246.1693+ | 2.83 | C12H24NO4+ | -4.9 | 37.3 (33.8-41.3) | 6.84 (6.2-7.5) |
| Isobutyrylcarnitine | 232.1531+ | 2.10 | C11H22NO4+ | -7.8 | 911 (708-1110) | 29.6 (28.5-30.6) |
indicates measurement obtained from serum clinical biochemistry analysis.
na, not applicable.
a = [Na+] adduct; b = [H+] protonated molecular ion; c = [M-H2O]+ fragmention
Identification of Biochemical Constituents in the Urine Metabolome with High Variable Importance Scores
Putative identities of ions described in Table 3 were arrived upon following searches of the Madison Metabolomics Consortium Database (MMCD) using a tolerance of 20 ppm.22 The ions 242.1003+, 220.1179+, and 202.1063+ ranked as the first, second, and third most important variables, respectively (Table 3). A search of the MMCD suggested 242.1003+ and 220.1179+ might be sodiated pantothenic acid and free pantothenic acid, respectively. Ion 202.1063+ was thought to be a fragment of 220.1179+ (-H2O). Ion 204.1233+ corresponded to acetylcarnitine. No significant matches were found for ions 308.1831+ and 319.1658+ and empirical formula calculations using Seven Golden Rules 23 yielded no biologically meaningful hits. Other notable biomarkers included serum cholesterol (mean rank day 0 vs. day 7, day 0 vs. day 14; 26.2, 15.1), free carnitine (28.2, 11.8), isovalerylcarnitine (37.3, 6.8), and isobutyrylcarnitine (911, 29.6).
The identities of pantothenic acid and acetylcarnitine were confirmed by comparison of retention time and tandem mass spectra from authentic compounds and the urinary constituent. Tandem MS of authentic pantothenic acid generated the following m/z fragments (numbers in parenthesis indicate relative percent): 220.1161+ (79.7), 202.1089+ (43.2), 184.0979+ (31.1), 142.0884+ (9.5), 124.0786+ (25.7), 90.0567+ (100), and 85.0679+ (9.5). Tandem MS of putative urinary pantothenic acid generated the following m/z fragments: 220.1185+ (78.4), 202.1125+ (38.5), 184.0981+ (27.0), 142.0946+ (4.1), 124.0786+ (23.0), 90.0580+ (100), and 84.0703+ (9.5). Tandem MS of authentic acetylcarnitine generated the following m/z fragments: 204.1115+ (22.1), 145.0479+ (11.8), and 85.0274+ (100). Tandem MS of putative urinary acetylcarnitine generated the following m/z fragments: 204.1135+ (39.7), 145.0487+ (16.2), and 85.0273+ (100).
Quantitation of Urinary Constituents in Human Volunteers
Leaving the metabolomics platform, the top biomarker identified by random forests, pantothenic acid, was quantitated using an ABI triple-quadrupole mass spectrometer which offers greater dynamic range as compared to ESI-QTOFMS. For each volunteer, a significant depletion of pantothenic acid from urine, when compared to day 0, was recorded on day 7 (p<0.001) and on day 14 (p<0.001) (Figure 2A). On average, there was a 5.5-fold depletion of urinary pantothenic acid on day 7 and on day 14 when compared with day 0. Volunteer 6 excreted greater quantities of urinary pantothenic acid on all days and upon using Grubb's test for outlier detection was considered a significant outlier on Day 14 (p<0.05) consistent with the random forests analysis (Figure 1B).
Figure 2.
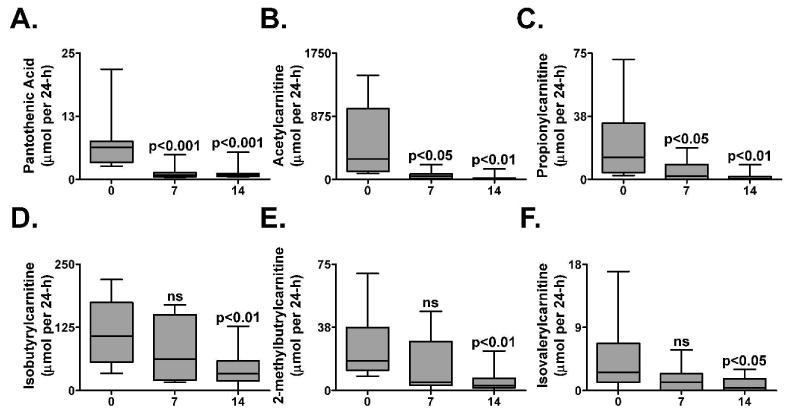
Quantitation of urinary pantothenic acid and acylcarnitines by comparison with standard curves of authentic compounds and serum clinical biochemistry. Results were adjusted by 24-h urine volumes and expressed as μmol per 24 h. (A) pantothenic acid (B) acetylcarnitine (C) propionylcarnitine (D) isobutyrylcarnitine (E) 2-methylbutyrylcarnitine and (F) isovalerylcarnitine. P-values were calculated using repeated measures ANOVA with Bonferroni's test for Multiple Comparisons. P-values represent comparison with day 0. Error bars represent standard error of the mean (SEM). ns, not significant.
Urinary acylcarnitines were best resolved by UPLC. Acetylcarnitine was identified as the second most important biomarker by random forests analysis (after excluding the in source fragments and sodium adducts of pantothenic acid). Urinary acetylcarnitine was significantly depleted on day 7 (p<0.05) and on day 14 (p<0.001) (Figure 2B). On day 7 there was a 8.1-fold depletion and on day 14 there was a 20.5-fold depletion compared to day 0. The 24-h excretion of other urinary acylcarnitines, including propionyl-, isobutyryl-, (S)-(+)-2-methylbutyryl-, and isovalerylcarnitine, was also measured (Figure 2C-F). Propionylcarnitine was found to be significantly depleted on day 7 (p<0.05) and all acylcarnitines were significantly depleted by day 14 (propionylcarnitine, p<0.01; isobutyrylcarnitine, p<0.01; (S)-(+)-2-methylbutyrylcarnitine, p<0.01; isovalerylcarnitine, p<0.05). Interestingly, the quantities of all measured acylcarnitines in the urine of Volunteer 9 increased, as opposed to the observed decreasing trend, on day 7 and on day 14 (data not shown). Using Grubb's test for outlier detection, Volunteer 9 was identified as a significant outlier on Day 14 (p<0.05).
Levels of fenofibric acid, the PPARα ligand from the pro-drug fenofibrate, in the urine on day 0, day 7 and day 14 were measured to verify each volunteer's compliance and to assess each volunteer's capacity to metabolize fenofibrate to the PPARα ligand, fenofibric acid (Figure 3). Each volunteer was 100% compliant on days 7 and 14 and no significant differences were observed with fenofibric acid levels.
Figure 3.
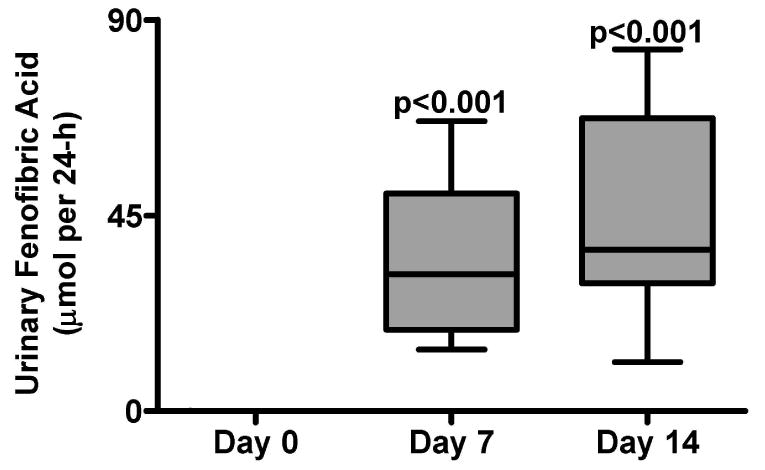
Fenofibric acid quantitation in urine from day 0, day 7, and day 14 in 10 human volunteers. Urine values were adjusted by the 24 h volume and are expressed as μmol per 24 h. P-values were calculated using repeated measures ANOVA with Bonferroni's test for Multiple Comparisons. P-values represent comparison with day 0. Error bars represent SEM.
Animal Studies
In order to verify that both pantothenic acid and acetylcarnitine, the top depleted human biomarkers after 14 days of fenofibrate treatment, were indeed specific to PPARα activation, male wild-type and Ppara-null mice were fed a diet containing 0.1% fenofibrate for 7 days and their urine was collected for 24 h both prior to and after 7 days on the diet. Pantothenic acid and acetylcarnitine were quantitated as above. Similar to the human volunteers taking oral fenofibrate, the wild-type mice on the 0.1% fenofibrate diet exhibited a significant and dramatic depletion of urinary pantothenic acid (40-fold less after 7 days, p<0.05, Figure 4A) and acetylcarnitine (88-fold less after 7 days, p<0.001, Figure 4C). However, the Ppara-null mice did not exhibit any appreciable change in either pantothenic acid or acetylcarnitine (Figure 4B and 4D, p>0.05) urine levels.
Figure 4.
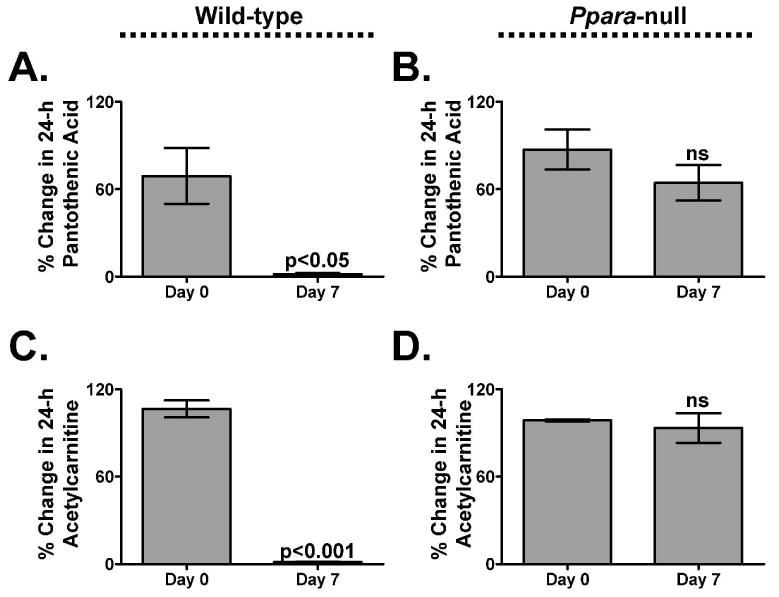
Percent change of urinary pantothenic acid (A-B) and acetylcarnitine (C-D) in male wild-type (n=3) and Ppara-null (n=3) mice. P-values were calculated using an unpaired t-test. Error bars represent SEM. ns, not significant.
Discussion
In this report, human urinary biomarkers of PPARα activation were identified using a metabolomics approach consisting of UPLC-ESI-QTOFMS profiling of the urine samples and random forests analysis. The inclusion of clinical variables into the data matrix proved to be useful as metabolomic-independent internal controls since serum uric acid has been reported to be depleted after fenofibrate treatment and was found to be highly ranked amongst the top urinary biomarkers in this study.24 Total cholesterol levels were also reduced although they were not ranked as highly (mean rank of 26.2 when comparing day 0 and day 7, mean rank of 15.1 when comparing day 0 and day 14) compared to other urinary biomarkers identified by metabolomic analysis. These observations alone strongly support the utility of metabolomics for drug efficacy studies and underscore this technique's potential for the identification of novel, non-traditional, biochemical indicators of drug effect.
Biomarker Rationalization
Activated in the presence of agonists such as fenofibrate, PPARα heterodimerizes with RXR, binds peroxisome proliferator response elements, and activates, among others, genes essential for β-oxidation of fatty acids in peroxisomes and in mitochondria.25 Among these genes, the pantothenate kinase gene, PANK1, as well as an array of genes involved in carnitine transport and synthesis, such as the carnitine palmitoyltransferase genes, CPT1 and CPT2, are strongly and specifically induced in the presence of the ligand-bound PPARα/RXR heterodimer.26, 27 Pantothenate kinase phosphorylates pantothenic acid, an essential B vitamin, converting it to 4′-phosphopantothenate in the initial step towards CoA synthesis. Therefore the dramatic depletion of urinary pantothenic acid and acetylcarnitine clearly reflect an increase in β-oxidation as surplus pools of pantothenic acid and short-chain acylcarnitines are redirected from the urinary excretory pathway to the mitochondria. Additionally, the depletion of these metabolites points to a unique biochemical step (e.g., pantothenic acid and CoA synthesis) as opposed to the depletion of traditional, clinical indicators such as total cholesterol and triglycerides where only less specific biochemical pathway information can be gleaned.
The observation that short- and branched-chain acylcarnitines were depleted after fenofibrate treatment points to a specific induction of hepatic mitochondrial short/branched chain specific acyl-CoA dehydrogenase (SBCAD; EC 1.3.99.3) that is responsible for the catabolism of leucine, isoleucine and valine, together with propionate and β-alanine. The human gene ACADSB encoding SBCAD has not previously been demonstrated to be a PPARα target gene, although the corresponding gene in the mouse appears to be upregulated by PPARα.26 Thus, treatment of patients with drugs that can activate PPARα might be expected to cause an increased catabolism of the branched-chain amino acids, β-alanine, and propionate.
Previous studies in rodents (Table 1) identified nicotinamide, nicotinamide 1-oxide, 1-methylnicotinamide, 1-methyl-2-pyridone-5-carboxamide (2PY), 1-methyl-4-pyridone-3-carboxamide (4PY), 11β,20-dihydroxy-3-oxopregn-4-en-21-oic acid (DHOPA), and 11β-hydroxy-3,20-dioxopregn-4-en-21-oic acid (HDOPA) as potential markers of PPARα activation. 13-16 However, in the clinical study reported here, none of those previously identified rodent markers were among the top highly important variables identified by random forests. Furthermore, none of the other rodent biomarkers was found to be altered by fenofibrate treatment of volunteers (data not shown). However, this study has clearly demonstrated through observations of fenofibrate associated changes in pantothenic acid and acetylcarnitine in both human and rodent models that biomarkers can transcend species.
Fenofibrate Pharmacometabolomics
Pharmacogenomics generally focuses on genetic influences on drug metabolism, efficacy, and toxicity. Similarly, pharmacometabolomics, or the use of small molecule profiling to predict and understand drug efficacy and toxicity, is a logical extension of pharmacogenomics but has the added advantage of being able to account for influences such as diet and environment.28 The power and sensitivity of pharmacometabolomics is exemplified in the metabolomic and clinical profiles of volunteers 6 and 9. There were no discernible differences in individual fenofibric acid excretion by any volunteer over the two week study (Figure 3) indicating 100% compliance, yet the MDS plot comparing day 0 and day 14 (Figure 1B) identified two potential outliers (volunteer 6 and 9) that were not apparent when comparing day 0 and day 7 (Figure 1A). While there was nothing abnormal regarding the clinical parameters (Table 2) for volunteer 6; total cholesterol concentrations in volunteer 9 increased slightly in concentration over the two week study (4.63 to 4.8 mmol/l) as opposed to the expected decrease in total cholesterol measured in the other 9 volunteers (mean ± standard deviation; 4.27 ± 0.25 to 3.64 ± 0.32 mmol/l). Interestingly, urinary acylcarnitine concentrations for volunteer 9 also increased over the 14-day study, yet pantothenic acid depleted, consistent with the other volunteers. These observations may suggest the presence of a subtle defect associated with β-oxidation. However, defects in pantothenic acid-dependent CoA synthesis are unlikely since pantothenic acid levels were depleted consistent with the other volunteers. Volunteer 6, while exhibiting a normal reduction of total cholesterol and triglycerides as well as acylcarnitines and pantothenic acid, showed higher quantities of urinary pantothenic acid (nearly 4-fold greater on day 0, day 7, and day 14) yet still exhibited the same trend for pantothenic acid depletion. This may reflect reduced enzyme activity of pantothenate kinase due to a polymorphic PANK1 gene or more simply a diet rich in pantothenic acid.
Conclusion
This study highlights the importance of metabolomics as a tool for uncovering metabolic phenotypes and for understanding drug efficacy. It also exemplifies the utility of metabolomics for understanding and/or predicting an individual's response to drug treatment. In combination with traditional diagnostics (e.g., clinical chemistry), the application of metabolomic profiling throughout the drug discovery and development process and extending into the clinic is likely to lead to improved pharmacotherapy by supporting individualized drug treatment. In this regard, determination of urinary pantothenic acid and short-chain acylcarnitines may prove useful monitors of PPARα activation by fibrate drugs.
Acknowledgments
This work was supported by a grant from the Czech Ministry of Education, VZ MSM0021620820 and in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. ADP is supported by a Pharmacology Research Associate in Training Fellowship from the National Institute of General Medical Sciences. JRI is grateful to the U.S. Smokeless Tobacco Company for a grant for collaborative research.
References
- 1.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O'Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58(4):726–41. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 2.Peters JM, Hennuyer N, Staels B, Fruchart JC, Fievet C, Gonzalez FJ, Auwerx J. Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor alpha-deficient mice. J Biol Chem. 1997;272(43):27307–12. doi: 10.1074/jbc.272.43.27307. [DOI] [PubMed] [Google Scholar]
- 3.Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, Auwerx J. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95(2):705–12. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kersten S, Mandard S, Escher P, Gonzalez FJ, Tafuri S, Desvergne B, Wahli W. The peroxisome proliferator-activated receptor alpha regulates amino acid metabolism. Faseb J. 2001;15(11):1971–8. doi: 10.1096/fj.01-0147com. [DOI] [PubMed] [Google Scholar]
- 5.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103(11):1489–98. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15(6):3012–22. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filippatos T, Milionis HJ. Treatment of hyperlipidaemia with fenofibrate and related fibrates. Expert Opin Investig Drugs. 2008;17(10):1599–614. doi: 10.1517/13543784.17.10.1599. [DOI] [PubMed] [Google Scholar]
- 8.Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, Herbert JM, Winegar DA, Willson TM, Fruchart JC, Berge RK, Staels B. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem. 2000;275(22):16638–42. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- 9.Keun HC, Athersuch TJ. Application of metabonomics in drug development. Pharmacogenomics. 2007;8(7):731–41. doi: 10.2217/14622416.8.7.731. [DOI] [PubMed] [Google Scholar]
- 10.Lindon JC, Holmes E, Nicholson JK. Metabonomics in pharmaceutical R&D. Febs J. 2007;274(5):1140–51. doi: 10.1111/j.1742-4658.2007.05673.x. [DOI] [PubMed] [Google Scholar]
- 11.Kaddurah-Daouk R, Kristal BS, Weinshilboum RM. Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol. 2008;48:653–83. doi: 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- 12.Enot DP, Lin W, Beckmann M, Parker D, Overy DP, Draper J. Preprocessing, classification modeling and feature selection using flow injection electrospray mass spectrometry metabolite fingerprint data. Nat Protoc. 2008;3(3):446–70. doi: 10.1038/nprot.2007.511. [DOI] [PubMed] [Google Scholar]
- 13.Zhen Y, Krausz KW, Chen C, Idle JR, Gonzalez FJ. Metabolomic and genetic analysis of biomarkers for peroxisome proliferator-activated receptor alpha expression and activation. Mol Endocrinol. 2007;21(9):2136–51. doi: 10.1210/me.2007-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delaney J, Hodson MP, Thakkar H, Connor SC, Sweatman BC, Kenny SP, McGill PJ, Holder JC, Hutton KA, Haselden JN, Waterfield CJ. Tryptophan-NAD+ pathway metabolites as putative biomarkers and predictors of peroxisome proliferation. Arch Toxicol. 2005;79(4):208–23. doi: 10.1007/s00204-004-0625-5. [DOI] [PubMed] [Google Scholar]
- 15.Connor SC, Hodson MP, Ringeissen S, Sweatman BC, McGill PJ, Waterfield CJ, Haselden JN. Development of a multivariate statistical model to predict peroxisome proliferation in the rat, based on urinary 1H-NMR spectral patterns. Biomarkers. 2004;9(45):364–85. doi: 10.1080/13547500400006005. [DOI] [PubMed] [Google Scholar]
- 16.Ringeissen S, Connor SC, Brown HR, Sweatman BC, Hodson MP, Kenny SP, Haworth RI, McGill P, Price MA, Aylott MC, Nunez DJ, Haselden JN, Waterfield CJ. Potential urinary and plasma biomarkers of peroxisome proliferation in the rat: identification of N-methylnicotinamide and N-methyl-4-pyridone-3-carboxamide by 1H nuclear magnetic resonance and high performance liquid chromatography. Biomarkers. 2003;8(34):240–71. doi: 10.1080/1354750031000149124. [DOI] [PubMed] [Google Scholar]
- 17.Patterson AD, Li H, Eichler GS, Krausz KW, Weinstein JN, Fornace AJ, Jr, Gonzalez FJ, Idle JR. UPLC-ESI-TOFMS-based metabolomics and gene expression dynamics inspector self-organizing metabolomic maps as tools for understanding the cellular response to ionizing radiation. Anal Chem. 2008;80(3):665–74. doi: 10.1021/ac701807v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyburski JB, Patterson AD, Krausz KW, Slavik J, Fornace AJ, Gonzalez FJ, Idle JR. Radiation metabolomics. 1. Identification of minimally invasive urine biomarkers for gamma-radiation exposure in mice. Radiat Res. 2008;170(1):1–14. doi: 10.1667/RR1265.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breiman L. Random Forests. Machine Learning. 2001;45(1):5–32. [Google Scholar]
- 20.Eichler GS, Reimers M, Kane D, Weinstein JN. The LeFE algorithm: embracing the complexity of gene expression in the interpretation of microarray data. Genome Biol. 2007;8(9):R187. doi: 10.1186/gb-2007-8-9-r187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enot DP, Beckmann M, Overy D, Draper J. Predicting interpretability of metabolome models based on behavior, putative identity, and biological relevance of explanatory signals. Proc Natl Acad Sci U S A. 2006;103(40):14865–70. doi: 10.1073/pnas.0605152103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Q, Lewis IA, Hegeman AD, Anderson ME, Li J, Schulte CF, Westler WM, Eghbalnia HR, Sussman MR, Markley JL. Metabolite identification via the Madison Metabolomics Consortium Database. Nat Biotechnol. 2008;26(2):162–4. doi: 10.1038/nbt0208-162. [DOI] [PubMed] [Google Scholar]
- 23.Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:105. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiortsis DN, Elisaf MS. Serum uric acid levels: a useful but not absolute marker of compliance with fenofibrate treatment. Fundam Clin Pharmacol. 2001;15(6):401–3. doi: 10.1046/j.1472-8206.2001.00049.x. [DOI] [PubMed] [Google Scholar]
- 25.Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51(2):85–94. doi: 10.1016/j.phrs.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 26.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61(4):393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramaswamy G, Karim MA, Murti KG, Jackowski S. PPARalpha controls the intracellular coenzyme A concentration via regulation of PANK1alpha gene expression. J Lipid Res. 2004;45(1):17–31. doi: 10.1194/jlr.M300279-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Ryals J, Lawton K, Stevens D, Milburn M, Metabolon, Inc. Pharmacogenomics. 2007;8(7):863–6. doi: 10.2217/14622416.8.7.863. [DOI] [PubMed] [Google Scholar]